
Enhanced Antitumor Activity by the Combination of Dasatinib and Selinexor in Chronic Myeloid Leukemia
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
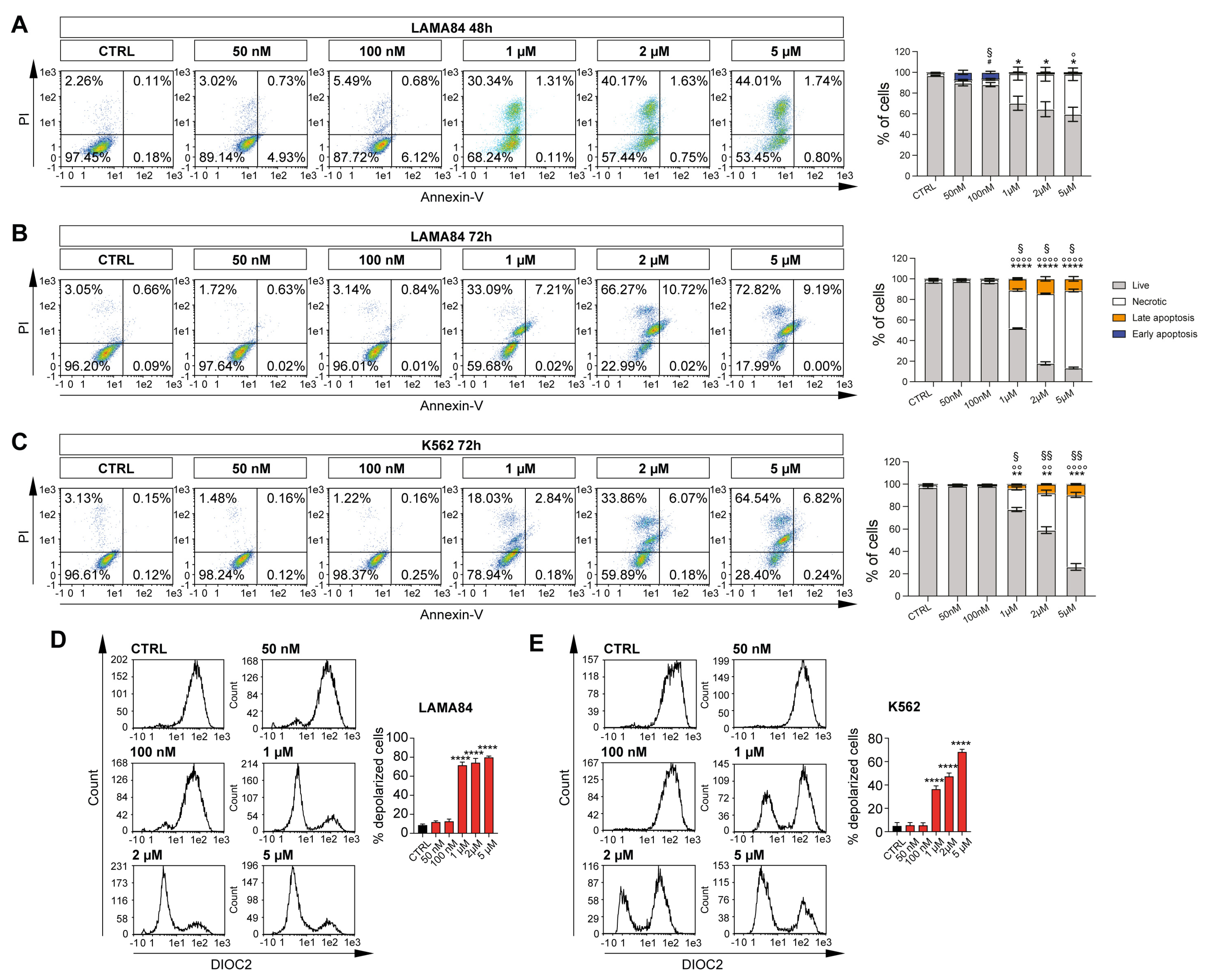
2.1. Selinexor Induces Apoptosis in CML Cell Lines by Mitochondrial Depolarization
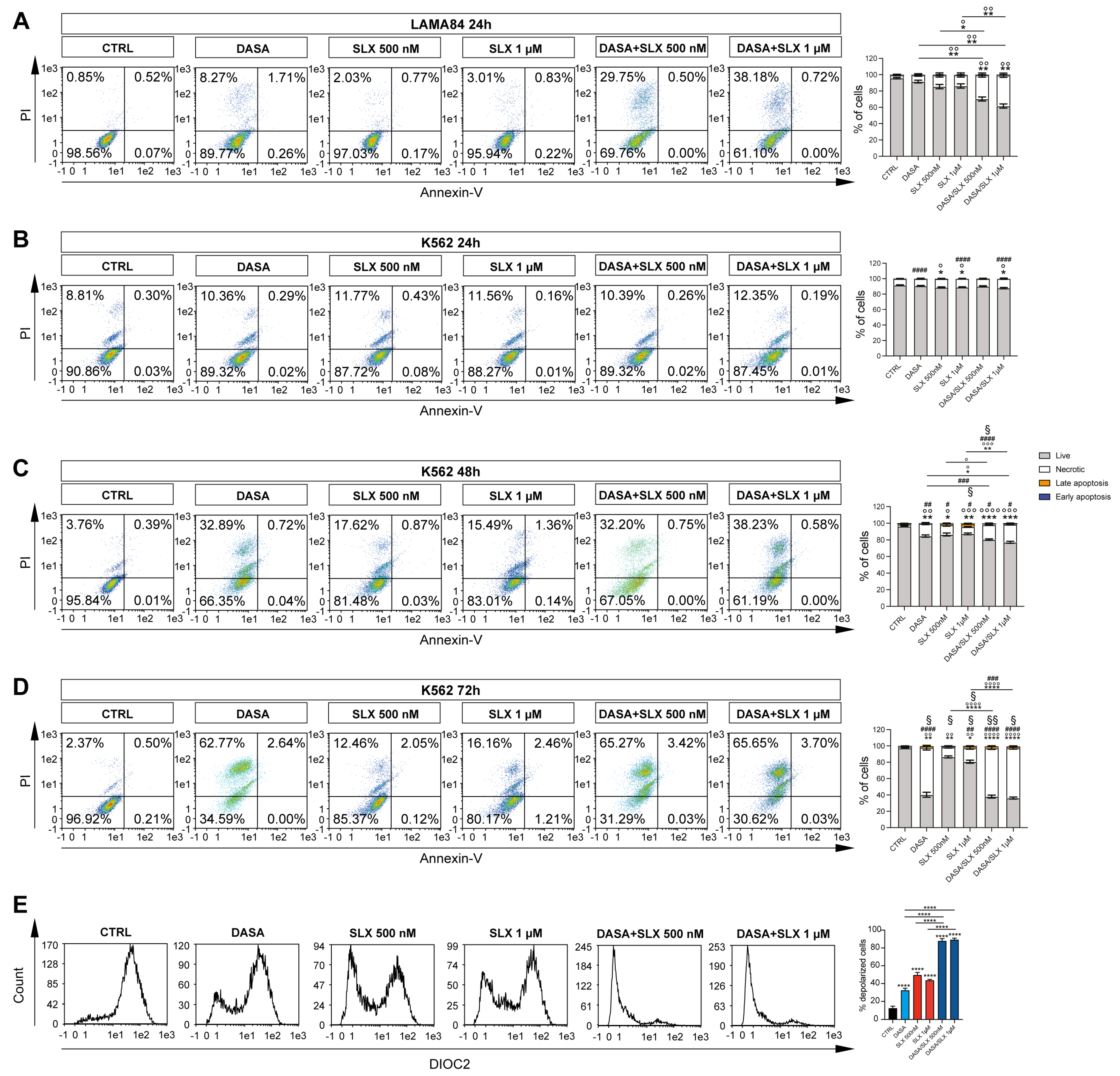
2.2. Selinexor Increases Cytotoxicity of Dasatinib in LAMA84 Cell Line
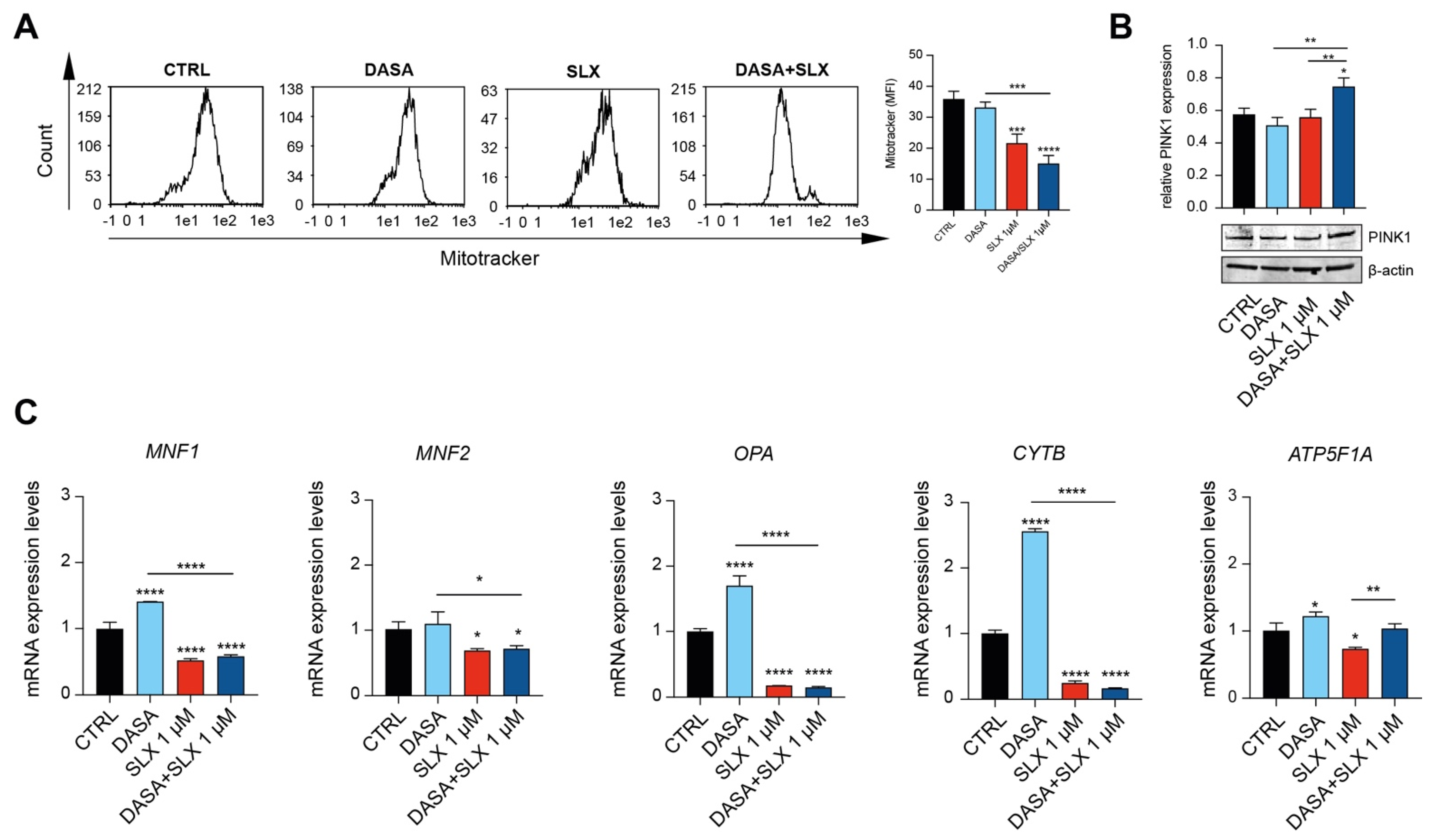
2.3. Dasatinib/Selinexor Combination Decreases CML Mitochondrial Efficiency
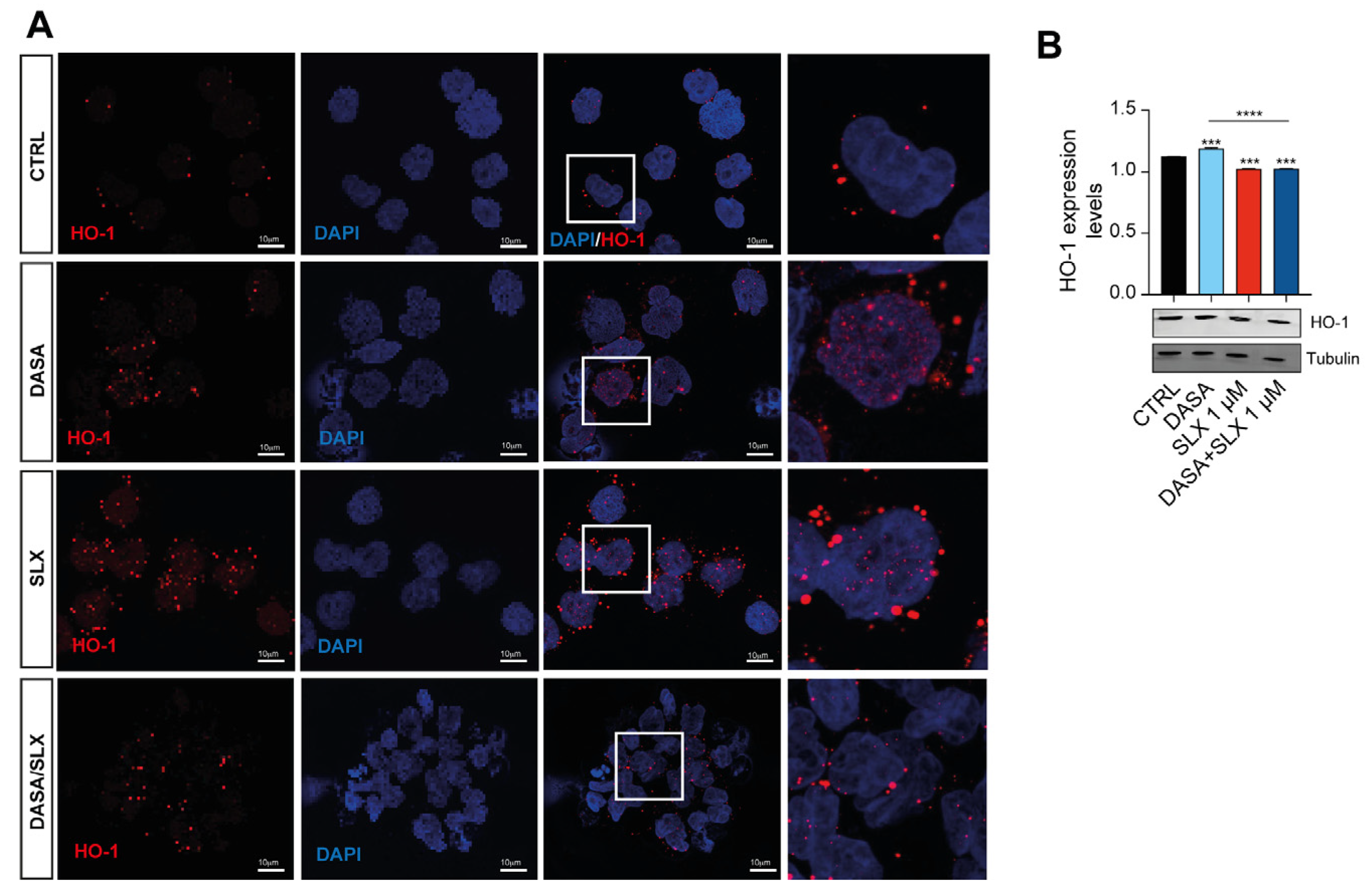
2.4. Dasatinib/Selinexor Combination Decreases HO-1 Nuclear Translocation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Flow Cytometry
4.3. Real-Time RT-PCR
4.4. Western Blot Analysis
4.5. Immunofluorescence
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giallongo, C.; Parrinello, N.L.; La Cava, P.; Camiolo, G.; Romano, A.; Scalia, M.; Stagno, F.; Palumbo, G.A.; Avola, R.; Li Volti, G.; et al. Monocytic myeloid-derived suppressor cells as prognostic factor in chronic myeloid leukaemia patients treated with dasatinib. J. Cell Mol. Med. 2018, 22, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.; Hochhaus, A.; Schultheis, B.; Hehlmann, R. Chronic myelogenous leukemia: Molecular and cellular aspects. J. Cancer Res. Clin. Oncol. 1998, 124, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Shah, N.P.; Hochhaus, A.; Cortes, J.; Shah, S.; Ayala, M.; Moiraghi, B.; Shen, Z.; Mayer, J.; Pasquini, R.; et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2010, 362, 2260–2270. