
Kynurenines and Inflammation: A Remarkable Axis for Multiple Sclerosis Treatment


 , , , and
, , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Epidemiology
3. Clinical Features
4. Diagnosis
5. Treatment
6. Cellular Dysfunctions Induced by MS
6.1. Loss of Myelin Sheet on Axons
6.2. Macrophage Responses
6.3. Induction of Inflammatory Cytokines
7. Kynurenines
7.1. Tryptophan Metabolism
7.2. Dysregulated Generation of Kynurenines
7.3. Effects of the Metabolites of Kynurenine on Brain Cells
7.4. Activation of Intracellular Pathways by Inflammatory Process
7.5. Kynurenine’s Receptors on Inflammatory Cells
7.6. T-Reg Responses to Kynurenine Signaling
8. Pharmacological Modulation of Kynurenine-Inflammation Axis
Drugs Repositioning for Multiple Sclerosis Treatment
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Olek, M.J. Multiple Sclerosis. Ann. Intern. Med. 2021, 174, ITC81–ITC96. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple sclerosis. A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef]
- Feigin, V.L.; Vos, T.; Nichols, E.; O Owolabi, M.; Carroll, W.M.; Dichgans, M.; Deuschl, G.; Parmar, P.; Brainin, M.; Murray, C. The global burden of neurological disorders: Translating evidence into policy. Lancet Neurol. 2020, 19, 255–265. [Google Scholar] [CrossRef]
- Haki, M.; Al-Biati, H.A.; Al-Tameemi, Z.S.; Ali, I.S.; Al-Hussaniy, H.A. Review of multiple sclerosis: Epidemiology, etiology, pathophysiology, and treatment. Medicine 2024, 103, e37297. [Google Scholar] [CrossRef]
- Yang, J.H.; Rempe, T.; Whitmire, N.; Dunn-Pirio, A.; Graves, J.S. Therapeutic Advances in Multiple Sclerosis. Front. Neurol. 2022, 13, 824926. [Google Scholar] [CrossRef]
- Bou Rjeily, N.; Mowry, E.M.; Ontaneda, D.; Carlson, A.K. Highly Effective Therapy vs. Escalation Approaches in Early Multiple Sclerosis: What Is the Future of Multiple Sclerosis Treatment? Neurol. Clin. 2024, 42, 185–201. [Google Scholar] [CrossRef]
- Fathi, M.; Vakili, K.; Yaghoobpoor, S.; Tavasol, A.; Jazi, K.; Hajibeygi, R.; Shool, S.; Sodeifian, F.; Klegeris, A.; McElhinney, A.; et al. Dynamic changes in metabolites of the kynurenine pathway in Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: A systematic Review and meta-analysis. Front. Immunol. 2022, 13, 997240. [Google Scholar] [CrossRef]
- Fathi, M.; Vakili, K.; Yaghoobpoor, S.; Tavasol, A.; Jazi, K.; Mohamadkhani, A.; Klegeris, A.; McElhinney, A.; Mafi, Z.; Hajiesmaeili, M.; et al. Dynamic changes in kynurenine pathway metabolites in multiple sclerosis: A systematic review. Front. Immunol. 2022, 13, 1013784. [Google Scholar] [CrossRef]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; Van Der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef]
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef]
- Koch-Henriksen, N.; Magyari, M. Apparent changes in the epidemiology and severity of multiple sclerosis. Nat. Rev. Neurol. 2021, 17, 676–688. [Google Scholar] [CrossRef]
- Langer-Gould, A.M.; Gonzales, E.G.; Smith, J.B.; Li, B.H.; Nelson, L.M. Racial and Ethnic Disparities in Multiple Sclerosis Preva-lence. Neurology 2022, 98, e1818–e1827. [Google Scholar] [CrossRef] [PubMed]
- Yamout, B.; Sahraian, M.; Bohlega, S.; Al-Jumah, M.; Goueider, R.; Dahdaleh, M.; Inshasi, J.; Hashem, S.; Alsharoqi, I.; Khoury, S.; et al. Consensus recommendations for the diagnosis and treatment of multiple sclerosis: 2019 revisions to the MENACTRIMS guidelines. Mult. Scler. Relat. Disord. 2020, 37, 101459. [Google Scholar] [CrossRef]
- Pozzilli, C.; Pugliatti, M.; Vermersch, P.; Grigoriadis, N.; Alkhawajah, M.; Airas, L.; Oreja-Guevara, C. Diagnosis and treatment of progressive multiple sclerosis: A position paper. Eur. J. Neurol. 2023, 30, 9–21. [Google Scholar] [CrossRef]
- Mey, G.M.; Mahajan, K.R.; DeSilva, T.M. Neurodegeneration in multiple sclerosis. WIREs Mech. Dis. 2023, 15, e1583. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Solomon, A.J.; Arrambide, G.; Brownlee, W.J.; Flanagan, E.P.; Amato, M.P.; Amezcua, L.; Banwell, B.L.; Barkhof, F.; Corboy, J.R.; Correale, J.; et al. Differential diagnosis of suspected multiple sclerosis: An updated consensus approach. Lancet Neurol. 2023, 22, 750–768. [Google Scholar] [CrossRef]
- Montalban, X.; Gold, R.; Thompson, A.J.; Otero-Romero, S.; Amato, M.P.; Chandraratna, D.; Clanet, M.; Comi, G.; Derfuss, T.; Fazekas, F.; et al. ECTRIMS/EAN Guideline on the phar-macological treatment of people with multiple sclerosis. Mult. Scler. 2018, 24, 96–120. [Google Scholar] [CrossRef]
