

Positive Allosteric Modulators of Trk Receptors for the Treatment of Alzheimer’s Disease
 ,
,
Abstract
:1. Introduction
2. Neurotrophins and the Trk Receptor Family
2.1. Brain Expression Pattern and Function of the Neurotrophins and Their Receptors
2.2. BDNF-Val66Met Polymorphism
2.3. Cellular Signaling of Trk Receptors
2.4. Processing of Pro-Neurotrophins
3. Physiological and Pathological Role of Neurotrophins
4. Past, Present, and Future Treatment Paradigms of Neurotrophins and Trk Receptors
4.1. Small-Molecule Positive Allosteric Modulators of Trk Receptors
4.2. Previously Described Modulators of Trk Receptors
4.3. Novel Small-Molecule Positive Allosteric Modulators of Trk Receptors
4.4. E2511, a Selective TrkA-PAM
4.5. ACD856, a Pan-Trk PAM
5. Clinical Trials with Modulators of Neurotrophin Signaling for the Treatment of Alzheimer’s Disease
6. Discussion
7. Conclusions
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mintun, M.A.; Lo, A.C.; Evans, C.D.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2022, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Dhadda, S.; Kanekiyo, M.; Li, D.; Swanson, C.J.; Irizarry, M.; Berry, S.; Kramer, L.D.; Berry, D.A. Consistency of Efficacy Results across Various Clinical Measures and Statistical Methods in the Lecanemab Phase 2 Trial of Early Alzheimer’s Disease. Alzheimer’s Res. Ther. 2022, 14, 182. [Google Scholar] [CrossRef]
- McDade, E.; Cummings, J.L.; Dhadda, S.; Swanson, C.J.; Reyderman, L.; Kanekiyo, M.; Koyama, A.; Irizarry, M.; Kramer, L.D.; Bateman, R.J. Lecanemab in Patients with Early Alzheimer’s Disease: Detailed Results on Biomarker, Cognitive, and Clinical Effects from the Randomized and Open-Label Extension of the Phase 2 Proof-of-Concept Study. Alzheimers Res. Ther. 2022, 14, 191. [Google Scholar] [CrossRef] [PubMed]
- Dickson, S.P.; Wessels, A.M.; Dowsett, S.A.; Mallinckrodt, C.; Sparks, J.D.; Chatterjee, S.; Hendrix, S. “Time Saved” As a Demonstration of Clinical Meaningfulness and Illustrated Using the Donanemab TRAILBLAZER-ALZ Study Findings. J. Prev. Alzheimers Dis. 2023, 10, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R. Effects of Mouse Tumor Transplantation on the Nervous System. Ann. N. Y. Acad. Sci. 1952, 55, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Gray, A.; Berman, C.; Dull, T.J. Human β-Nerve Growth Factor Gene Sequence Highly Homologous to That of Mouse. Nature 1983, 303, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.; Selby, M.; Urdea, M.; Quiroga, M.; Bell, G.I.; Rutter, W.J. Isolation and Nucleotide Sequence of a CDNA Encoding the Precursor of Mouse Nerve Growth Factor. Nature 1983, 302, 538–540. [Google Scholar] [CrossRef] [PubMed]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a New Neurotrophic Factor from Mammalian Brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef]
- Ernfors, P.; Ibáñez, C.F.; Ebendal, T.; Olson, L.; Persson, H. Molecular Cloning and Neurotrophic Activities of a Protein with Structural Similarities to Nerve Growth Factor: Developmental and Topographical Expression in the Brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5454–5458. [Google Scholar] [CrossRef]
- Jones, K.R.; Reichardt, L.F. Molecular Cloning of a Human Gene That Is a Member of the Nerve Growth Factor Family. Proc. Natl. Acad. Sci. USA 1990, 87, 8060–8064. [Google Scholar] [CrossRef]
- Hohn, A.; Leibrock, J.; Bailey, K.; Barde, Y.-A. Identification and Characterization of a Novel Member of the Nerve Growth Factor/Brain-Derived Neurotrophic Factor Family. Nature 1990, 344, 339–341. [Google Scholar] [CrossRef]
- Berkemeier, L.R.; Winslow, J.W.; Kaplan, D.R.; Nikolics, K.; Goeddel, D.V.; Rosenthal, A. Neurotrophin-5: A Novel Neurotrophic Factor That Activates Trk and TrkB. Neuron 1991, 7, 857–866. [Google Scholar] [CrossRef]
- Hallböök, F.; Ibáñez, C.F.; Persson, H. Evolutionary Studies of the Nerve Growth Factor Family Reveal a Novel Member Abundantly Expressed in Xenopus Ovary. Neuron 1991, 6, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Ip, N.Y.; Ibáñez, C.F.; Nye, S.H.; McClain, J.; Jones, P.F.; Gies, D.R.; Belluscio, L.; Beau, M.M.L.; Espinosa, R.; Squinto, S.P. Mammalian Neurotrophin-4: Structure, Chromosomal Localization, Tissue Distribution, and Receptor Specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 3060–3064. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Jing, S.; Nanduri, V.; O’Rourke, E.; Barbacid, M. The Trk Proto-Oncogene Encodes a Receptor for Nerve Growth Factor. Cell 1991, 65, 189–197. [Google Scholar] [CrossRef]
- Klein, R.; Nanduri, V.; Jing, S.; Lamballe, F.; Tapley, P.; Bryant, S.; Cordon-Cardo, C.; Jones, K.R.; Reichardt, L.F.; Barbacid, M. The TrkB Tyrosine Protein Kinase Is a Receptor for Brain-Derived Neurotrophic Factor and Neurotrophin-3. Cell 1991, 66, 395–403. [Google Scholar] [CrossRef] [PubMed]
- McGregor, L.M.; Baylin, S.B.; Griffin, C.A.; Hawkins, A.L.; Nelkin, B.D. Molecular Cloning of the CDNA for Human TrkC (NTRK3), Chromosomal Assignment, and Evidence for a Splice Variant. Genomics 1994, 22, 267–272. [Google Scholar] [CrossRef]
- Chao, M.V. The P75 Neurotrophin Receptor. J. Neurobiol. 1994, 25, 1373–1385. [Google Scholar] [CrossRef]
- Lanave, C.; Colangelo, A.M.; Saccone, C.; Alberghina, L. Molecular Evolution of the Neurotrophin Family Members and Their Trk Receptors. Gene 2007, 394, 1–12. [Google Scholar] [CrossRef]
- Hallböök, F.; Wilson, K.; Thorndyke, M.; Olinski, R. Formation and Evolution of the Chordate Neurotrophin and Trk Receptor Genes. Brain Behav. Evol. 2006, 68, 133–144. [Google Scholar] [CrossRef]
- Zhu, B.; Pennack, J.A.; McQuilton, P.; Forero, M.G.; Mizuguchi, K.; Sutcliffe, B.; Gu, C.J.; Fenton, J.C.; Hidalgo, A. Drosophila Neurotrophins Reveal a Common Mechanism for Nervous System Formation. PLoS Biol. 2008, 6, e284. [Google Scholar] [CrossRef] [PubMed]