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.; Goncalves, A.C.; Rutella, S.; Almeida, A.M.; De Las Rivas, J.; Trougakos, I.P.; Sarmento Ribeiro, A.B. Resistance to Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia-From Molecular Mechanisms to Clinical Relevance. Cancers 2021, 13, 4820. [Google Scholar] [CrossRef] [PubMed]
- Sawyers, C.L. Molecular consequences of the BCR-ABL translocation in chronic myelogenous leukemia. Leuk. Lymphoma 1993, 11 (Suppl. 2), 101–103. [Google Scholar] [CrossRef] [PubMed]
- Torres-Barrera, P.; Mayani, H.; Chavez-Gonzalez, A. Understanding the hematopoietic microenvironment in chronic myeloid leukemia: A concise review. Curr. Res. Transl. Med. 2021, 69, 103295. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.B.; Nemkov, T.; D’Alessandro, A.; Welner, R.S. Deciphering Metabolic Adaptability of Leukemic Stem Cells. Front. Oncol. 2022, 12, 846149. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef]
- Pianigiani, G.; Gagliardi, A.; Mezzasoma, F.; Rocchio, F.; Tini, V.; Bigerna, B.; Sportoletti, P.; Caruso, S.; Marra, A.; Peruzzi, S.; et al. Prolonged XPO1 inhibition is essential for optimal antileukemic activity in NPM1-mutated AML. Blood Adv. 2022, 6, 5938–5949. [Google Scholar] [CrossRef]
- Peterson, T.J.; Orozco, J.; Buege, M. Selinexor: A First-in-Class Nuclear Export Inhibitor for Management of Multiply Relapsed Multiple Myeloma. Ann. Pharmacother. 