- Samjoo, I.A.; Worthington, E.; Drudge, C.; Zhao, M.; Cameron, C.; Häring, D.A.; Stoneman, D.; Klotz, L.; Adlard, N. Efficacy classification of modern therapies in multiple sclerosis. J. Comp. Eff. Res. 2021, 10, 495–507. [Google Scholar] [CrossRef]
- Baldassari, L.E.; Fox, R.J. Therapeutic Advances and Challenges in the Treatment of Progressive Multiple Sclerosis. Drugs 2018, 78, 1549–1566. [Google Scholar] [CrossRef]
- Yong, H.Y.F.; Yong, V.W. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat. Rev. Neurol. 2022, 18, 40–55. [Google Scholar] [CrossRef]
- Butt, A.M.; Dinsdale, J. Fibroblast growth factor 2 induces loss of adult oligodendrocytes and myelin in vivo. Exp. Neurol. 2005, 192, 125–133. [Google Scholar] [CrossRef]
- Butt, A.M.; Dinsdale, J. Fibroblast growth factor 2 mediated disruption of myelin-forming oligodendrocytes in vivo is associated with increased tau immunoreactivity. Neurosci. Lett. 2005, 375, 28–32. [Google Scholar] [CrossRef]
- Liu, R.; Du, S.; Zhao, L.; Jain, S.; Sahay, K.; Rizvanov, A.; Lezhnyova, V.; Khaibullin, T.; Martynova, E.; Khaiboullina, S.; et al. Autoreactive lymphocytes in multiple sclerosis: Pathogenesis and treatment target. Front. Immunol. 2022, 13, 996469. [Google Scholar] [CrossRef]
- Prineas, J.W.; Kwon, E.E.; Cho, E.S.; Sharer, L.R.; Barnett, M.H.; Oleszak, E.L.; Hoffman, B.; Morgan, B.P. Immuno-pathology of secondary-progressive multiple sclerosis. Ann. Neurol. 2001, 50, 646–657. [Google Scholar] [CrossRef]
- Lovelace, M.D.; Varney, B.; Sundaram, G.; Franco, N.F.; Ng, M.L.; Pai, S.; Lim, C.K.; Guillemin, G.J.; Brew, B.J. Current Evidence for a Role of the Kynurenine Pathway of Tryptophan Metabolism in Multiple Sclerosis. Front. Immunol. 2016, 7, 246. [Google Scholar] [CrossRef]
- Gaitán, M.I.; Shea, C.D.; Evangelou, I.E.; Stone, R.D.; Fenton, K.M.; Bielekova, B.; Massacesi, L.; Reich, D.S. Evolution of the blood–brain barrier in newly forming multiple sclerosis lesions. Ann. Neurol. 2011, 70, 22–29. [Google Scholar] [CrossRef]
- Fox, E.J. Immunopathology of multiple sclerosis. Neurology 2004, 63, S3–S7. [Google Scholar] [CrossRef]
- Chiarugi, A.; Cozzi, A.; Ballerini, C.; Massacesi, L.; Moroni, F. Kynurenine 3-mono-oxygenase activity and neurotoxic kynurenine metabolites increase in the spinal cord of rats with experimental allergic encephalomyelitis. Neuroscience 2001, 102, 687–695. [Google Scholar] [CrossRef]
- Mancuso, R.; Hernis, A.; Agostini, S.; Rovaris, M.; Caputo, D.; Fuchs, D.; Clerici, M. Indoleamine 2,3 Dioxygenase (IDO) Expression and Activity in Relapsing- Remitting Multiple Sclerosis. PLoS ONE 2015, 10, e0130715. [Google Scholar] [CrossRef]
- Rajda, C.; Galla, Z.; Polyák, H.; Maróti, Z.; Babarczy, K.; Pukoli, D.; Vécsei, L. Cerebrospinal Fluid Neurofilament Light Chain Is Associated with Kynurenine Pathway Metabolite Changes in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 2665. [Google Scholar] [CrossRef] [PubMed]
- Kwidzinski, E.; Bunse, J.; Aktas, O.; Richter, D.; Mutlu, L.; Zipp, F.; Nitsch, R.; Bechmann, I. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J. 2005, 19, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.M.; Schwarz, M.J.; Steinbusch, H.W.; Leonard, B.E. Neuropsychiatric disorders related to interferon and interleukins treatment. Metab. Brain Dis. 2009, 24, 55–68. [Google Scholar] [CrossRef]
- Correale, J.; Villa, A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. Ann. Neurol. 2010, 67, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Kreft, K.L.; Verbraak, E.; Wierenga-Wolf, A.F.; Laman, J.D.; Hintzen, R.Q. Role of CD8 regulatory T-cells in multiple sclerosis. Ann. Neurol. 2011, 69, 593. [Google Scholar] [CrossRef]
- Kallaur, A.P.; Lopes, J.; Oliveira, S.R.; Simão, A.N.; Reiche, E.M.; de Almeida, E.R.D.; Morimoto, H.K.; de Pereira, W.L.; Alfieri, D.F.; Borelli, S.D.; et al. Immune-Inflammatory and Oxidative and Nitrosative Stress Biomarkers of Depression Symptoms in Subjects with Multiple Sclerosis: Increased Peripheral Inflammation but Less Acute Neuroinflammation. Mol. Neurobiol. 2016, 53, 5191–5202. [Google Scholar] [CrossRef]
- Amirkhani, A.; Rajda, C.; Arvidsson, B.; Bencsik, K.; Boda, K.; Seres, E.; Markides, K.E.; Vécsei, L.; Bergquist, J. Interferon-beta affects the tryptophan metabolism in multiple sclerosis patients. Eur. J. Neurol. 2005, 12, 625–631. [Google Scholar] [CrossRef]