- Sobreviela, T.; Clary, D.O.; Reichardt, L.F.; Brandabur, M.M.; Kordower, J.H.; Mufson, E.J. TrkA-Immunoreactive Profiles in the Central Nervous System: Colocalization with Neurons Containing P75 Nerve Growth Factor Receptor, Choline Acetyltransferase, and Serotonin. J. Comp. Neurol. 1994, 350, 587–611. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Kilbridge, J.; Li, Y.; Cunningham, E., Jr.; Lenn, N.J.; Clary, D.O.; Reichardt, L.F.; Mobley, W.C. TrkA Expression in the CNS: Evidence for the Existence of Several Novel NGF-Responsive CNS Neurons. J. Neurosci. 1995, 15, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Muragaki, Y.; Timothy, N.; Leight, S.; Hempstead, B.L.; Chao, M.V.; Trojanowski, J.Q.; Lee, V.M. Expression of Trk Receptors in the Developing and Adult Human Central and Peripheral Nervous System. J. Comp. Neurol. 1995, 356, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Josephson, A.; Widenfalk, J.; Trifunovski, A.; Widmer, H.-R.; Olson, L.; Spenger, C. GDNF and NGF Family Members and Receptors in Human Fetal and Adult Spinal Cord and Dorsal Root Ganglia. J. Comp. Neurol. 2001, 440, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Vega, J.A.; Vazquez, E.; Naves, F.J.; Valle, M.E.D.; Calzada, B.; Represa, J.J. Immunohistochemical Localization of the High-Affinity NGF Receptor (Gp 140-TrkA) in the Adult Human Dorsal Root and Sympathetic Ganglia and in the Nerves and Sensory Corpuscles Supplying Digital Skin. Anat. Rec. 1994, 240, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.C.; Sozmen, E.G.; Baeza-Raja, B.; Moan, N.L.; Akassoglou, K.; Schachtrup, C. In Vivo Functions of P75NTR: Challenges and Opportunities for an Emerging Therapeutic Target. Trends Pharmacol. Sci. 2021, 42, 772–788. [Google Scholar] [CrossRef]
- Yang, C.; Liang, R.; Liu, Y.; Meng, F.; Zhou, F.; Zhang, X.; Ning, L.; Wang, Z.; Liu, S.; Zhou, X. Upregulation of ProBDNF/P75NTR Signaling in Immune Cells and Its Correlation with Inflammatory Markers in Patients with Major Depression. FASEB J. 2024, 38, e23312. [Google Scholar] [CrossRef]
- Zhao, L.; Lai, Y.; Jiao, H.; Huang, J. Nerve Growth Factor Receptor Limits Inflammation to Promote Remodeling and Repair of Osteoarthritic Joints. Nat. Commun. 2024, 15, 3225. [Google Scholar] [CrossRef]
- Vega, J.A.; Garcia-Suarez, O.; Hannestad, J.; Perez-Perez, M.; Germana, A. Neurotrophins and the Immune System. J. Anat. 2003, 203, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Minnone, G.; Benedetti, F.D.; Bracci-Laudiero, L. NGF and Its Receptors in the Regulation of Inflammatory Response. Int. J. Mol. Sci. 2017, 18, 1028. [Google Scholar] [CrossRef] [PubMed]
- Bonni, A.; Greenberg, M.E. Neurotrophin Regulation of Gene Expression. Can. J. Neurol. Sci. J. Can. Des Sci. Neurol. 1997, 24, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-O.; Skarnes, W.C.; Minsk, B.; Palmieri, S.; Jackson-Grusby, L.; Wagner, J.A. Nerve Growth Factor Regulates Gene Expression by Several Distinct Mechanisms. Mol. Cell. Biol. 1989, 9, 135–143. [Google Scholar] [CrossRef]
- Biarc, J.; Chalkley, R.J.; Burlingame, A.L.; Bradshaw, R.A. Dissecting the Roles of Tyrosines 490 and 785 of TrkA Protein in the Induction of Downstream Protein Phosphorylation Using Chimeric Receptors. J. Biol. Chem. 2013, 288, 16606–16618. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R.; Blum, R.; Kafitz, K.W.; Kovalchuk, Y.; Konnerth, A. From Modulator to Mediator: Rapid Effects of BDNF on Ion Channels. BioEssays 2004, 26, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Bonnington, J.K.; McNaughton, P.A. Signalling Pathways Involved in the Sensitisation of Mouse Nociceptive Neurones by Nerve Growth Factor. J. Physiol. 2003, 551, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Lesser, S.S.; Sherwood, N.T.; Lo, D.C. Neurotrophins Differentially Regulate Voltage-Gated Ion Channels. Mol. Cell Neurosci. 1997, 10, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Watson, J.J.; Dawbarn, D. The Neurotrophins and Their Role in Alzheimer’s Disease. Curr. Neuropharmacol. 2011, 9, 559–573. [Google Scholar] [CrossRef]
- Castrén, E. Neurotrophins and Psychiatric Disorders. In Neurotrophic Factors; Springer: Berlin/Heidelberg, Germany, 2014; pp. 461–479. [Google Scholar]
- Kaplan, G.B.; Vasterling, J.J.; Vedak, P.C. Brain-Derived Neurotrophic Factor in Traumatic Brain Injury, Post-Traumatic Stress Disorder, and Their Comorbid Conditions: Role in Pathogenesis and Treatment. Behav. Pharmacol. 2010, 21, 427. [Google Scholar] [CrossRef]
- Montagnoli, C.; Tiribuzi, R.; Crispoltoni, L.; Pistilli, A.; Stabile, A.M.; Manfreda, F.; Placella, G.; Rende, M.; Cerulli, G.G. β-NGF and β-NGF Receptor Upregulation in Blood and Synovial Fluid in Osteoarthritis. Biol. Chem. 2017, 398, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Bracci-Laudiero, L.; Aloe, L.; Levi-Montalcini, R.; Galeazzi, M.; Schilter, D.; Scully, J.L.; Otten, U. Increased Levels of NGF in Sera of Systemic Lupus Erythematosus Patients. NeuroReport 1993, 4, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Laudiero, L.B.; Aloe, L.; Levi-Montalcini, R.; Buttinelli, C.; Schilter, D.; Gillessen, S.; Otten, U. Multiple Sclerosis Patients Express Increased Levels of β-Nerve Growth Factor in Cerebrospinal Fluid. Neurosci. Lett. 1992, 147, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Bonini, S.; Lambiase, A.; Bonini, S.; Angelucci, F.; Magrini, L.; Manni, L.; Aloe, L. Circulating Nerve Growth Factor Levels Are Increased in Humans with Allergic Diseases and Asthma. Proc. Natl. Acad. Sci. USA 1996, 93, 10955–10960. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF Val66met Polymorphism Affects Activity-Dependent Secretion of BDNF and Human Memory and Hippocampal Function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Hariri, A.R.; Goldberg, T.E.; Mattay, V.S.; Kolachana, B.S.; Callicott, J.H.; Egan, M.F.; Weinberger, D.R. Brain-Derived Neurotrophic Factor Val66met Polymorphism Affects Human Memory-Related Hippocampal Activity and Predicts Memory Performance. J. Neurosci. 2003, 23, 6690–6694. [Google Scholar] [CrossRef] [PubMed]
- Boots, E.A.; Schultz, S.A.; Clark, L.R.; Racine, A.M.; Darst, B.F.; Koscik, R.L.; Carlsson, C.M.; Gallagher, C.L.; Hogan, K.J.; Bendlin, B.B.; et al. BDNF Val66Met Predicts Cognitive Decline in the Wisconsin Registry for Alzheimer’s Prevention. Neurology 2017, 88, 2098–2106. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Villemagne, V.L.; Laws, S.M.; Ames, D.; Pietrzak, R.H.; Ellis, K.A.; Harrington, K.; Bourgeat, P.; Bush, A.I.; Martins, R.N.; et al. Effect of BDNF Val66Met on Memory Decline and Hippocampal Atrophy in Prodromal Alzheimer’s Disease: A Preliminary Study. PLoS ONE 2014, 9, e86498. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Y.; Maruff, P.; Barthélemy, N.R.; Goate, A.; Hassenstab, J.; Sato, C.; Fagan, A.M.; Benzinger, T.L.S.; Xiong, C.; Cruchaga, C.; et al. Association of BDNF Val66Met with Tau Hyperphosphorylation and Cognition in Dominantly Inherited Alzheimer Disease. JAMA Neurol. 2022, 79, 261–270. [Google Scholar] [CrossRef]
- Holmes, S.E.; Esterlis, I.; Mazure, C.M.; Lim, Y.; Ames, D.; Rainey-Smith, S.; Martins, R.N.; Salvado, O.; Dore, V.; Villemagne, V.L.; et al. β-Amyloid, APOE and BDNF Genotype, and Depressive and Anxiety Symptoms in Cognitively Normal Older Women and Men. Am. J. Geriatr. Psychiatry 2016, 24, 1191–1195. [Google Scholar] [CrossRef]