2020, 54, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Kanojia, D.; Mayakonda, A.; Said, J.W.; Doan, N.B.; Chien, W.; Ganesan, T.S.; Chuang, L.S.; Venkatachalam, N.; Baloglu, E.; et al. Molecular mechanism and therapeutic implications of selinexor (KPT-330) in liposarcoma. Oncotarget 2017, 8, 7521–7532. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, T.; Argueta, C.; Aboukameel, A.; Unger, T.J.; Klebanov, B.; Mohammad, R.M.; Muqbil, I.; Azmi, A.S.; Drolen, C.; Senapedis, W.; et al. Selinexor, a Selective Inhibitor of Nuclear Export (SINE) compound, acts through NF-kappaB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death. Oncotarget 2016, 7, 78883–78895. [Google Scholar] [CrossRef] [PubMed]
- Galinski, B.; Luxemburg, M.; Landesman, Y.; Pawel, B.; Johnson, K.J.; Master, S.R.; Freeman, K.W.; Loeb, D.M.; Hebert, J.M.; Weiser, D.A. XPO1 inhibition with selinexor synergizes with proteasome inhibition in neuroblastoma by targeting nuclear export of IkB. Transl. Oncol. 2021, 14, 101114. [Google Scholar] [CrossRef]
- Keskin, D.; Sadri, S.; Eskazan, A.E. Dasatinib for the treatment of chronic myeloid leukemia: Patient selection and special considerations. Drug Des. Devel Ther. 2016, 10, 3355–3361. [Google Scholar] [CrossRef]
- Scandura, G.; Giallongo, C.; Puglisi, F.; Romano, A.; Parrinello, N.L.; Zuppelli, T.; Longhitano, L.; Giallongo, S.; Di Rosa, M.; Musumeci, G.; et al. TLR4 Signaling and Heme Oxygenase-1/Carbon Monoxide Pathway Crosstalk Induces Resiliency of Myeloma Plasma Cells to Bortezomib Treatment. Antioxidants 2022, 11, 767. [Google Scholar] [CrossRef]