- Joisten, N.; Rademacher, A.; Bloch, W.; Schenk, A.; Oberste, M.; Dalgas, U.; Langdon, D.; Caminada, D.; Purde, M.-T.; Gonzenbach, R.; et al. Influence of different rehabilitative aerobic exercise programs on (anti-) inflammatory immune signalling, cognitive and functional capacity in persons with MS—Study protocol of a randomized controlled trial. BMC Neurol. 2019, 19, 37. [Google Scholar] [CrossRef]
- Müller, N. Neuroprogression in Schizophrenia and Psychotic Disorders: The Possible Role of Inflammation. Mod. Trends Pharmacopsychiatry 2017, 31, 1–9. [Google Scholar]
- Maes, M.; Leonard, B.E.; Myint, A.M.; Kubera, M.; Verkerk, R. The new ‘5-HT’ hypothesis of depression: Cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 702–721. [Google Scholar] [CrossRef]
- Morris, G.; Reiche, E.M.V.; Murru, A.; Carvalho, A.F.; Maes, M.; Berk, M.; Puri, B.K. Multiple Immune-Inflammatory and Oxidative and Nitrosative Stress Pathways Explain the Frequent Presence of Depression in Multiple Sclerosis. Mol. Neurobiol. 2018, 55, 6282–6306. [Google Scholar] [CrossRef]
- Oxenkrug, G.F. Genetic and hormonal regulation of tryptophan kynurenine metabolism: Implications for vascular cognitive impairment, major depressive disorder, and aging. Ann. N. Y. Acad. Sci. 2007, 1122, 35–49. [Google Scholar] [CrossRef]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Ormstad, H.; Simonsen, C.S.; Broch, L.; Maes, D.M.; Anderson, G.; Celius, E.G. Chronic fatigue and depression due to multiple sclerosis: Immune-inflammatory pathways, tryptophan catabolites and the gut-brain axis as possible shared pathways. Mult. Scler. Relat. Disord. 2020, 46, 102533. [Google Scholar] [CrossRef]
- Hurley, L.L.; Tizabi, Y. Neuroinflammation, neurodegeneration, and depression. Neurotox. Res. 2013, 23, 131–144. [Google Scholar] [CrossRef]
- Maes, M.; Rief, W. Diagnostic classifications in depression and somatization should include biomarkers, such as disorders in the tryptophan catabolite (TRYCAT) pathway. Psychiatry Res. 2012, 196, 243–249. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M. Myalgic encephalomyelitis/chronic fatigue syndrome and encephalomyelitis disseminata/multiple sclerosis show remarkable levels of similarity in phenomenology and neuroimmune characteristics. BMC Med. 2013, 11, 205. [Google Scholar] [CrossRef]
- Wichers, M.C.; Koek, G.H.; Robaeys, G.; Verkerk, R.; Scharpé, S.; Maes, M. IDO and interferon-α-induced depressive symptoms: A shift in hypothesis from tryptophan depletion to neurotoxicity. Mol. Psychiatry 2005, 10, 538–544. [Google Scholar] [CrossRef]
- Tan, L.S.Y.; Francis, H.M.; Lim, C.K. Exploring the roles of tryptophan metabolism in MS beyond neuroinflammation and neurodegeneration: A paradigm shift to neuropsychiatric symptoms. Brain Behav. Immun. Health 2021, 12, 100201. [Google Scholar] [CrossRef]
- Dang, H.; Castro-Portuguez, R.; Espejo, L.; Backer, G.; Freitas, S.; Spence, E.; Meyers, J.; Shuck, K.; Gardea, E.A.; Chang, L.M.; et al. On the benefits of the tryptophan metabolite 3-hydroxyanthranilic acid in Caenorhabditis elegans and mouse aging. Nat. Commun. 2023, 14, 8338. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef]
- Sathyasaikumar, K.V.; Pérez de la Cruz, V.; Pineda, B.; Vázquez Cervantes, G.I.; Ramírez Ortega, D.; Donley, D.W.; Severson, P.L.; West, B.L.; Giorgini, F.; Fox, J.H.; et al. Cellular Localization of Kynurenine 3-Monooxygenase in the Brain: Challenging the Dogma. Antioxidants 2022, 11, 315. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Smith, D.G.; Kerr, S.J.; Smythe, G.A.; Kapoor, V.; Armati, P.J.; Brew, B.J. Characterisation of kynurenine pathway metabolism in human astrocytes and implications in neuropathogenesis. Redox Rep. 2000, 5, 108–111. [Google Scholar] [CrossRef]
- Kanai, M.; Funakoshi, H.; Takahashi, H.; Hayakawa, T.; Mizuno, S.; Matsumoto, K.; Nakamura, T. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol. Brain 2009, 2, 8. [Google Scholar] [CrossRef]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794. [Google Scholar] [CrossRef]
- Trombetti, S.; Sessa, R.; Catapano, R.; Rinaldi, L.; Lo Bianco, A.; Feliciello, A.; Izzo, P.; Grosso, M. Exploring the Leukemogenic Potential of GATA-1(S), the Shorter Isoform of GATA-1: Novel Insights into Mechanisms Hampering Respiratory Chain Complex II Activity and Limiting Oxidative Phosphorylation Efficiency. Antioxidants 2021, 10, 1603. [Google Scholar] [CrossRef]