- Lim, Y.; Villemagne, V.L.; Laws, S.M.; Ames, D.; Pietrzak, R.H.; Ellis, K.A.; Harrington, K.D.; Bourgeat, P.; Salvado, O.; Darby, D.; et al. BDNF Val66Met, Aβ Amyloid, and Cognitive Decline in Preclinical Alzheimer’s Disease. Neurobiol. Aging 2013, 34, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Franzmeier, N.; Ren, J.; Damm, A.; Monté-Rubio, G.; Boada, M.; Ruiz, A.; Ramirez, A.; Jessen, F.; Düzel, E.; Gómez, O.R.; et al. The BDNF(Val66Met) SNP Modulates the Association between Beta-Amyloid and Hippocampal Disconnection in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 614–628. [Google Scholar] [CrossRef]
- Ward, D.D.; Summers, M.J.; Saunders, N.L.; Janssen, P.; Stuart, K.E.; Vickers, J.C. APOE and BDNF Val66Met Polymorphisms Combine to Influence Episodic Memory Function in Older Adults. Behav. Brain Res. 2014, 271, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Y.; Villemagne, V.L.; Laws, S.M.; Pietrzak, R.H.; Snyder, P.J.; Ames, D.; Ellis, K.A.; Harrington, K.; Rembach, A.; Martins, R.N.; et al. APOE and BDNF Polymorphisms Moderate Amyloid β-Related Cognitive Decline in Preclinical Alzheimer’s Disease. Mol. Psychiatry 2015, 20, 1322–1328. [Google Scholar] [CrossRef]
- Stonnington, C.M.; Velgos, S.N.; Chen, Y.; Syed, S.; Huentelman, M.; Thiyyagura, P.; Lee, W.; Richholt, R.; Caselli, R.J.; Locke, D.E.C.; et al. Interaction Between BDNF Val66Met and APOE4 on Biomarkers of Alzheimer’s Disease and Cognitive Decline. J. Alzheimers Dis. 2020, 78, 721–734. [Google Scholar] [CrossRef]
- Kennedy, K.M.; Reese, E.D.; Horn, M.M.; Sizemore, A.N.; Unni, A.K.; Meerbrey, M.E.; Kalich, A.G.; Rodrigue, K.M. BDNF Val66met Polymorphism Affects Aging of Multiple Types of Memory. Brain Res. 2015, 1612, 104–117. [Google Scholar] [CrossRef]
- Azeredo, L.A.; Nardi, T.; Grassi-Oliveira, R. BDNF Val66Met Polymorphism and Memory Performance in Older Adults: The Met Carrier Effect Is More Complex than Previously Thought: Authors’ Reply. Rev. Bras. Psiquiatr. 2017, 39, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Ciampa, C.J.; Morin, T.M.; Murphy, A.; Joie, R.; Landau, S.M.; Berry, A.S. DAT1 and BDNF Polymorphisms Interact to Predict Aβ and Tau Pathology. Neurobiol. Aging 2024, 133, 115–124. [Google Scholar] [CrossRef]
- Hong, C.J.; Huo, S.J.; Yen, F.C.; Tung, C.L.; Pan, G.M.; Tsai, S.J. Association Study of a Brain-Derived Neurotrophic-Factor Genetic Polymorphism and Mood Disorders, Age of Onset and Suicidal Behavior. Neuropsychobiology 2003, 48, 186–189. [Google Scholar] [CrossRef]
- Stephens, R.M.; Loeb, D.M.; Copeland, T.D.; Pawson, T.; Greene, L.A.; Kaplan, D.R. Trk Receptors Use Redundant Signal Transduction Pathways Involving SHC and PLC-Γ1 to Mediate NGF Responses. Neuron 1994, 12, 691–705. [Google Scholar] [CrossRef]
- Baxter, R.M.; Cohen, P.; Obermeier, A.; Ullrich, A.; Downes, C.P.; Doza, Y.N. Phosphotyrosine Residues in the Nerve-Growth-Factor Receptor (Trk-A). Eur. J. Biochem. 1995, 234, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, A.; Lammers, R.; Wiesmüller, K.H.; Jung, G.; Schlessinger, J.; Ullrich, A. Identification of Trk Binding Sites for SHC and Phosphatidylinositol 3′-Kinase and Formation of a Multimeric Signaling Complex. J. Biol. Chem. 1993, 268, 22963–22966. [Google Scholar] [CrossRef] [PubMed]
- Minichiello, L. TrkB Signalling Pathways in LTP and Learning. Nat. Rev. Neurosci. 2009, 10, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Kiris, E.; Wang, T.; Yanpallewar, S.; Dorsey, S.G.; Becker, J.; Bavari, S.; Palko, M.E.; Coppola, V.; Tessarollo, L. TrkA in Vivo Function Is Negatively Regulated by Ubiquitination. J. Neurosci. 2014, 34, 4090–4098. [Google Scholar] [CrossRef]
- Barker, P.A.; Mantyh, P.; Arendt-Nielsen, L.; Viktrup, L.; Tive, L. Nerve Growth Factor Signaling and Its Contribution to Pain. J. Pain. Res. 2020, 13, 1223–1241. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of Cell Survival by Secreted Proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.A.; Tischler, A.S. Establishment of a Noradrenergic Clonal Line of Rat Adrenal Pheochromocytoma Cells Which Respond to Nerve Growth Factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF Signaling in Context: From Synaptic Regulation to Psychiatric Disorders. Cell 2022, 185, 62–76. [Google Scholar] [CrossRef]
- Swain, M.; Soman, S.K.; Tapia, K.; Dagda, R.Y.; Dagda, R.K. Brain-derived Neurotrophic Factor Protects Neurons by Stimulating Mitochondrial Function through Protein Kinase A. J. Neurochem. 2023, 167, 104–125. [Google Scholar] [CrossRef]
- Tong, Q.; Wang, F.; Zhou, H.; Sun, H.; Song, H.; Shu, Y.; Gong, Y.; Zhang, W.; Cai, T.; Yang, F.; et al. Structural and Functional Insights into Lipid-bound Nerve Growth Factors. FASEB J. 2012, 26, 3811–3821. [Google Scholar] [CrossRef] [PubMed]
- Monshipouri, M.; Jiang, H.; Lazarovici, P. NGF Stimulation of Erk Phosphorylation Is Impaired by a Point Mutation in the Transmembrane Domain of TrkA Receptor. J. Mol. Neurosci. 2000, 14, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Biarc, J.; Chalkley, R.J.; Burlingame, A.L.; Bradshaw, R.A. The Induction of Serine/Threonine Protein Phosphorylations by a PDGFR/TrkA Chimera in Stably Transfected PC12 Cells. Mol. Cell Proteom. 2012, 11, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Thoenen, H.; Lindholm, D. TrkA Tyrosine Residues Involved in NGF-induced Neurite Outgrowth of PC12 Cells. Eur. J. Neurosci. 1995, 7, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Leßmann, V.; Brigadski, T. Mechanisms, Locations, and Kinetics of Synaptic BDNF Secretion: An Update. Neurosci. Res. 2009, 65, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Suter, U.; Heymach, J.V.; Shooter, E.M. Two Conserved Domains in the NGF Propeptide Are Necessary and Sufficient for the Biosynthesis of Correctly Processed and Biologically Active NGF. EMBO J. 1991, 10, 2395–2400. [Google Scholar] [CrossRef] [PubMed]
- Al-Qudah, M.A.; Al-Dwairi, A. Mechanisms and Regulation of Neurotrophin Synthesis and Secretion. Neurosciences 2016, 21, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Benjannet, S.; Pareek, S.; Savaria, D.; Hamelin, J.; Goulet, B.; Laliberté, J.; Lazure, C.; Chrétien, M.; Murphy, R.A. Cellular Processing of the Nerve Growth Factor Precursor by the Mammalian Pro-Protein Convertases. Biochem. J. 1996, 314, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.A.; Cuello, A.C. Activity-Dependent Release of Precursor Nerve Growth Factor, Conversion to Mature Nerve Growth Factor, and Its Degradation by a Protease Cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 6735–6740. [Google Scholar] [CrossRef]