- Tibullo, D.; Barbagallo, I.; Giallongo, C.; La Cava, P.; Parrinello, N.; Vanella, L.; Stagno, F.; Palumbo, G.A.; Li Volti, G.; Di Raimondo, F. Nuclear translocation of heme oxygenase-1 confers resistance to imatinib in chronic myeloid leukemia cells. Curr. Pharm. Des. 2013, 19, 2765–2770. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gutierrez, V.; Hernandez-Boluda, J.C. Tyrosine Kinase Inhibitors Available for Chronic Myeloid Leukemia: Efficacy and Safety. Front. Oncol. 2019, 9, 603. [Google Scholar] [CrossRef] [PubMed]
- Panina, S.B.; Pei, J.; Kirienko, N.V. Mitochondrial metabolism as a target for acute myeloid leukemia treatment. Cancer Metab. 2021, 9, 17. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- de Beauchamp, L.; Himonas, E.; Helgason, G.V. Mitochondrial metabolism as a potential therapeutic target in myeloid leukaemia. Leukemia 2022, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Kashton, C.; Lichtensztejn, D.; Baloglu, E.; Senapedis, W.; Shacham, S.; Kauffman, M.G.; Kotb, R.; Mai, S. XPO1 Inhibition Preferentially Disrupts the 3D Nuclear Organization of Telomeres in Tumor Cells. J. Cell. Physiol. 2016, 231, 2711–2719. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target. Ther. 2022, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Green, A.; Hossain, T.; Eckmann, D.M. Mitochondrial dynamics involves molecular and mechanical events in motility, fusion and fission. Front. Cell Dev. Biol. 2022, 10, 1010232. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Fathi, M.; Gholizadeh Navashenaq, J.; Mohammadi, H.; Yousefi, M.; Hojjat-Farsangi, M.; Namdar, A.; Movasaghpour Akbari, A.A.; Jadidi-Niaragh, F. The prognostic and therapeutic potential of HO-1 in leukemia and MDS. Cell Commun. Signal 2023, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Carota, G.; Distefano, A.; Spampinato, M.; Giallongo, C.; Broggi, G.; Longhitano, L.; Palumbo, G.A.; Parenti, R.; Caltabiano, R.; Giallongo, S.; et al. Neuroprotective Role of alpha-Lipoic Acid in Iron-Overload-Mediated Toxicity and Inflammation in In Vitro and In Vivo Models. Antioxidants 2022, 11, 1596. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, S.; Rehakova, D.; Biagini, T.; Lo Re, O.; Raina, P.; Lochmanova, G.; Zdrahal, Z.; Resnick, I.; Pata, P.; Pata, I.; et al. Histone Variant macroH2A1.1 Enhances Nonhomologous End Joining-dependent DNA Double-strand-break Repair and Reprogramming Efficiency of Human iPSCs. Stem Cells 2022, 40, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, S.; Lo Re, O.; Lochmanova, G.; Parca, L.; Petrizzelli, F.; Zdrahal, Z.; Mazza, T.; Vinciguerra, M. Phosphorylation within Intrinsic Disordered Region Discriminates Histone Variant macroH2A1 Splicing Isoforms-macroH2A1.1 and macroH2A1.2. Biology 2021, 10, 659. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LAMA84 | K562 |
|---|---|
| Amelogenin: X | Amelogenin: X |
| CSF1P0: 11, 12 | CSF1P0: 9, 10 |
| D13S317: 11, 12 | D13S317: 8 |
| D16S539: 11 | D16S539: 11, 12 |
| D5S818: 11, 12 | D5S818: 11, 12 |
| D7S820: 11 | D7S820: 9, 11 |
| THO1: 6, 7 | THO1: 9.3 |
| TPOX: 10, 11 | TPOX: 8, 9 |