- Badawy, A.A.; Guillemin, G. The Plasma [Kynurenine]/[Tryptophan] Ratio and Indoleamine 2,3-Dioxygenase: Time for Appraisal. Int. J. Tryptophan Res. 2019, 12, 1178646919868978. [Google Scholar] [CrossRef]
- Orhan, F.; Schwieler, L.; Engberg, G.; Samuelsson, M. Kynurenine Metabolites in CSF and Plasma in Healthy Males. Int. J. Tryptophan Res. 2024, 17, 11786469241245323. [Google Scholar] [CrossRef]
- Kita, T.; Morrison, P.F.; Heyes, M.P.; Markey, S.P. Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the l-kynurenine and quinolinic acid pools in brain. J. Neurochem. 2002, 82, 258–268. [Google Scholar] [CrossRef]
- Biernacki, T.; Sandi, D.; Bencsik, K.; Vécsei, L. Kynurenines in the Pathogenesis of Multiple Sclerosis: Therapeutic Perspectives. Cells 2020, 9, 1564. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Carrillo-Mora, P.; Pedraza-Chaverrí, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzón, E.; Ortiz-Islas, E.; et al. On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine metabolism in multiple sclerosis. Acta Neurol. Scand. 2005, 112, 93–96. [Google Scholar] [CrossRef]
- Tavares, R.G.; Tasca, C.I.; Santos, C.E.; Alves, L.B.; Porciúncula, L.O.; Emanuelli, T.; Souza, D.O. Quinolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes. Neurochem. Int. 2002, 40, 621–627. [Google Scholar] [CrossRef]
- Perkins, M.N.; Stone, T.W. Quinolinic acid: Regional variations in neuronal sensitivity. Brain Res. 1983, 259, 172–176. [Google Scholar] [CrossRef]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef]
- Aarsland, T.I.M.; Instanes, J.T.; Posserud, M.-B.R.; Ulvik, A.; Kessler, U.; Haavik, J. Changes in Tryptophan-Kynurenine Metabolism in Patients with Depression Undergoing ECT—A Systematic Review. Pharmaceuticals 2022, 15, 1439. [Google Scholar] [CrossRef]
- Németh, H.; Toldi, J.; Vécsei, L. Role of kynurenines in the central and peripheral nervous systems. Curr. Neurovasc. Res. 2005, 2, 249–260. [Google Scholar]
- Wences Chirino, T.; Rangel López, E.; Luna Angulo, A.; Carrillo Mora, P.; Landa Solis, C.; Samudio Cruz, M.A.; Fuentes Bello, A.C.; Paniagua Pérez, R.; Ríos Martínez, J.; Sánchez Chapul, L. Crosstalk between Exercise-Derived Endocannabinoidome and Kynurenines: Potential Target Therapies for Obesity and Depression Symptoms. Pharmaceuticals 2023, 16, 1421. [Google Scholar] [CrossRef]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef]
- Lim, C.K.; Bilgin, A.; Lovejoy, D.B.; Tan, V.; Bustamante, S.; Taylor, B.V.; Bessede, A.; Brew, B.J.; Guillemin, G.J. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci. Rep. 2017, 7, 41473. [Google Scholar] [CrossRef]
- Lovelace, M.D.; Varney, B.; Sundaram, G.; Lennon, M.J.; Lim, C.K.; Jacobs, K.; Guillemin, G.J.; Brew, B.J. Recent evi-dence for an expanded role of the kynurenine pathway of tryptophan metabolism in neurological diseases. Neuropharmacology 2017, 112, 373–388. [Google Scholar] [CrossRef]
- Sakurai, K.; Zou, J.-P.; Tschetter, J.R.; Ward, J.M.; Shearer, G.M. Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002, 129, 186–196. [Google Scholar] [CrossRef]
- Pukoli, D.; Polyák, H.; Rajda, C.; Vécsei, L. Kynurenines and Neurofilament Light Chain in Multiple Sclerosis. Front. Neurosci. 2021, 15, 658202. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Adamczyk-Sowa, M.; Gajewska, A.; Bartosz, G. Oxidative modification of blood serum proteins in multiple sclerosis after interferon or mitoxantrone treatment. J. Neuroimmunol. 2014, 266, 67–74. [Google Scholar] [CrossRef]
- Rejdak, K.; Bartosik-Psujek, H.; Dobosz, B.; Kocki, T.; Grieb, P.; Giovannoni, G.; Turski, W.A.; Stelmasiak, Z. Decreased level of kynurenic acid in cerebrospinal fluid of relapsing-onset multiple sclerosis patients. Neurosci. Lett. 2002, 331, 63–65. [Google Scholar] [CrossRef]
- Kepplinger, B.; Baran, H.; Kainz, A.; Ferraz-Leite, H.; Newcombe, J.; Kalina, P. Age-related increase of kynurenic acid in human cerebrospinal fluid—IgG and beta2-microglobulin changes. Neurosignals 2005, 14, 126–135. [Google Scholar] [CrossRef]
- Rejdak, K.; Petzold, A.; Kocki, T.; Kurzepa, J.; Grieb, P.; Turski, W.A.; Stelmasiak, Z. Astrocytic activation in relation to inflammatory markers during clinical exacerbation of relapsing-remitting multiple sclerosis. J. Neural Transm. 2007, 114, 1011–1015. [Google Scholar] [CrossRef]