- Xue, B.; Waseem, S.M.A.; Zhu, Z.; Alshahrani, M.A.; Nazam, N.; Anjum, F.; Habib, A.H.; Rafeeq, M.M.; Nazam, F.; Sharma, M. Brain-Derived Neurotrophic Factor: A Connecting Link Between Nutrition, Lifestyle, and Alzheimer’s Disease. Front. Neurosci. 2022, 16, 925991. [Google Scholar] [CrossRef]
- Nandi, A.; Counts, N.; Chen, S.; Seligman, B.; Tortorice, D.; Vigo, D.; Bloom, D.E. Global and Regional Projections of the Economic Burden of Alzheimer’s Disease and Related Dementias from 2019 to 2050: A Value of Statistical Life Approach. eClinicalMedicine 2022, 51, 101580. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Aisen, P.S.; Cummings, J.; Detke, M.J.; Longo, F.M.; Raman, R.; Sabbagh, M.; Schneider, L.; Tanzi, R.; Tariot, P.; et al. Non-Amyloid Approaches to Disease Modification for Alzheimer’s Disease: An EU/US CTAD Task Force Report. J. Prev. Alzheimer’s Dis. 2020, 7, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Ruit, K.G.; Elliott, J.L.; Osborne, P.A.; Yan, Q.; Snider, W.D. Selective Dependence of Mammalian Dorsal Root Ganglion Neurons on Nerve Growth Factor during Embryonic Development. Neuron 1992, 8, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Smeyne, R.J.; Klein, R.; Schnapp, A.; Long, L.K.; Bryant, S.; Lewin, A.; Lira, S.A.; Barbacid, M. Severe Sensory and Sympathetic Neuropathies in Mice Carrying a Disrupted Trk/NGF Receptor Gene. Nature 1994, 368, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Crowley, C.; Spencer, S.D.; Nishimura, M.C.; Chen, K.S.; Pitts-Meek, S.; Armaninl, M.P.; Ling, L.H.; McMahon, S.B.; Shelton, D.L.; Levinson, A.D.; et al. Mice Lacking Nerve Growth Factor Display Perinatal Loss of Sensory and Sympathetic Neurons yet Develop Basal Forebrain Cholinergic Neurons. Cell 1994, 76, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Smeyne, R.J.; Wurst, W.; Long, L.K.; Auerbach, B.A.; Joyner, A.L.; Barbacid, M. Targeted Disruption of the TrkB Neurotrophin Receptor Gene Results in Nervous System Lesions and Neonatal Death. Cell 1993, 75, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Ernfors, P.; Lee, K.F.; Jaenisch, R. Mice Lacking Brain-Derived Neurotrophic Factor Develop with Sensory Deficits. Nature 1994, 368, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Linnarsson, S.; Björklund, A.; Ernfors, P. Learning Deficit in BDNF Mutant Mice. Eur. J. Neurosci. 1997, 9, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Endres, T.; Lessmann, V. Age-Dependent Deficits in Fear Learning in Heterozygous BDNF Knock-out Mice. Learn. Mem. 2012, 19, 561–570. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; García-Suarez, O.; Germanà, A.; Díaz-Esnal, B.; de Carlos, F.; Silos-Santiago, I.; del Valle, M.E.; Cobo, J.; Vega, J.A. Characterization of Sensory Deficits in TrkB Knockout Mice. Neurosci. Lett. 2008, 433, 43–47. [Google Scholar] [CrossRef]
- Erickson, J.T.; Conover, J.C.; Borday, V.; Champagnat, J.; Barbacid, M.; Yancopoulos, G.; Katz, D.M. Mice Lacking Brain-Derived Neurotrophic Factor Exhibit Visceral Sensory Neuron Losses Distinct from Mice Lacking NT4 and Display a Severe Developmental Deficit in Control of Breathing. J. Neurosci. 1996, 16, 5361–5371. [Google Scholar] [CrossRef] [PubMed]
- Rios, M.; Fan, G.; Fekete, C.; Kelly, J.; Bates, B.; Kuehn, R.; Lechan, R.M.; Jaenisch, R. Conditional Deletion Of Brain-Derived Neurotrophic Factor in the Postnatal Brain Leads to Obesity and Hyperactivity. Mol. Endocrinol. 2001, 15, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.S.H.; Hung, C.-C.; Rochford, J.; Keogh, J.; Gray, J.; Sivaramakrishnan, S.; O’Rahilly, S.; Farooqi, S.I. A de Novo Mutation Affecting Human TrkB Associated with Severe Obesity and Developmental Delay. Nat. Neurosci. 2004, 7, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- Kernie, S.G.; Liebl, D.J.; Parada, L.F. Reduction of Brain Derived Neurotrophic Factor Causes Obesity and Hyperactivity: Implications for WAGR Syndrome. Pediatr. Res. 1999, 45, 42. [Google Scholar] [CrossRef]
- Han, D.; Sun, D.; Xiu, M.; Su, X.; Wang, J.; Li, J.; Wang, D. Association between the Improvement in Depressive Symptoms and Serum BDNF Levels in Drug-Naive First Episode Patients with Schizophrenia: A Longitudinal Follow-Up. Psychoneuroendocrinology 2021, 133, 105392. [Google Scholar] [CrossRef] [PubMed]
- Fagan, A.M.; Zhang, H.; Landis, S.; Smeyne, R.J.; Silos-Santiago, I.; Barbacid, M. TrkA, but Not TrkC, Receptors Are Essential for Survival of Sympathetic Neurons in Vivo. J. Neurosci. 1996, 16, 6208–6218. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Silos-Santiago, I.; Smeyne, R.J.; Lira, S.A.; Brambilla, R.; Bryant, S.; Zhang, L.; Snider, W.D.; Barbacid, M. Disruption of the Neurotrophin-3 Receptor Gene TrkC Eliminates La Muscle Afferents and Results in Abnormal Movements. Nature 1994, 368, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.A.; Kumar, S.; Liebl, D.; Chang, R.; Parada, L.F.; Vellis, J.D. Mice Lacking NT-3, and Its Receptor TrkC, Exhibit Profound Deficiencies in CNS Glial Cells. Glia 1999, 26, 153–165. [Google Scholar] [CrossRef]
- Shimazu, K.; Zhao, M.; Sakata, K.; Akbarian, S.; Bates, B.; Jaenisch, R.; Lu, B. NT-3 Facilitates Hippocampal Plasticity and Learning and Memory by Regulating Neurogenesis. Learn. Mem. 2006, 13, 307–315. [Google Scholar] [CrossRef]
- Huang, Z.J.; Kirkwood, A.; Pizzorusso, T.; Porciatti, V.; Morales, B.; Bear, M.F.; Maffei, L.; Tonegawa, S. BDNF Regulates the Maturation of Inhibition and the Critical Period of Plasticity in Mouse Visual Cortex. Cell 1999, 98, 739–755. [Google Scholar] [CrossRef]
- Cunha, C.; Angelucci, A.; D’Antoni, A.; Dobrossy, M.D.; Dunnett, S.B.; Berardi, N.; Brambilla, R. Brain-Derived Neurotrophic Factor (BDNF) Overexpression in the Forebrain Results in Learning and Memory Impairments. Neurobiol. Dis. 2009, 33, 358–368. [Google Scholar] [CrossRef]
- Gharami, K.; Xie, Y.; An, J.J.; Tonegawa, S.; Xu, B. Brain-Derived Neurotrophic Factor over-Expression in the Forebrain Ameliorates Huntington’s Disease Phenotypes in Mice. J. Neurochem. 2008, 105, 369–379. [Google Scholar] [CrossRef]