| vWA: 14, 17 | vWA: 16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spampinato, M.; Zuppelli, T.; Dulcamare, I.; Longhitano, L.; Sambataro, D.; Santisi, A.; Alanazi, A.M.; Barbagallo, I.A.; Vicario, N.; Parenti, R.; et al. Enhanced Antitumor Activity by the Combination of Dasatinib and Selinexor in Chronic Myeloid Leukemia. Pharmaceuticals 2024, 17, 894. https://doi.org/10.3390/ph17070894
Spampinato M, Zuppelli T, Dulcamare I, Longhitano L, Sambataro D, Santisi A, Alanazi AM, Barbagallo IA, Vicario N, Parenti R, et al. Enhanced Antitumor Activity by the Combination of Dasatinib and Selinexor in Chronic Myeloid Leukemia. Pharmaceuticals. 2024; 17(7):894. https://doi.org/10.3390/ph17070894
Chicago/Turabian StyleSpampinato, Mariarita, Tatiana Zuppelli, Ilaria Dulcamare, Lucia Longhitano, Domenico Sambataro, Annalisa Santisi, Amer M. Alanazi, Ignazio A. Barbagallo, Nunzio Vicario, Rosalba Parenti, and et al. 2024. "Enhanced Antitumor Activity by the Combination of Dasatinib and Selinexor in Chronic Myeloid Leukemia" Pharmaceuticals 17, no. 7: 894. https://doi.org/10.3390/ph17070894
APA StyleSpampinato, M., Zuppelli, T., Dulcamare, I., Longhitano, L., Sambataro, D., Santisi, A., Alanazi, A. M., Barbagallo, I. A., Vicario, N., Parenti, R., Romano, A., Musumeci, G., Li Volti, G., Palumbo, G. A., Di Raimondo, F., Nicolosi, A., Giallongo, S., & Del Fabro, V. (2024). Enhanced Antitumor Activity by the Combination of Dasatinib and Selinexor in Chronic Myeloid Leukemia. Pharmaceuticals, 17(7), 894. https://doi.org/10.3390/ph17070894