- Aeinehband, S.; Brenner, P.; Ståhl, S.; Bhat, M.; Fidock, M.D.; Khademi, M.; Olsson, T.; Engberg, G.; Jokinen, J.; Erhardt, S.; et al. Cerebrospinal fluid kynurenines in multiple sclerosis; relation to disease course and neurocognitive symp-toms. Brain Behav. Immun. 2016, 51, 47–55. [Google Scholar] [CrossRef]
- Petty, M.A.; Lo, E.H. Junctional complexes of the blood-brain barrier: Permeability changes in neuroinflammation. Prog. Neurobiol. 2002, 68, 311–323. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv. Exp. Med. Biol. 2003, 527, 105–112. [Google Scholar]
- Hughes, T.D.; Güner, O.F.; Iradukunda, E.C.; Phillips, R.S.; Bowen, J.P. The Kynurenine Pathway and Kynurenine 3-Monooxygenase Inhibitors. Molecules 2022, 27, 273. [Google Scholar] [CrossRef]
- Suzuki, H.; Ohgidani, M.; Kuwano, N.; Chrétien, F.; Lorin de la Grandmaison, G.; Onaya, M.; Tominaga, I.; Setoyama, D.; Kang, D.; Mimura, M.; et al. Suicide and Microglia: Recent Findings and Future Perspectives Based on Human Studies. Front. Cell. Neurosci. 2019, 13, 31. [Google Scholar] [CrossRef]
- Newton, A.; McCann, L.; Huo, L.; Liu, A. Kynurenine Pathway Regulation at Its Critical Junctions with Fluctuation of Tryptophan. Metabolites 2023, 13, 500. [Google Scholar] [CrossRef]
- Chiarugi, A.; Dello Sbarba, P.; Paccagnini, A.; Donnini, S.; Filippi, S.; Moroni, F. Combined inhibition of indoleamine 2,3-dioxygenase and nitric oxide synthase modulates neurotoxin release by interferon-gamma-activated macrophages. J. Leukoc. Biol. 2000, 68, 260–266. [Google Scholar] [CrossRef]
- Buchanan, J.L.; Rauckhorst, A.J.; Taylor, E.B. 3-hydroxykynurenine is a ROS-inducing cytotoxic tryptophan metabolite that disrupts the TCA cycle. bioRxiv 2023. [Google Scholar]
- Vazquez, S.; Garner, B.; Sheil, M.M.; Truscott, R.J. Characterisation of the major autoxidation products of 3-hydroxykynurenine under physiological conditions. Free Radic. Res. 2000, 32, 11–23. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Zhu, D.; Tan, K.S.; Zhang, X.; Sun, A.Y.; Sun, G.Y.; Lee, J.C. Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J. Cell Sci. 2005, 118, 3695–3703. [Google Scholar] [CrossRef]
- Feng, W.; Wang, Y.; Liu, Z.Q.; Zhang, X.; Han, R.; Miao, Y.Z.; Qin, Z.H. Microglia activation contributes to quinolinic ac-id-induced neuronal excitotoxicity through TNF-α. Apoptosis 2017, 22, 696–709. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Hestad, K.; Alexander, J.; Rootwelt, H.; Aaseth, J.O. The Role of Tryptophan Dysmetabolism and Quinolinic Acid in Depressive and Neurodegenerative Diseases. Biomolecules 2022, 12, 998. [Google Scholar] [CrossRef]
- Khaitin, A. Calcium in Neuronal and Glial Response to Axotomy. Int. J. Mol. Sci. 2021, 22, 13344. [Google Scholar] [CrossRef]
- Hidalgo, C.; Núñez, M.T. Calcium, iron and neuronal function. IUBMB Life 2007, 59, 280–285. [Google Scholar] [CrossRef]
- Kułak, W.; Sobaniec, W. Molecular mechanisms of brain plasticity: Neurophysiologic and neuroimaging studies in the developing patients. Rocz. Akad. Med. Bialymst. 2004, 49, 227–236. [Google Scholar]
- Braidy, N.; Grant, R.; Adams, S.; Brew, B.J.; Guillemin, G.J. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox. Res. 2009, 16, 77–86. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de La Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxidative Med. Cell Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Raven, E.L. A short history of heme dioxygenases: Rise, fall and rise again. J. Biol. Inorg. Chem. 2017, 22, 175–183. [Google Scholar] [CrossRef]
- Dai, X.; Zhu, B.T. Indoleamine 2,3-dioxygenase tissue distribution and cellular localization in mice: Implications for its biological functions. J. Histochem. Cytochem. 2010, 58, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Widner, B.; Ledochowski, M.; Fuchs, D. Interferon-gamma-induced tryptophan degradation: Neuropsychiatric and immunological consequences. Curr. Drug Metab. 2000, 1, 193–204. [Google Scholar] [CrossRef]
- Dang, Y.; Dale, W.E.; Brown, O.R. Comparative effects of oxygen on indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase of the kynurenine pathway. Free Radic. Biol. Med. 2000, 28, 615–624. [Google Scholar] [CrossRef]
- Robinson, A.P.; Harp, C.T.; Noronha, A.; Miller, S.D. The experimental autoimmune encephalomyelitis (EAE) model of MS: Utility for understanding disease pathophysiology and treatment. Handb. Clin. Neurol. 2014, 122, 173–189. [Google Scholar] [PubMed]