- Tolwani, R.J.; Buckmaster, P.S.; Varma, S.; Cosgaya, J.M.; Wu, Y.; Suri, C.; Shooter, E.M. BDNF Overexpression Increases Dendrite Complexity in Hippocampal Dentate Gyrus. Neuroscience 2002, 114, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Tolwani, R.J.; Cosgaya, J.M.; Varma, S.; Jacob, R.; Kuo, L.E.; Shooter, E.M. BDNF Overexpression Produces a Long-Term Increase in Myelin Formation in the Peripheral Nervous System. J. Neurosci. Res. 2004, 77, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Cappoli, N.; Tabolacci, E.; Aceto, P.; Russo, C.D. The Emerging Role of the BDNF-TrkB Signaling Pathway in the Modulation of Pain Perception. J. Neuroimmunol. 2020, 349, 577406. [Google Scholar] [CrossRef] [PubMed]
- Eu, W.Z.; Chen, Y.-J.; Chen, W.-T.; Wu, K.-Y.; Tsai, C.-Y.; Cheng, S.-J.; Carter, R.N.; Huang, G.-J. The Effect of Nerve Growth Factor on Supporting Spatial Memory Depends upon Hippocampal Cholinergic Innervation. Transl. Psychiatry 2021, 11, 162. [Google Scholar] [CrossRef]
- Khan, N.; Smith, M.T. Neurotrophins and Neuropathic Pain: Role in Pathobiology. Molecules 2015, 20, 10657–10688. [Google Scholar] [CrossRef]
- Mitre, M.; Mariga, A.; Chao, M.V. Neurotrophin Signalling: Novel Insights into Mechanisms and Pathophysiology. Clin. Sci. 2016, 131, 13–23. [Google Scholar] [CrossRef]
- Gärtner, A.; Polnau, D.G.; Staiger, V.; Sciarretta, C.; Minichiello, L.; Thoenen, H.; Bonhoeffer, T.; Korte, M. Hippocampal Long-Term Potentiation Is Supported by Presynaptic and Postsynaptic Tyrosine Receptor Kinase B-Mediated Phospholipase Cγ Signaling. J. Neurosci. 2006, 26, 3496–3504. [Google Scholar] [CrossRef]
- Guyon, N.; Zacharias, L.R.; van Lunteren, J.A.; Immenschuh, J.; Fuzik, J.; Märtin, A.; Xuan, Y.; Zilberter, M.; Kim, H.; Meletis, K.; et al. Adult TrkB Signaling in Parvalbumin Interneurons Is Essential to Prefrontal Network Dynamics. J. Neurosci. 2021, 41, 3120–3141. [Google Scholar] [CrossRef]
- Laske, C.; Stellos, K.; Hoffmann, N.; Stransky, E.; Straten, G.; Eschweiler, G.W.; Leyhe, T. Higher BDNF Serum Levels Predict Slower Cognitive Decline in Alzheimer’s Disease Patients. Int. J. Neuropsychopharmacol. 2011, 14, 399–404. [Google Scholar] [CrossRef]
- Lärkfors, L.; Ebendal, T.; Whittemore, S.R.; Persson, H.; Hoffer, B.; Olson, L. Decreased Level of Nerve Growth Factor (NGF) and Its Messenger RNA in the Aged Rat Brain. Mol. Brain Res. 1987, 3, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Odaka, H. The Role of Neurotrophin Signaling in Age-Related Cognitive Decline and Cognitive Diseases. Int. J. Mol. Sci. 2022, 23, 7726. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Nie, Z.; Shu, H.; Kuang, Y.; Chen, X.; Cheng, J.; Yu, S.; Liu, H. The Role of BDNF on Neural Plasticity in Depression. Front. Cell. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Mosiołek, A.; Mosiołek, J.; Jakima, S.; Pięta, A.; Szulc, A. Effects of Antidepressant Treatment on Neurotrophic Factors (BDNF and IGF-1) in Patients with Major Depressive Disorder (MDD). J. Clin. Med. 2021, 10, 3377. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK Fusion-Positive Cancers and TRK Inhibitor Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, C.B.; Krebs, M.G.; Tourneau, C.L.; Sokol, E.S.; Maund, S.L.; Wilson, T.R.; Jin, D.X.; Newberg, J.Y.; Fabrizio, D.; Veronese, L.; et al. Genomic Context of NTRK1/2/3 Fusion-Positive Tumours from a Large Real-World Population. npj Precis. Oncol. 2021, 5, 69. [Google Scholar] [CrossRef]
- Jönhagen, M.E.; Nordberg, A.; Amberla, K.; Bäckman, L.; Ebendal, T.; Meyerson, B.; Olson, L.; Seiger; Shigeta, M.; Theodorsson, E.; et al. Intracerebroventricular Infusion of Nerve Growth Factor in Three Patients with Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 1998, 9, 246–257. [Google Scholar] [CrossRef]
- Chiaretti, A.; Antonelli, A.; Genovese, O.; Fernandez, E.; Giuda, D.; Mariotti, P.; Riccardi, R. Intraventricular Nerve Growth Factor Infusion Improves Cerebral Blood Flow and Stimulates Doublecortin Expression in Two Infants with Hypoxic-Ischemic Brain Injury. Neurol. Res. 2008, 30, 223–228. [Google Scholar] [CrossRef]
- Chiaretti, A.; Conti, G.; Falsini, B.; Buonsenso, D.; Crasti, M.; Manni, L.; Soligo, M.; Fantacci, C.; Genovese, O.; Calcagni, M.L.; et al. Intranasal Nerve Growth Factor Administration Improves Cerebral Functions in a Child with Severe Traumatic Brain Injury: A Case Report. Brain Inj. 2017, 31, 1538–1547. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; U, H.S.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; et al. A Phase 1 Clinical Trial of Nerve Growth Factor Gene Therapy for Alzheimer Disease. Nat. Med. 2005, 11, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Eriksdotter-Jönhagen, M.; Linderoth, B.; Lind, G.; Aladellie, L.; Almkvist, O.; Andreasen, N.; Blennow, K.; Bogdanovic, N.; Jelic, V.; Kadir, A.; et al. Encapsulated Cell Biodelivery of Nerve Growth Factor to the Basal Forebrain in Patients with Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2012, 33, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S.; Baumann, T.L.; Bakay, R.A.; Ostrove, J.M.; Siffert, J.; Fleisher, A.S.; Herzog, C.D.; Barba, D.; Pay, M.; Salmon, D.P.; et al. A Phase1 Study of Stereotactic Gene Delivery of AAV2-NGF for Alzheimer’s Disease. Alzheimers Dement. 2014, 10, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S.; Tuszynski, M.H.; Thomas, R.G.; Barba, D.; Brewer, J.B.; Rissman, R.A.; Siffert, J.; Aisen, P.S.; Mintzer, J.; Lerner, A.; et al. Adeno-Associated Viral Vector (Serotype 2)–Nerve Growth Factor for Patients with Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Karami, A.; Eyjolfsdottir, H.; Vijayaraghavan, S.; Lind, G.; Almqvist, P.; Kadir, A.; Linderoth, B.; Andreasen, N.; Blennow, K.; Wall, A.; et al. Changes in CSF Cholinergic Biomarkers in Response to Cell Therapy with NGF in Patients with Alzheimer’s Disease. Alzheimers Dement. 2015, 11, 1316–1328. [Google Scholar] [CrossRef] [PubMed]
- Group, T.B.S. A Controlled Trial of Recombinant Methionyl Human BDNF in ALS. Neurology 1999, 52, 1427. [Google Scholar] [CrossRef]
- Wellmer, A.; Misra, V.P.; Sharief, M.K.; Kopelman, P.G.; Anand, P. A Double-Blind Placebo-Controlled Clinical Trial of Recombinant Human Brain-Derived Neurotrophic Factor (RhBDNF) in Diabetic Polyneuropathy. J. Peripher. Nerv. Syst. 2001, 6, 204–210. [Google Scholar] [CrossRef]
- Sahenk, Z.; Nagaraja, H.N.; McCracken, B.S.; King, W.M.; Freimer, M.L.; Cedarbaum, J.M.; Mendell, J.R. NT-3 Promotes Nerve Regeneration and Sensory Improvement in CMT1A Mouse Models and in Patients. Neurology 2005, 65, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Sahenk, Z.; Galloway, G.; Clark, K.R.; Malik, V.; Rodino-Klapac, L.R.; Kaspar, B.K.; Chen, L.; Braganza, C.; Montgomery, C.; Mendell, J.R. AAV1.NT-3 Gene Therapy for Charcot–Marie–Tooth Neuropathy. Mol. Ther. 2014, 22, 511–521. [Google Scholar] [CrossRef]