- Depboylu, C.; Reinhart, T.A.; Takikawa, O.; Imai, Y.; Maeda, H.; Mitsuya, H.; Rausch, D.; Eiden, L.E.; Weihe, E. Brain virus burden and indoleamine-2,3-dioxygenase expression during lentiviral infection of rhesus monkey are concomitantly lowered by 6-chloro-2’,3’-dideoxyguanosine. Eur. J. Neurosci. 2004, 19, 2997–3005. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Kerr, S.J.; Pemberton, L.A.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. IFN-beta1b induces kynurenine pathway metabolism in human macrophages: Potential implications for multiple sclerosis treatment. J. Interferon Cytokine Res. 2001, 21, 1097–1101. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2005, 49, 15–23. [Google Scholar] [CrossRef]
- Pamart, G.; Gosset, P.; Le Rouzic, O.; Pichavant, M.; Poulain-Godefroy, O. Kynurenine Pathway in Respiratory Diseases. Int. J. Tryptophan Res. 2024, 17, 11786469241232871. [Google Scholar] [CrossRef] [PubMed]
- Bach, E.A.; Aguet, M.; Schreiber, R.D. The IFN gamma receptor: A paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 1997, 15, 563–591. [Google Scholar] [CrossRef]
- Yadav, M.C.; Burudi, E.M.; Alirezaei, M.; Flynn, C.C.; Watry, D.D.; Lanigan, C.M.; Fox, H.S. IFN-gamma-induced IDO and WRS expression in microglia is differentially regulated by IL-4. Glia 2007, 55, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Rock, R.B.; Hu, S.; Deshpande, A.; Munir, S.; May, B.J.; Baker, C.A.; Peterson, P.K.; Kapur, V. Transcriptional response of human microglial cells to interferon-gamma. Genes Immun. 2005, 6, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Brockie, S.; Zhou, C.; Fehlings, M.G. Resident immune responses to spinal cord injury: Role of astrocytes and microglia. Neural Regen. Res. 2024, 19, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Lefauconnier, J.-M.; Trouvé, R. Developmental changes in the pattern of amino acid transport at the blood-brain barrier in rats. Dev. Brain Res. 1983, 6, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Cascio, L.; Chen, C.; Pauly, R.; Srikanth, S.; Jones, K.; Skinner, C.D.; Stevenson, R.E.; Schwartz, C.E.; Boccuto, L. Abnormalities in the genes that encode Large Amino Acid Transporters increase the risk of Autism Spectrum Disorder. Mol. Genet. Genom. Med. 2020, 8, e1036. [Google Scholar] [CrossRef] [PubMed]
- Taslimifar, M.; Faltys, M.; Kurtcuoglu, V.; Verrey, F.; Makrides, V. Analysis of L-leucine amino acid transporter species activity and gene expression by human blood brain barrier hCMEC/D3 model reveal potential LAT1, LAT4, B(0)AT2 and y(+)LAT1 functional cooperation. J. Cereb. Blood Flow Metab. 2022, 42, 90–103. [Google Scholar] [CrossRef]
- Duelli, R.; Enerson, B.E.; Gerhart, D.Z.; Drewes, L.R. Expression of large amino acid transporter LAT1 in rat brain endothelium. J Cereb. Blood Flow Metab. 2000, 20, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, A.; Kang, Y.-S. Pretreatment Effect of Inflammatory Stimuli and Characteristics of Tryptophan Transport on Brain Capillary Endothelial (TR-BBB) and Motor Neuron Like (NSC-34) Cell Lines. Biomedicines 2020, 9, 9. [Google Scholar] [CrossRef]
- Jones, S.P.; Franco, N.F.; Varney, B.; Sundaram, G.; Brown, D.A.; de Bie, J.; Lim, C.K.; Guillemin, G.J.; Brew, B.J. Expression of the Kynurenine Pathway in Human Peripheral Blood Mononuclear Cells: Implications for Inflammatory and Neurodegenerative Disease. PLoS ONE 2015, 10, e0131389. [Google Scholar] [CrossRef]
- Sinclair, L.V.; Neyens, D.; Ramsay, G.; Taylor, P.M.; Cantrell, D.A. Single cell analysis of kynurenine and System L amino acid transport in T cells. Nat. Commun. 2018, 9, 1981. [Google Scholar] [CrossRef]
- Riaz, F.; Wei, P.; Pan, F. Fine-tuning of regulatory T cells is indispensable for the metabolic steatosis-related hepatocellular carcinoma: A review. Front. Cell Dev. Biol. 2022, 10, 949603. [Google Scholar] [CrossRef]
- Belladonna, M.L.; Orabona, C.; Grohmann, U.; Puccetti, P. TGF-beta and kynurenines as the key to infectious tolerance. Trends Mol. Med. 2009, 15, 41–49. [Google Scholar] [CrossRef]
- Walczak, K.; Wnorowski, A.; Turski, W.A.; Plech, T. Kynurenic acid and cancer: Facts and controversies. Cell Mol. Life Sci. 2020, 77, 1531–1550. [Google Scholar] [CrossRef]
- Wirthgen, E.; Hoeflich, A.; Rebl, A.; Günther, J. Kynurenic Acid: The Janus-Faced Role of an Immunomodulatory Tryptophan Metabolite and Its Link to Pathological Conditions. Front. Immunol. 2017, 8, 1957. [Google Scholar] [CrossRef]
- Mándi, Y.; Vécsei, L. The kynurenine system and immunoregulation. J. Neural Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef]
- Seo, S.-K.; Kwon, B. Immune regulation through tryptophan metabolism. Exp. Mol. Med. 2023, 55, 1371–1379. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Correale, J. Immunosuppressive Amino-Acid Catabolizing Enzymes in Multiple Sclerosis. Front. Immunol. 2020, 11, 600428. [Google Scholar] [CrossRef]
- Pires, A.S.; Sundaram, G.; Heng, B.; Krishnamurthy, S.; Brew, B.J.; Guillemin, G.J. Recent advances in clinical trials targeting the kynurenine pathway. Pharmacol. Ther. 2022, 236, 108055. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, G.X.; Gran, B.; Fallarino, F.; Yu, S.; Li, H.; Cullimore, M.L.; Rostami, A.; Xu, H. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J. Immunol. 2010, 185, 5953–5961. [Google Scholar] [CrossRef]
- Bo, L.; Guojun, T.; Li, G. An Expanded Neuroimmunomodulation Axis: sCD83-Indoleamine 2,3-Dioxygenase-Kynurenine Pathway and Updates of Kynurenine Pathway in Neurologic Diseases. Front. Immunol. 2018, 9, 1363. [Google Scholar] [CrossRef]
- Lemos, H.; Mohamed, E.; Ou, R.; McCardle, C.; Zheng, X.; McGuire, K.; Homer, N.Z.M.; Mole, D.J.; Huang, L.; Mellor, A.L. Co-treatments to Boost IDO Activity and Inhibit Production of Downstream Catabolites Induce Durable Suppression of Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2020, 11, 1256. [Google Scholar] [CrossRef]
- Sundaram, G.; Lim, C.K.; Brew, B.J.; Guillemin, G.J. Kynurenine pathway modulation reverses the experimental auto-immune encephalomyelitis mouse disease progression. J. Neuroinflamm. 2020, 17, 176. [Google Scholar] [CrossRef]
- Álvarez-Sánchez, N.; Cruz-Chamorro, I.; López-González, A.; Utrilla, J.C.; Fernández-Santos, J.M.; Martínez-López, A.; Lardone, P.J.; Guerrero, J.M.; Carrillo-Vico, A. Melatonin controls experimental autoimmune encephalomyelitis by altering the T effector/regulatory balance. Brain Behav. Immun. 2015, 50, 101–114. [Google Scholar] [CrossRef]
- Escribano, B.M.; Muñoz-Jurado, A.; Caballero-Villarraso, J.; Valdelvira, M.E.; Giraldo, A.I.; Paz-Rojas, E.; Gascón, F.; Santamaría, A.; Agüera, E.; Túnez, I. Protective effects of melatonin on changes occurring in the experimental autoimmune encephalomyelitis model of multiple sclerosis. Mult. Scler. Relat. Disord. 2022, 58, 103520. [Google Scholar] [CrossRef]
- Razmaray, H.; Nasiri, E.; Vakilipour, P.; Morsali, S.; Moradi, A.; Ebrahimian, A.; Rashidi, S.; Mosaddeghi-Heris, R.; Sadigh-Eteghad, S.; Naseri, A. The effects of melatonin supplementation on neurobehavioral outcomes and clinical severity in rodent models of multiple sclerosis; a systematic review and meta-analysis. Inflammopharmacology 2024, 32, 927–944. [Google Scholar] [CrossRef]
- Jand, Y.; Ghahremani, M.H.; Ghanbari, A.; Ejtemaei-Mehr, S.; Guillemin, G.J.; Ghazi-Khansari, M. Melatonin ameliorates disease severity in a mouse model of multiple sclerosis by modulating the kynurenine pathway. Sci. Rep. 2022, 12, 15963. [Google Scholar] [CrossRef]
- Sundaram, G.; Bessede, A.; Gilot, D.; Staats Pires, A.; Sherman, L.S.; Brew, B.J.; Guillemin, G.J. Prophylactic and Therapeutic Effect of Kynurenine for Experimental Autoimmune Encephalomyelitis (EAE) Disease. Int. J. Tryptophan Res. 2022, 15, 11786469221118657. [Google Scholar] [CrossRef]
- Aharoni, R.; Saada, R.; Eilam, R.; Hayardeny, L.; Sela, M.; Arnon, R. Oral treatment with laquinimod augments regulatory T-cells and brain-derived neurotrophic factor expression and reduces injury in the CNS of mice with experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2012, 251, 14–24. [Google Scholar] [CrossRef]
- Comi, G.; Dadon, Y.; Sasson, N.; Steinerman, J.R.; Knappertz, V.; Vollmer, T.L.; Boyko, A.; Vermersch, P.; Ziemssen, T.; Montalban, X.; et al. Concerto: A randomized, placebo-controlled trial of oral laquinimod in relapsing-remitting multiple sclerosis. Mult. Scler. 2022, 28, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Cunniffe, N.; Vuong, K.A.; Ainslie, D.; Baker, D.; Beveridge, J.; Bickley, S.; Camilleri, P.; Craner, M.; Fitzgerald, D.; de la Fuente, A.G.; et al. Systematic approach to selecting licensed drugs for repurposing in the treatment of progressive multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 295–302. [Google Scholar] [CrossRef]