- Smet, F.D.; Christopoulos, A.; Carmeliet, P. Allosteric Targeting of Receptor Tyrosine Kinases. Nat. Biotechnol. 2014, 32, 1113–1120. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, K.; Yu, Z. Drug Development in Channelopathies: Allosteric Modulation of Ligand-Gated and Voltage-Gated Ion Channels. J. Med. Chem. 2020, 63, 15258–15278. [Google Scholar] [CrossRef] [PubMed]
- Girmaw, F. Review on Allosteric Modulators of Dopamine Receptors so Far. Heal. Sci. Rep. 2024, 7, e1984. [Google Scholar] [CrossRef] [PubMed]
- Bagal, S.K.; Omoto, K.; Blakemore, D.C.; Bungay, P.J.; Bilsland, J.G.; Clarke, P.J.; Corbett, M.S.; Cronin, C.N.; Cui, J.J.; Dias, R.; et al. Discovery of Allosteric, Potent, Subtype Selective, and Peripherally Restricted TrkA Kinase Inhibitors. J. Med. Chem. 2019, 62, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Furuya, N.; Momose, T.; Katsuno, K.; Fushimi, N.; Muranaka, H.; Handa, C.; Ozawa, T.; Kinoshita, T. The Juxtamembrane Region of TrkA Kinase Is Critical for Inhibitor Selectivity. Bioorg Med. Chem. Lett. 2017, 27, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, H.W.; Girard, B.M.; Ratkovits, L.; Campbell, S.E.; Vizzard, M.A. Effects of Pharmacological Neurotrophin Receptor Inhibition on Bladder Function in Female Mice with Cyclophosphamide-Induced Cystitis. Front. Urol. 2022, 2, 1037511. [Google Scholar] [CrossRef] [PubMed]
- Dahlström, M.; Madjid, N.; Nordvall, G.; Halldin, M.M.; Vazquez-Juarez, E.; Lindskog, M.; Sandin, J.; Winblad, B.; Eriksdotter, M.; Forsell, P. Identification of Novel Positive Allosteric Modulators of Neurotrophin Receptors for the Treatment of Cognitive Dysfunction. Cells 2021, 10, 1871. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, T.; Moriyama, Y.; Hiramatsu, N.; Kosasa, T.; Kondo, K.; Wakita, H. E2511, a Novel Small Compound TrkA Allosteric Modulator, Induces a Specific Trophic Signaling via Direct Binding to TrkA, and Can Reverse the Loss of Choline Acetyltransferase (ChAT) Positive Neurons in Transgenic Models of AD. Alzheimer’s Dement. 2021, 17, e051985. [Google Scholar] [CrossRef]
- Laske, C.; Stransky, E.; Leyhe, T.; Eschweiler, G.W.; Wittorf, A.; Richartz, E.; Bartels, M.; Buchkremer, G.; Schott, K. Stage-Dependent BDNF Serum Concentrations in Alzheimer’s Disease. J. Neural Transm. 2006, 113, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Massa, S.M.; Tran, K.C.; Simmons, D.A.; Rajadas, J.; Zeng, A.Y.; Jang, T.; Carsanaro, S.; Longo, F.M. A Small Molecule TrkB/TrkC Neurotrophin Receptor Co-Activator with Distinctive Effects on Neuronal Survival and Process Outgrowth. Neuropharmacology 2016, 110, 343–361. [Google Scholar] [CrossRef]
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant Drugs Act by Directly Binding to TRKB Neurotrophin Receptors. Cell 2021, 184, 1299–1313.e19. [Google Scholar] [CrossRef]
- Moliner, R.; Girych, M.; Brunello, C.A.; Kovaleva, V.; Biojone, C.; Enkavi, G.; Antenucci, L.; Kot, E.F.; Goncharuk, S.A.; Kaurinkoski, K.; et al. Psychedelics Promote Plasticity by Directly Binding to BDNF Receptor TrkB. Nat. Neurosci. 2023, 26, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.P.; Juric, S.; Backlund, M.; Dahlström, M.; Madjid, N.; Lidell, V.; Rasti, A.; Sandin, J.; Nordvall, G.; Forsell, P. Neuroprotective and Disease-Modifying Effects of the Triazinetrione ACD856, a Positive Allosteric Modulator of Trk-Receptors for the Treatment of Cognitive Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 11159. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, T.; Moriyama, Y.; Hiramatsu, N.; Kosasa, T. E2511, a Novel Small Compound TrkA Biased Positive Allosteric Modulator, Reinnervates Cholinergic Neuron via Enhancement of Specific Trophic Signaling of TrkA in Non-Clinical. Alzheimer’s Dement. 2023, 19, e062590. [Google Scholar] [CrossRef]
- Jang, S.-W.; Okada, M.; Sayeed, I.; Xiao, G.; Stein, D.; Jin, P.; Ye, K. Gambogic Amide, a Selective Agonist for TrkA Receptor That Possesses Robust Neurotrophic Activity, Prevents Neuronal Cell Death. Proc. Natl. Acad. Sci. USA 2007, 104, 16329–16334. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.-W.; Liu, X.; Chan, C.; France, S.A.; Sayeed, I.; Tang, W.; Lin, X.; Xiao, G.; Andero, R.; Chang, Q.; et al. Deoxygedunin, a Natural Product with Potent Neurotrophic Activity in Mice. PLoS ONE 2010, 5, e11528. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A Selective TrkB Agonist with Potent Neurotrophic Activities by 7,8-Dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Li, R.; Lama, T.; Saragovi, H.U.; Cumberlidge, G.; Meerovitch, K. An NGF Mimetic, MIM-D3, Stimulates Conjunctival Cell Glycoconjugate Secretion and Demonstrates Therapeutic Efficacy in a Rat Model of Dry Eye. Exp. Eye Res. 2011, 93, 503–512. [Google Scholar] [CrossRef]
- Yu, Z.; Joy, S.; Mi, T.; Yazdanpanah, G.; Burgess, K.; de Paiva, C.S. New, Potent, Small Molecule Agonists of Tyrosine Kinase Receptors Attenuate Dry Eye Disease. Front. Med. 2022, 9, 937142. [Google Scholar] [CrossRef]
- Lazaridis, I.; Charalampopoulos, I.; Alexaki, V.-I.; Avlonitis, N.; Pediaditakis, I.; Efstathopoulos, P.; Calogeropoulou, T.; Castanas, E.; Gravanis, A. Neurosteroid Dehydroepiandrosterone Interacts with Nerve Growth Factor (NGF) Receptors, Preventing Neuronal Apoptosis. PLoS Biol. 2011, 9, e1001051. [Google Scholar] [CrossRef]
- Pediaditakis, I.; Kourgiantaki, A.; Prousis, K.C.; Potamitis, C.; Xanthopoulos, K.P.; Zervou, M.; Calogeropoulou, T.; Charalampopoulos, I.; Gravanis, A. BNN27, a 17-Spiroepoxy Steroid Derivative, Interacts with and Activates P75 Neurotrophin Receptor, Rescuing Cerebellar Granule Neurons from Apoptosis. Front. Pharmacol. 2016, 7, 512. [Google Scholar] [CrossRef]
- Zagrebelsky, M.; Korte, M. Are TrkB Receptor Agonists the Right Tool to Fulfill the Promises for a Therapeutic Value of the Brain-Derived Neurotrophic Factor? Neural Regen. Res. 2023, 19, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Pediaditakis, I.; Efstathopoulos, P.; Prousis, K.C.; Zervou, M.; Arévalo, J.; Alexaki, V.I.; Nikoletopoulou, V.; Karagianni, E.; Potamitis, C.; Tavernarakis, N.; et al. Selective and Differential Interactions of BNN27, a Novel C17-Spiroepoxy Steroid Derivative, with TrkA Receptors, Regulating Neuronal Survival and Differentiation. Neuropharmacology 2016, 111, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.L.; Dill, L.K.; Wood, R.J.; Wang, S.; Robertson, K.; Murray, S.S.; Zamani, A.; Semple, B.D. Acute Treatment with TrkB Agonist LM22A-4 Confers Neuroprotection and Preserves Myelin Integrity in a Mouse Model of Pediatric Traumatic Brain Injury. Exp. Neurol. 2021, 339, 113652. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ahn, E.H.; Liu, X.; Wang, Z.H.; Luo, S.; Liao, J.; Ye, K. Optimized TrkB Agonist Ameliorates Alzheimer’s Disease Pathologies and Improves Cognitive Functions via Inhibiting Delta-Secretase. ACS Chem. Neurosci. 2021, 12, 2448–2461. [Google Scholar] [CrossRef] [PubMed]