- Tóth, F.; Cseh, E.K.; Vécsei, L. Natural Molecules and Neuroprotection: Kynurenic Acid, Pantethine and α-Lipoic Acid. Int. J. Mol. Sci. 2021, 22, 403. [Google Scholar] [CrossRef]
- Castro-Portuguez, R.; Sutphin, G.L. Kynurenine pathway, NAD(+) synthesis, and mitochondrial function: Targeting tryp-tophan metabolism to promote longevity and healthspan. Exp. Gerontol. 2020, 132, 110841. [Google Scholar] [CrossRef]
- Muzik, O.; Burghardt, P.; Yi, Z.; Kumar, A.; Seyoum, B. Successful metformin treatment of insulin resistance is associated with down-regulation of the kynurenine pathway. Biochem. Biophys. Res. Commun. 2017, 488, 29–32. [Google Scholar] [CrossRef]
- Vesterinen, H.M.; Connick, P.; Irvine, C.M.; Sena, E.S.; Egan, K.J.; Carmichael, G.G.; Tariq, A.; Pavitt, S.; Chataway, J.; Macleod, M.R.; et al. Drug repurposing: A systematic approach to evaluate candidate oral neuroprotective interventions for secondary progressive multiple sclerosis. PLoS ONE 2015, 10, e0117705. [Google Scholar] [CrossRef]
- Fois, A.G.; Sotgiu, E.; Scano, V.; Negri, S.; Mellino, S.; Zinellu, E.; Pirina, P.; Pintus, G.; Carru, C.; Mangoni, A.A.; et al. Effects of Pirfenidone and Nintedanib on Markers of Systemic Oxidative Stress and Inflammation in Patients with Idiopathic Pulmonary Fibrosis: A Preliminary Report. Antioxidants 2020, 9, 1064. [Google Scholar] [CrossRef]
- Yang, Z.; Li, Z.; Guo, Z.; Ren, Y.; Zhou, T.; Xiao, Z.; Duan, J.; Han, C.; Cheng, Y.; Xu, F. Antitumor Effect of Fluoxetine on Chronic Stress-Promoted Lung Cancer Growth via Suppressing Kynurenine Pathway and Enhancing Cellular Immunity. Front. Pharmacol. 2021, 12, 685898. [Google Scholar] [CrossRef]
- Shevtsov, A.; Raevskiy, M.; Stupnikov, A.; Medvedeva, Y. In Silico Drug Repurposing in Multiple Sclerosis Using scRNA-Seq Data. Int. J. Mol. Sci. 2023, 24, 985. [Google Scholar] [CrossRef]
- Yin, X.; Rang, X.; Hong, X.; Zhou, Y.; Xu, C.; Fu, J. Immune cells transcriptome-based drug repositioning for multiple sclerosis. Front. Immunol. 2022, 13, 1020721. [Google Scholar] [CrossRef]
- Gouasmi, R.; Ferraro-Peyret, C.; Nancey, S.; Coste, I.; Renno, T.; Chaveroux, C.; Aznar, N.; Ansieau, S. The Kynurenine Pathway and Cancer: Why Keep It Simple When You Can Make It Complicated. Cancers 2022, 14, 2793. [Google Scholar] [CrossRef]
- Fakan, B.; Szalardy, L.; Vecsei, L. Exploiting the Therapeutic Potential of Endogenous Immunomodulatory Systems in Multiple Sclerosis—Special Focus on the Peroxisome Proliferator-Activated Receptors (PPARs) and the Kynurenines. Int. J. Mol. Sci. 2019, 20, 426. [Google Scholar] [CrossRef] [PubMed]
- Venters, H.D.; Dantzer, R.; Kelley, K.W. A new concept in neurodegeneration: TNFalpha is a silencer of survival signals. Trends Neurosci. 2000, 23, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Donia, S.A.; Allison, D.J.; Gammage, K.L.; Ditor, D.S. The effects of acute aerobic exercise on mood and inflammation in individuals with multiple sclerosis and incomplete spinal cord injury. NeuroRehabilitation 2019, 45, 117–124. [Google Scholar] [CrossRef]
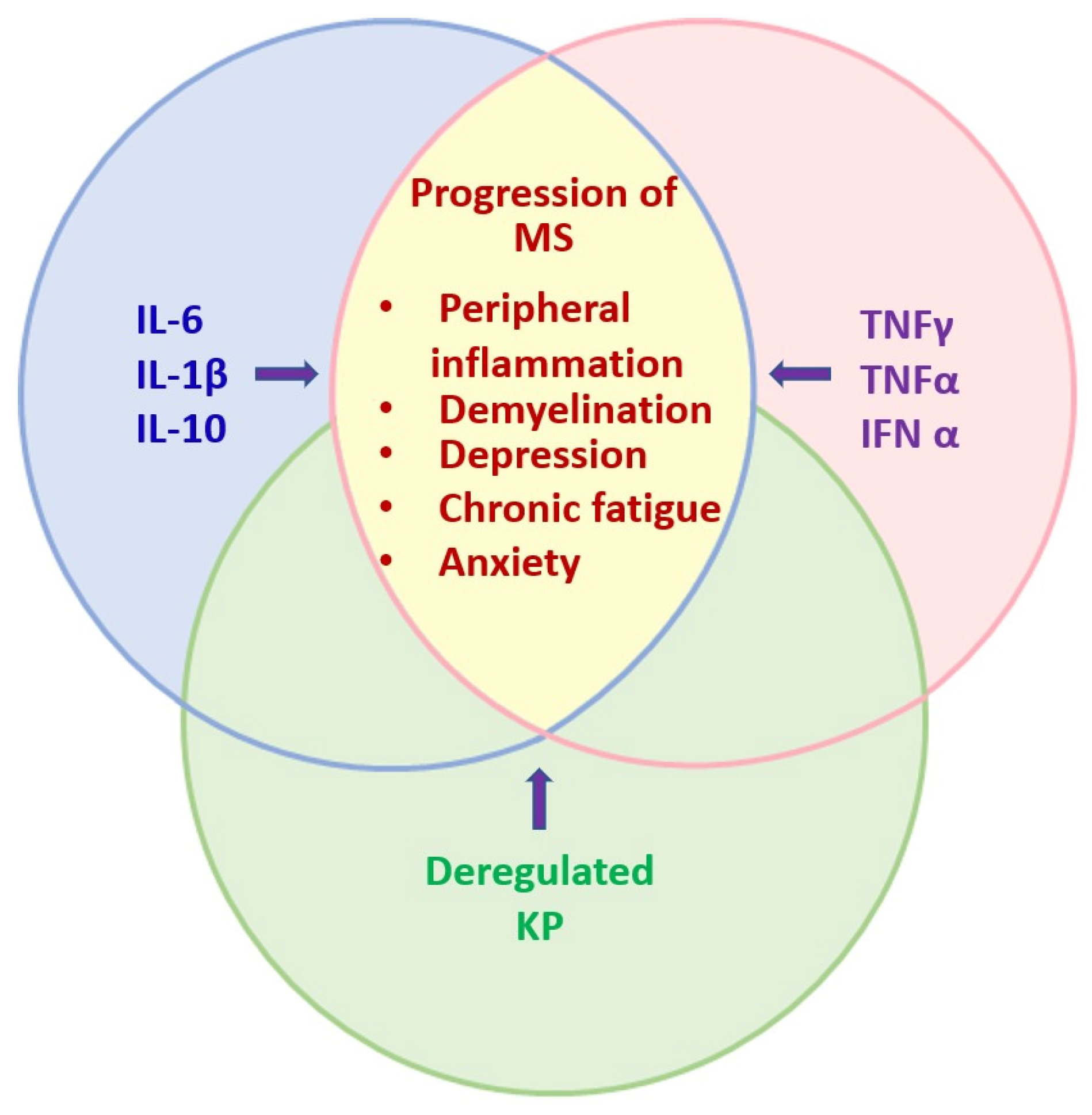
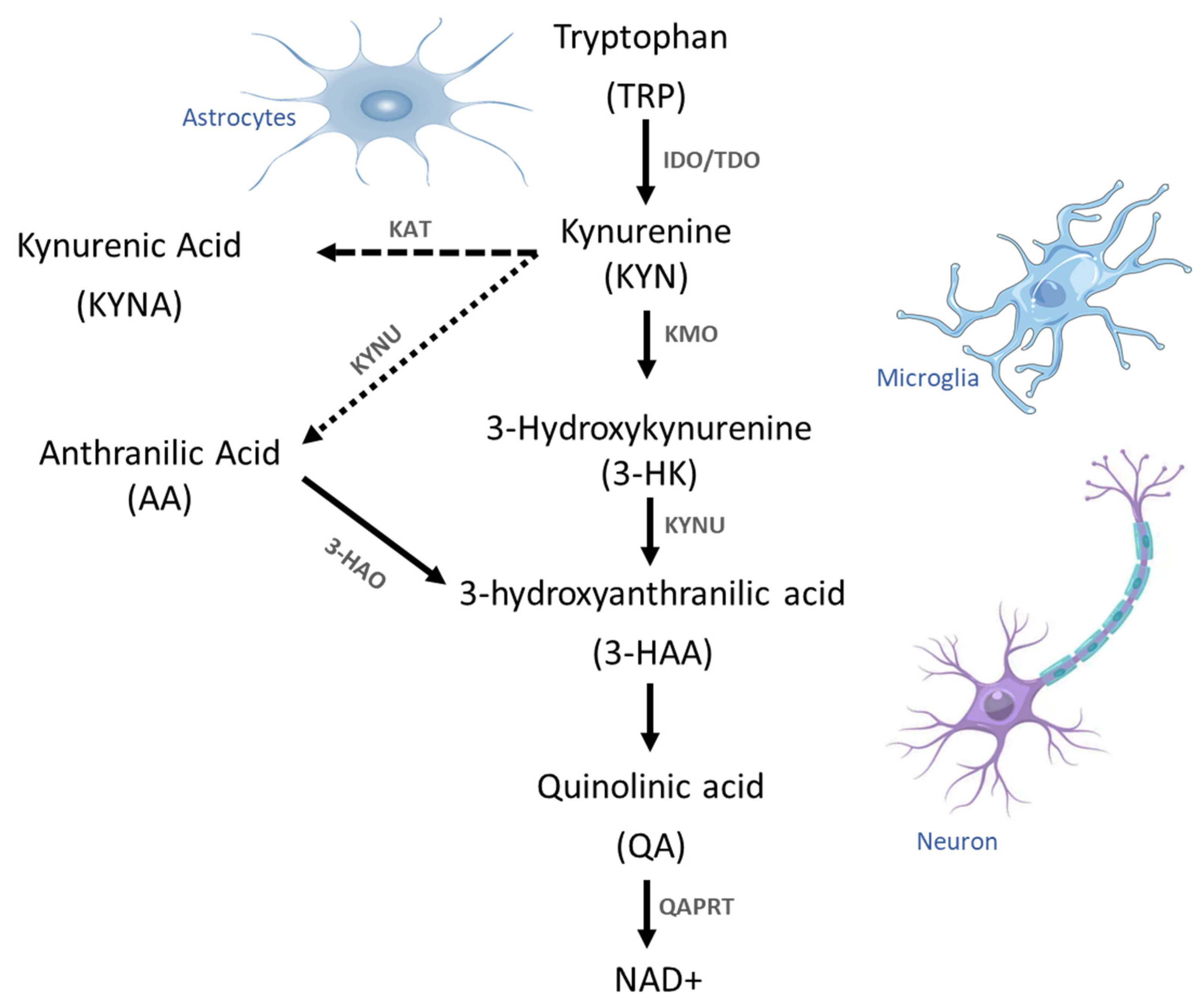
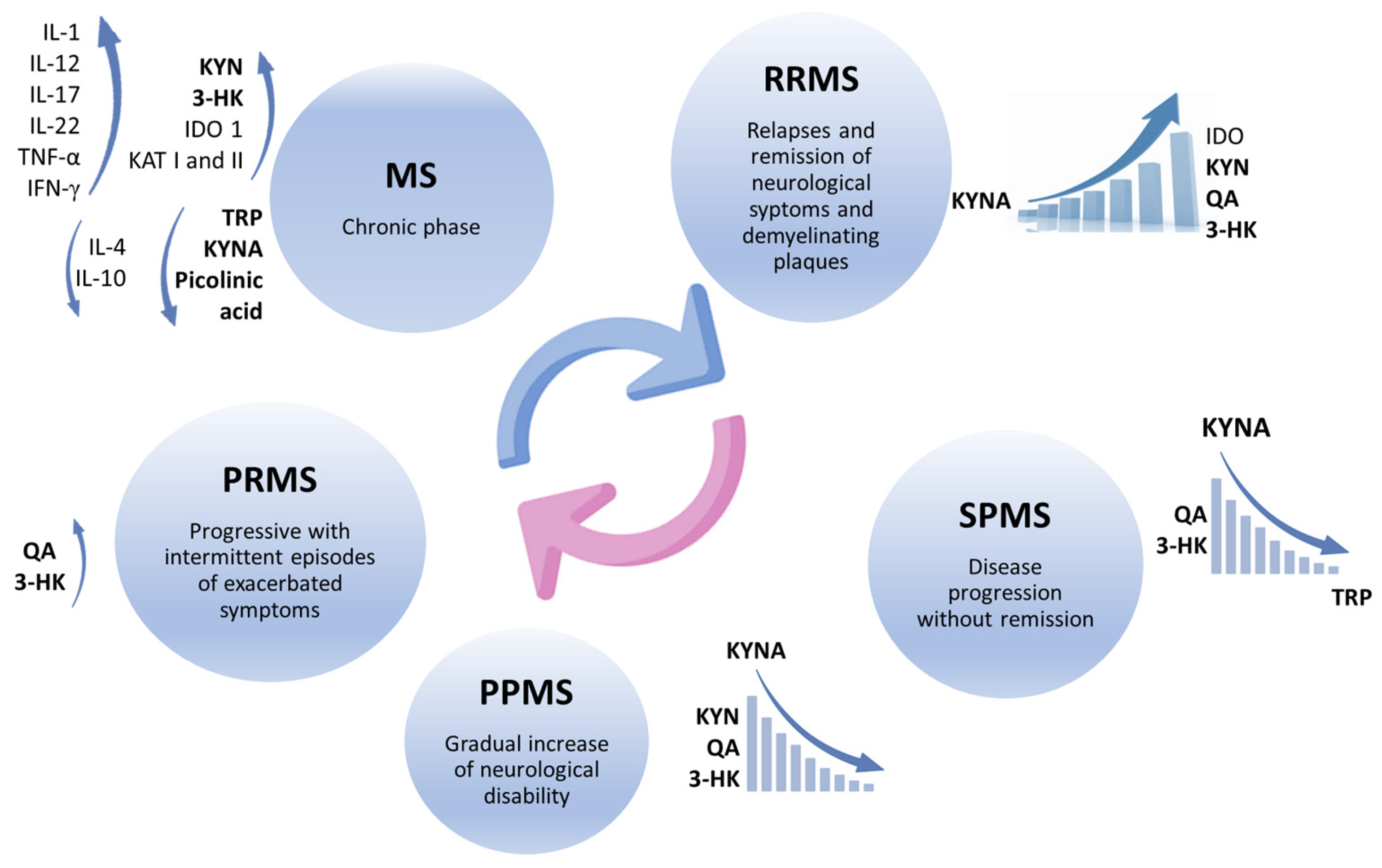

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrillo-Mora, P.; Landa-Solís, C.; Valle-Garcia, D.; Luna-Angulo, A.; Avilés-Arnaut, H.; Robles-Bañuelos, B.; Sánchez-Chapul, L.; Rangel-López, E. Kynurenines and Inflammation: A Remarkable Axis for Multiple Sclerosis Treatment. Pharmaceuticals 2024, 17, 983. https://doi.org/10.3390/ph17080983
Carrillo-Mora P, Landa-Solís C, Valle-Garcia D, Luna-Angulo A, Avilés-Arnaut H, Robles-Bañuelos B, Sánchez-Chapul L, Rangel-López E. Kynurenines and Inflammation: A Remarkable Axis for Multiple Sclerosis Treatment. Pharmaceuticals. 2024; 17(8):983. https://doi.org/10.3390/ph17080983
Chicago/Turabian StyleCarrillo-Mora, Paul, Carlos Landa-Solís, David Valle-Garcia, Alexandra Luna-Angulo, Hamlet Avilés-Arnaut, Benjamín Robles-Bañuelos, Laura Sánchez-Chapul, and Edgar Rangel-López. 2024. "Kynurenines and Inflammation: A Remarkable Axis for Multiple Sclerosis Treatment" Pharmaceuticals 17, no. 8: 983. https://doi.org/10.3390/ph17080983
APA StyleCarrillo-Mora, P., Landa-Solís, C., Valle-Garcia, D., Luna-Angulo, A., Avilés-Arnaut, H., Robles-Bañuelos, B., Sánchez-Chapul, L., & Rangel-López, E. (2024). Kynurenines and Inflammation: A Remarkable Axis for Multiple Sclerosis Treatment. Pharmaceuticals, 17(8), 983. https://doi.org/10.3390/ph17080983