- Charou, D.; Rogdakis, T.; Latorrata, A.; Valcarcel, M.; Papadogiannis, V.; Athanasiou, C.; Tsengenes, A.; Papadopoulou, M.A.; Lypitkas, D.; Lavigne, M.D.; et al. Comprehensive Characterization of the Neurogenic and Neuroprotective Action of a Novel TrkB Agonist Using Mouse and Human Stem Cell Models of Alzheimer’s Disease. Stem Cell Res. Ther. 2024, 15, 200. [Google Scholar] [CrossRef]
- Rogdakis, T.; Charou, D.; Latorrata, A.; Papadimitriou, E.; Tsengenes, A.; Athanasiou, C.; Papadopoulou, M.; Chalikiopoulou, C.; Katsila, T.; Ramos, I.; et al. Development and Biological Characterization of a Novel Selective TrkA Agonist with Neuroprotective Properties against Amyloid Toxicity. Biomedicines 2022, 10, 614. [Google Scholar] [CrossRef]
- Gonzalez, S.; McHugh, T.L.M.; Yang, T.; Syriani, W.; Massa, S.M.; Longo, F.M.; Simmons, D.A. Small Molecule Modulation of TrkB and TrkC Neurotrophin Receptors Prevents Cholinergic Neuron Atrophy in an Alzheimer’s Disease Mouse Model at an Advanced Pathological Stage. Neurobiol. Dis. 2022, 162, 105563. [Google Scholar] [CrossRef]
- Latif-Hernandez, A.; Yang, T.; Raymond-Butler, R.; Losada, P.M.; Minhas, P.S.; White, H.; Tran, K.C.; Liu, H.; Simmons, D.A.; Langness, V.; et al. A TrkB and TrkC Partial Agonist Restores Deficits in Synaptic Function and Promotes Activity-dependent Synaptic and Microglial Transcriptomic Changes in a Late-stage Alzheimer’s Mouse Model. Alzheimer’s Dement. 2024, 20, 4434–4460. [Google Scholar] [CrossRef]
- Antonijevic, M.; Charou, D.; Davis, A.; Curel, T.; Valcarcel, M.; Ramos, I.; Villacé, P.; Claeysen, S.; Dallemagne, P.; Gravanis, A.; et al. Development of Pleiotropic TrkB and 5-HT4 Receptor Ligands as Neuroprotective Agents. Molecules 2024, 29, 515. [Google Scholar] [CrossRef]
- Todd, D.; Gowers, I.; Dowler, S.J.; Wall, M.D.; McAllister, G.; Fischer, D.F.; Dijkstra, S.; Fratantoni, S.A.; van de Bospoort, R.; Veenman-Koepke, J.; et al. A Monoclonal Antibody TrkB Receptor Agonist as a Potential Therapeutic for Huntington’s Disease. PLoS ONE 2014, 9, e87923. [Google Scholar] [CrossRef]
- Boltaev, U.; Meyer, Y.; Tolibzoda, F.; Jacques, T.; Gassaway, M.; Xu, Q.; Wagner, F.; Zhang, Y.L.; Palmer, M.; Holson, E.; et al. Multiplex Quantitative Assays Indicate a Need for Reevaluating Reported Small-Molecule TrkB Agonists. Sci. Signal 2017, 10, eaal1670. [Google Scholar] [CrossRef]
- Pankiewicz, P.; Szybiński, M.; Kisielewska, K.; Gołębiowski, F.; Krzemiński, P.; Rutkowska-Włodarczyk, I.; Moszczyński-Pętkowski, R.; Gurba-Bryśkiewicz, L.; Delis, M.; Mulewski, K.; et al. Do Small Molecules Activate the TrkB Receptor in the Same Manner as BDNF? Limitations of Published TrkB Low Molecular Agonists and Screening for Novel TrkB Orthosteric Agonists. Pharmaceuticals 2021, 14, 704. [Google Scholar] [CrossRef]
- Chen, J.; Chua, K.-W.; Chua, C.C.; Yu, H.; Pei, A.; Chua, B.H.L.; Hamdy, R.C.; Xu, X.; Liu, C.-F. Antioxidant Activity of 7,8-Dihydroxyflavone Provides Neuroprotection against Glutamate-Induced Toxicity. Neurosci. Lett. 2011, 499, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Jakaria, M.; Belaidi, A.A.; Southon, A.; Dent, K.A.; Lane, D.J.R.; Bush, A.I.; Ayton, S. Receptor-Independent Anti-Ferroptotic Activity of TrkB Modulators. Int. J. Mol. Sci. 2022, 23, 16205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhu, L.; Lu, S.; Li, M.; Bai, M.; Li, Y.; Xu, E. The Antidepressant-like Effect of Formononetin on Chronic Corticosterone-Treated Mice. Brain Res. 2022, 1783, 147844. [Google Scholar] [CrossRef] [PubMed]
- Narducci, D.; Charou, D.; Rogdakis, T.; Zota, I.; Bafiti, V.; Zervou, M.; Katsila, T.; Gravanis, A.; Prousis, K.C.; Charalampopoulos, I.; et al. A Quest for the Stereo-Electronic Requirements for Selective Agonism for the Neurotrophin Receptors TrkA and TrkB in 17-Spirocyclic-Dehydroepiandrosterone Derivatives. Front. Mol. Neurosci. 2023, 16, 1244133. [Google Scholar] [CrossRef]
- Shanks, H.R.C.; Chen, K.; Reiman, E.M.; Blennow, K.; Cummings, J.L.; Massa, S.M.; Longo, F.M.; Börjesson-Hanson, A.; Windisch, M.; Schmitz, T.W. P75 Neurotrophin Receptor Modulation in Mild to Moderate Alzheimer Disease: A Randomized, Placebo-Controlled Phase 2a Trial. Nat. Med. 2024, 30, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Diniz, C.R.A.F.; Crestani, A.P.; Casarotto, P.C.; Biojone, C.; Cannarozzo, C.; Winkel, F.; Prozorov, M.A.; Kot, E.F.; Goncharuk, S.A.; Marques, D.B.; et al. Fluoxetine and Ketamine Trigger the P75NTR Proteolytic Pathway and Enhance Extinction Memory and Brain Plasticity through P75NTR. Biol. Psychiatry, 2024; in press. [Google Scholar] [CrossRef]
- Ohashi, Y.; Norimine, Y.; Hoshikawa, T.; Yoshida, Y.; Kobayashi, Y.; Sato, N.; Hagiwara, K. Pentacyclic Compounds. U.S. Patent US10239889B1, 26 March 2019. [Google Scholar]
- Aceves, P.; Hall, N.; Dayal, S.; Yagi, T.; Chang, J.; Mikamoto, M.; Ringheim, G.E.; Takesuya, T.; Hiramatsu, N.; Gordon, R.; et al. First-in-Human (FIH), Single- and Multiple-Ascending-Dose (SAD/MAD) Studies in Healthy Subjects of E2511, a Novel Tropomyosin Receptor Kinase a (TrkA) Positive Allosteric Modulator (PAM). Alzheimer’s Dement. 2023, 19, e066208. [Google Scholar] [CrossRef]
- Nordvall, G.; Forsell, P. Triazine Derivatives for Treating Diseases Relating to Neurotrophins. WO2019162702A1, 26 February 2019. [Google Scholar]
- Nordvall, G.; Forsell, P. 4-Substituted Phenyl-1,3,5-triazine Derivatives as Modulators of Trk Receptors. WO2020002950A1, 28 June 2019. [Google Scholar]
- Nordvall, G.; Forsell, P. Triazine Derivatives for Treating Diseases Relating to Neurotrophins. WO2021038241A1, 28 August 2020. [Google Scholar]
- Nordvall, G.; Forsell, P. 4-Substituted Phenyl-1,3,5-triazine Derivatives as Modulators of Trk Receptors. WO2020002949A1, 28 June 2019. [Google Scholar]
- Nordvall, G.; Forsell, P.; Sandin, J. Triazinetrione Derivatives and Their Use as Modulators of Neurotrophin Receptor and Receptor Tyrosine Kinases. WO2018115891A1, 21 December 2017. 71p. [Google Scholar]
- Nilsson, B.; Bylund, J.; Halldin, M.M.; Rother, M.; Rein-Hedin, E.; Önnestam, K.; Segerdahl, M. ACD856, a Novel Positive Allosteric Modulator of Trk Receptors, Single Ascending Doses in Healthy Subjects: Safety and Pharmacokinetics. Eur. J. Clin. Pharmacol. 2024, 80, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Önnestam, K.; Nilsson, B.; Rother, M.; Rein-Hedin, E.; Bylund, J.; Anderer, P.; Kemethofer, M.; Halldin, M.M.; Sandin, J.; Segerdahl, M. Safety, Tolerability, Pharmacokinetics and Quantitative Electroencephalography Assessment of ACD856, a Novel Positive Allosteric Modulator of Trk-Receptors Following Multiple Doses in Healthy Subjects. J. Prev. Alzheimer’s Dis. 2023, 10, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, R.B. Modulation of Tropomyosin Receptor Kinase for the Treatment of Neurotrophin Diseases: Alzheimer’s, Huntington’s and Parkinson’s. ACS Med. Chem. Lett. 2019, 10, 1590–1591. [Google Scholar] [CrossRef]
- Cheng, P.-L.; Song, A.-H.; Wong, Y.-H.; Wang, S.; Zhang, X.; Poo, M.-M. Self-Amplifying Autocrine Actions of BDNF in Axon Development. Proc. Natl. Acad. Sci. USA 2011, 108, 18430–18435. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Numakawa, T.; Adachi, N.; Ooshima, Y.; Odaka, H.; Yoshimura, A.; Kunugi, H. Self-Amplified BDNF Transcription Is a Regulatory System for Synaptic Maturation in Cultured Cortical Neurons. Neurochem. Int. 2015, 91, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Madjid, N.; Lidell, V.; Nordvall, G.; Lindskog, M.; Ögren, S.-O.; Forsell, P.; Sandin, J. Antidepressant Effects of Novel Positive Allosteric Modulators of Trk-Receptor Mediated Signaling—A Potential Therapeutic Concept? Psychopharmacology 2023, 240, 1789–1804. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Ye, Y.; Sasaki, K.; Kamakura, T.; Ringheim, G.; Giorgi, L.; Penner, N.; Horie, K.; Devanarayan, V.; Sachdev, P. E2511, a Novel TrkA Modulator, Engages its CNS Cholinergic Target in a Phase 1 Clinical Study. In Proceedings of the 16th Clinical Trials on Alzheimer’s Disease (CTAD), Boston, MA, USA, 24–27 October 2023; Volume 10, pp. 4–55. [Google Scholar] [CrossRef]
- DiBenedetti, D.B.; Slota, C.; Wronski, S.L.; Vradenburg, G.; Comer, M.; Callahan, L.F.; Winfield, J.; Rubino, I.; Krasa, H.B.; Hartry, A.; et al. Assessing What Matters Most to Patients with or at Risk for Alzheimer’s and Care Partners: A Qualitative Study Evaluating Symptoms, Impacts, and Outcomes. Alzheimers Res. Ther. 2020, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Arosio, B.; Guerini, F.R.; Voshaar, R.C.O.; Aprahamian, I. Blood Brain-Derived Neurotrophic Factor (BDNF) and Major Depression: Do We Have a Translational Perspective? Front. Behav. Neurosci. 2021, 15, 626906. [Google Scholar] [CrossRef]
- Wang, Y.; Li, O.; Li, N.; Sha, Z.; Zhao, Z.; Xu, J. Association between the BDNF Val66Met Polymorphism and Major Depressive Disorder: A Systematic Review and Meta-Analysis. Front. Psychiatry 2023, 14, 1143833. [Google Scholar] [CrossRef]
- Białecka, M.; Kurzawski, M.; Roszmann, A.; Robowski, P.; Sitek, E.J.; Honczarenko, K.; Mak, M.; Deptuła-Jarosz, M.; Gołąb-Janowska, M.; Droździk, M.; et al. BDNF G196A (Val66Met) Polymorphism Associated with Cognitive Impairment in Parkinson’s Disease. Neurosci. Lett. 2014, 561, 86–90. [Google Scholar] [CrossRef]
- Ferrer, I.; Goutan, E.; Marín, C.; Rey, M.J.; Ribalta, T. Brain-Derived Neurotrophic Factor in Huntington Disease. Brain Res. 2000, 866, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Deng, X.-H.; Lao, H.; Wu, Z.-C.; Chen, Y.; Luo, Y.; Wu, H.; Wang, J.; Fu, Q.-L.; Xiong, H. BDNF-Enriched Small Extracellular Vesicles Protect against Noise-Induced Hearing Loss in Mice. J. Control. Release 2023, 364, 546–561. [Google Scholar] [CrossRef] [PubMed]
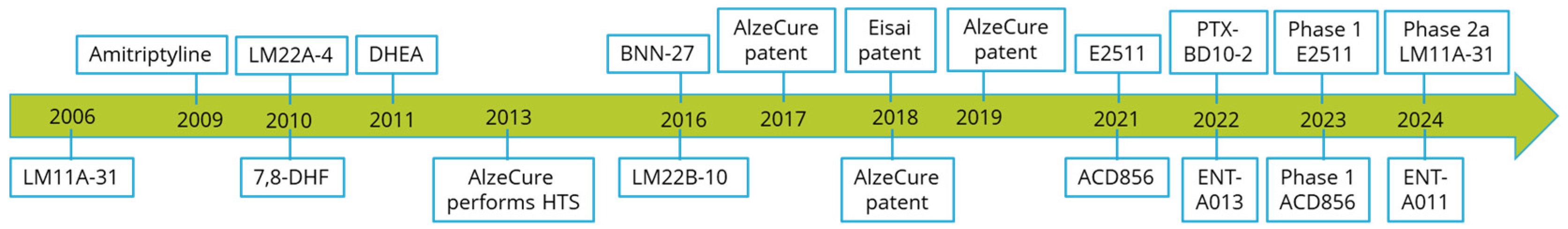


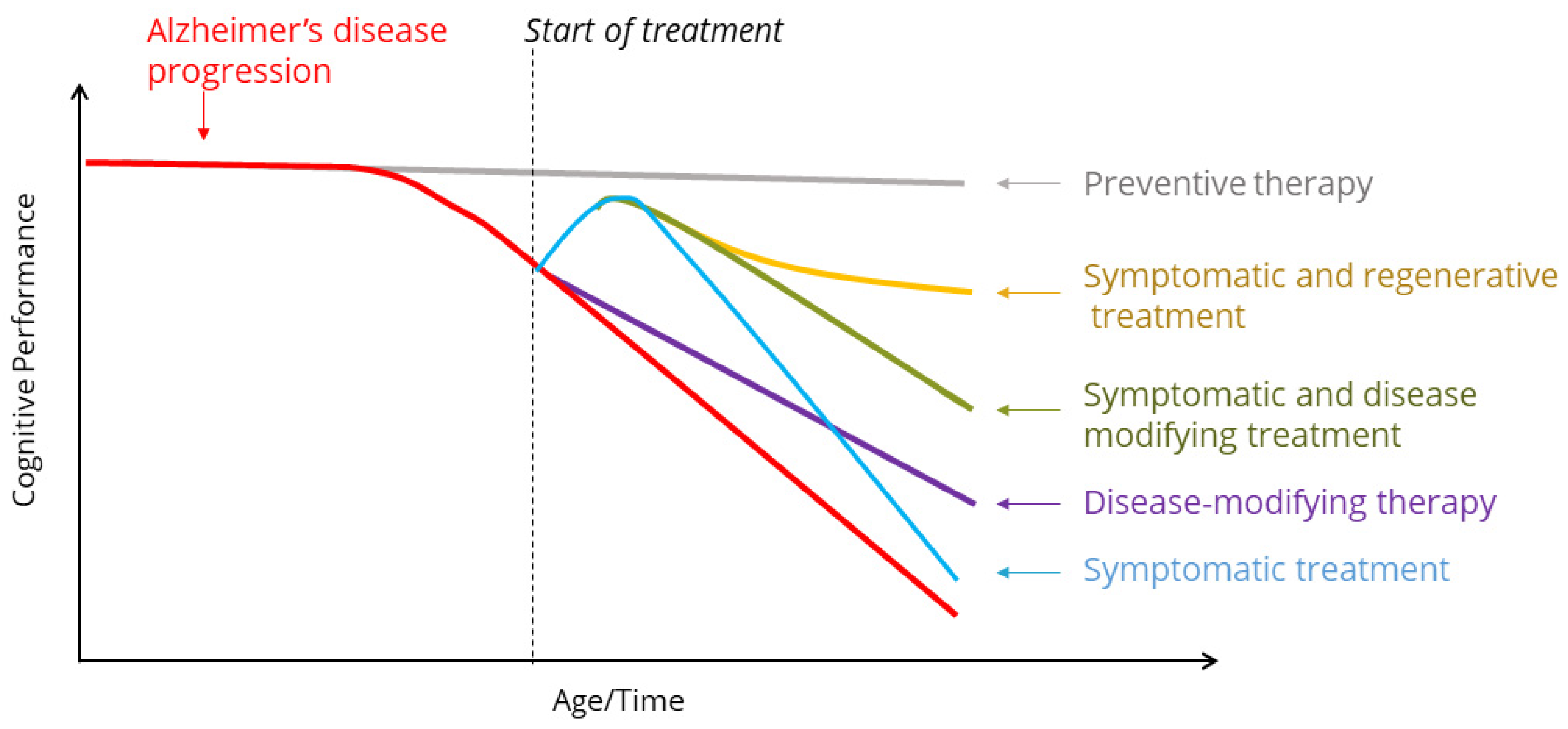
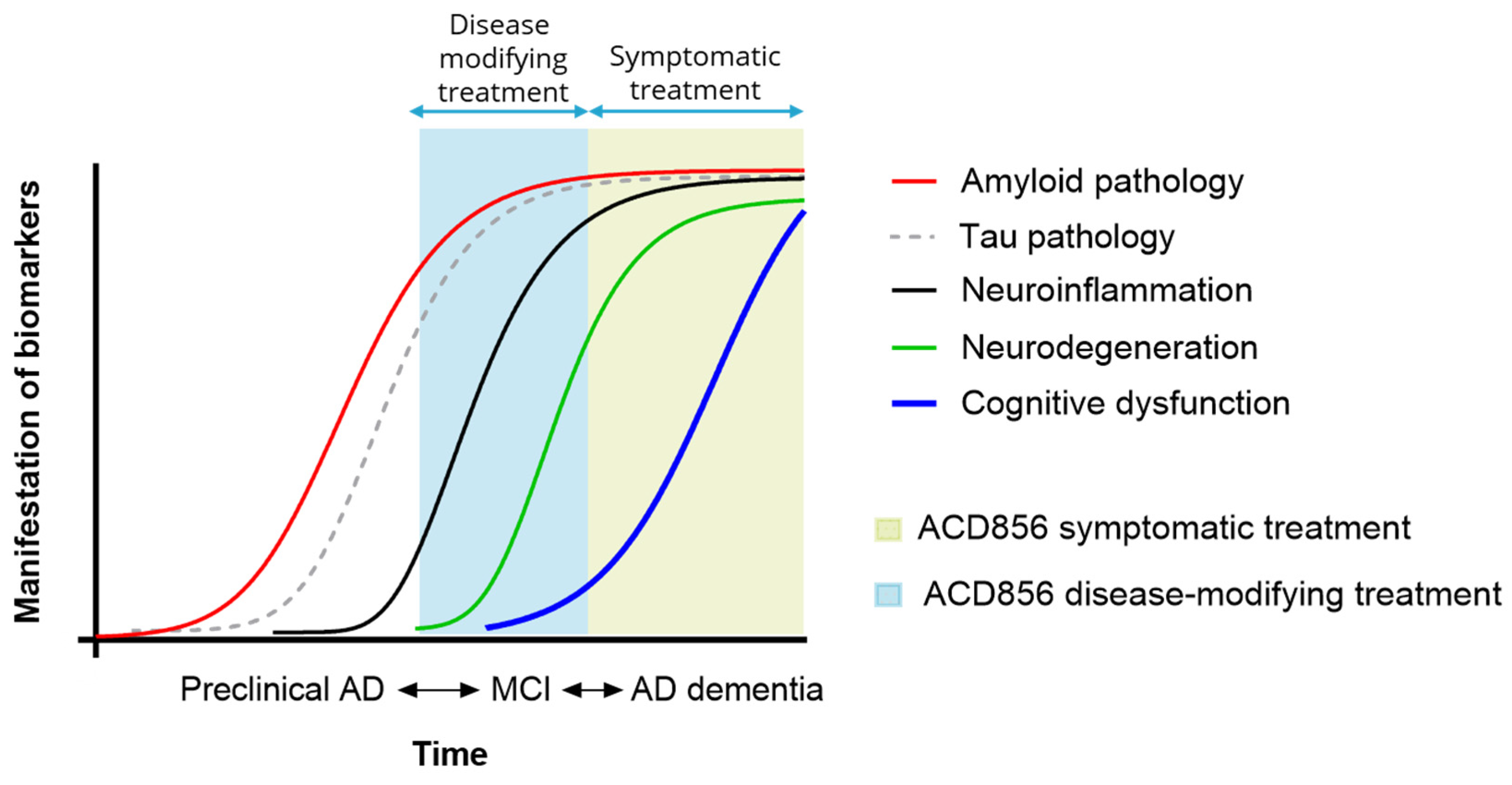


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACD856 | Single dose | Multiple dose—7 d | |||||||||||||||
| Placebo | ACD856 (mg) | Total | Placebo | ACD856 (mg) | Total | ||||||||||||
| 1 | 3 | 10 | 20 | 40 | 75 | 150 | Total | 10 | 30 | 90 | Total | ||||||
| N | 14 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 42 | 56 | 6 | 6 | 6 | 6 | 18 | 24 | |
| Sex (M/F) | 11/3 | 5/1 | 5/1 | 5/1 | 5/1 | 5/1 | 6/0 | 6/0 | 37/5 | 5/1 | 6/0 | 5/1 | 5/1 | 16/2 | 21/3 | ||
| Age, years (mean[SD]) | 43.9 (12.8) | 38.5 (13) | 39.2 (17) | 35.3 (11) | 33.0 (13) | 44.0 (16) | 32 (6) | 30 (6) | 36 | 40.5 | 46.3 (14) | 29.5 (7.1) | 41.3 (16) | 45 (15) | 38.6 | 40.0 | |
| Adverse events (AE) | No dose-dependent, serious or severe treatment-related AEs. Most common AE was headache due to lumbar punctures. | ||||||||||||||||
| Safety | No significant findings in vital signs, ECG, labs, EEG (MAD only), or physical examinations were reported | ||||||||||||||||
| E2511 | Single dose | Multiple dose—14 d | |||||||||||||||
| Placebo | E2511 (mg) | Total | Placebo | E2511 (mg) | Total | ||||||||||||
| 5 | 10 | 20 | 40 | 80 | Total | 10 | 30 | 90 | Total | ||||||||
| N | 10 | 6 | 6 | 6 | 6 | 6 | 30 | 40 | 6 | 6 | 6 | 6 | 18 | 24 | |||
| Sex (M/F) | 6/4 | 3/3 | 4/2 | 5/1 | 4/2 | 4/2 | 20/10 | 26/14 | 6/0 | 4/2 | 3/3 | 4/2 | 11/7 | 17/7 | |||
| Age, years (mean[SD]) | 34 (9) | 38 (13) | 40 (13) | 34 (8) | 35 (9) | 36 (9) | 36 (10) | 36 (10) | 45 (6) | 27 (4) | 36 (9) | 32 (5) | 32 (7) | 35 (8) | |||
| Adverse events (AE) | No dose-dependent, serious or severe treatment-related AEs. Most common AEs was headache due to lumbar punctures. | ||||||||||||||||
| Safety | No significant findings in vital signs, ECG, labs, EEG, or physical examinations were reported | ||||||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forsell, P.; Parrado Fernández, C.; Nilsson, B.; Sandin, J.; Nordvall, G.; Segerdahl, M. Positive Allosteric Modulators of Trk Receptors for the Treatment of Alzheimer’s Disease. Pharmaceuticals 2024, 17, 997. https://doi.org/10.3390/ph17080997
Forsell P, Parrado Fernández C, Nilsson B, Sandin J, Nordvall G, Segerdahl M. Positive Allosteric Modulators of Trk Receptors for the Treatment of Alzheimer’s Disease. Pharmaceuticals. 2024; 17(8):997. https://doi.org/10.3390/ph17080997
Chicago/Turabian StyleForsell, Pontus, Cristina Parrado Fernández, Boel Nilsson, Johan Sandin, Gunnar Nordvall, and Märta Segerdahl. 2024. "Positive Allosteric Modulators of Trk Receptors for the Treatment of Alzheimer’s Disease" Pharmaceuticals 17, no. 8: 997. https://doi.org/10.3390/ph17080997
APA StyleForsell, P., Parrado Fernández, C., Nilsson, B., Sandin, J., Nordvall, G., & Segerdahl, M. (2024). Positive Allosteric Modulators of Trk Receptors for the Treatment of Alzheimer’s Disease. Pharmaceuticals, 17(8), 997. https://doi.org/10.3390/ph17080997