


Pharmacophore-Based Study: An In Silico Perspective for the Identification of Potential New Delhi Metallo-β-lactamase-1 (NDM-1) Inhibitors


Abstract

1. Introduction
2. Results and Discussion
2.1. Protein Structure and Docking
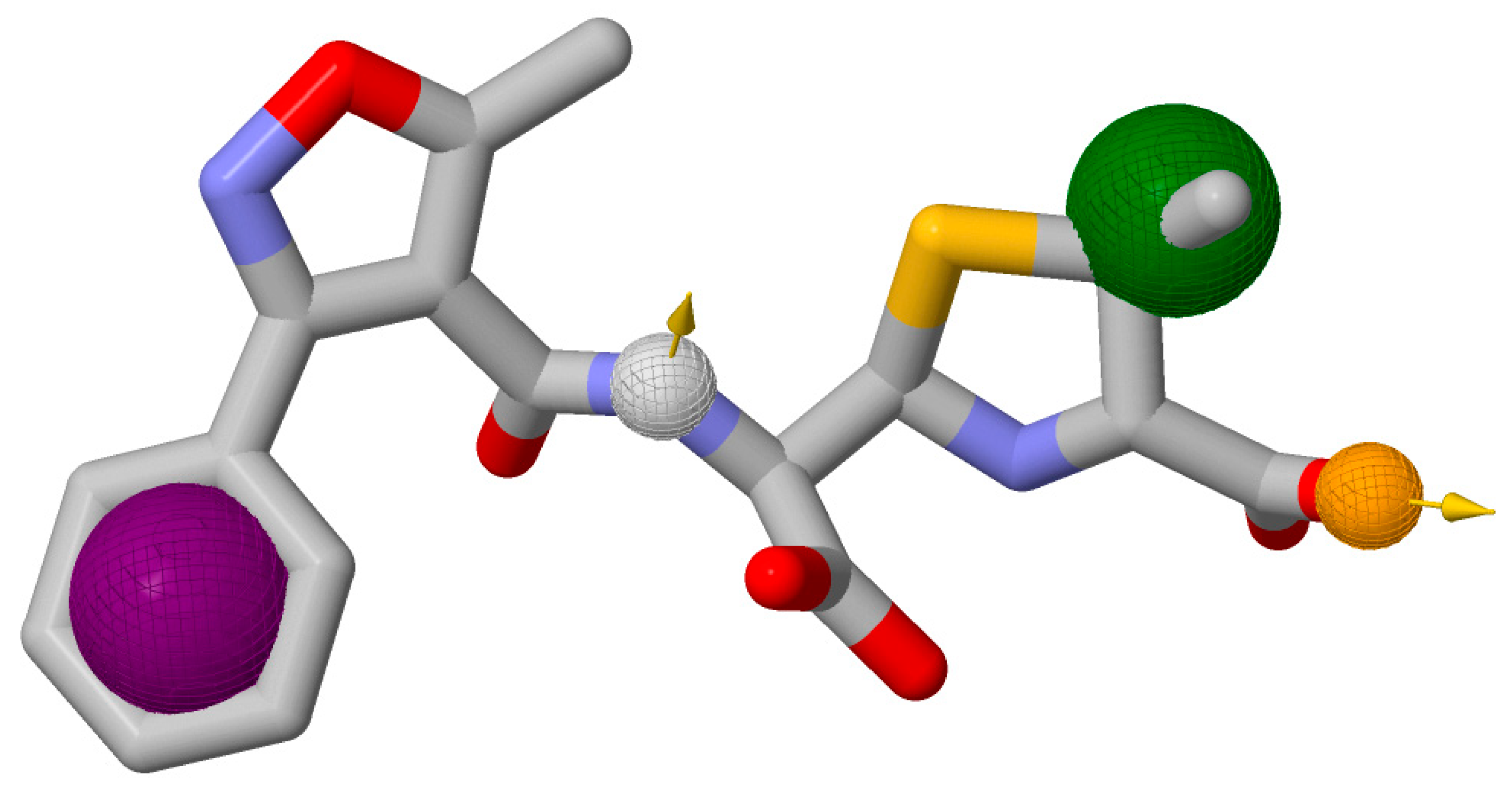
2.2. Pharmacophore
2.3. Virtual Screening
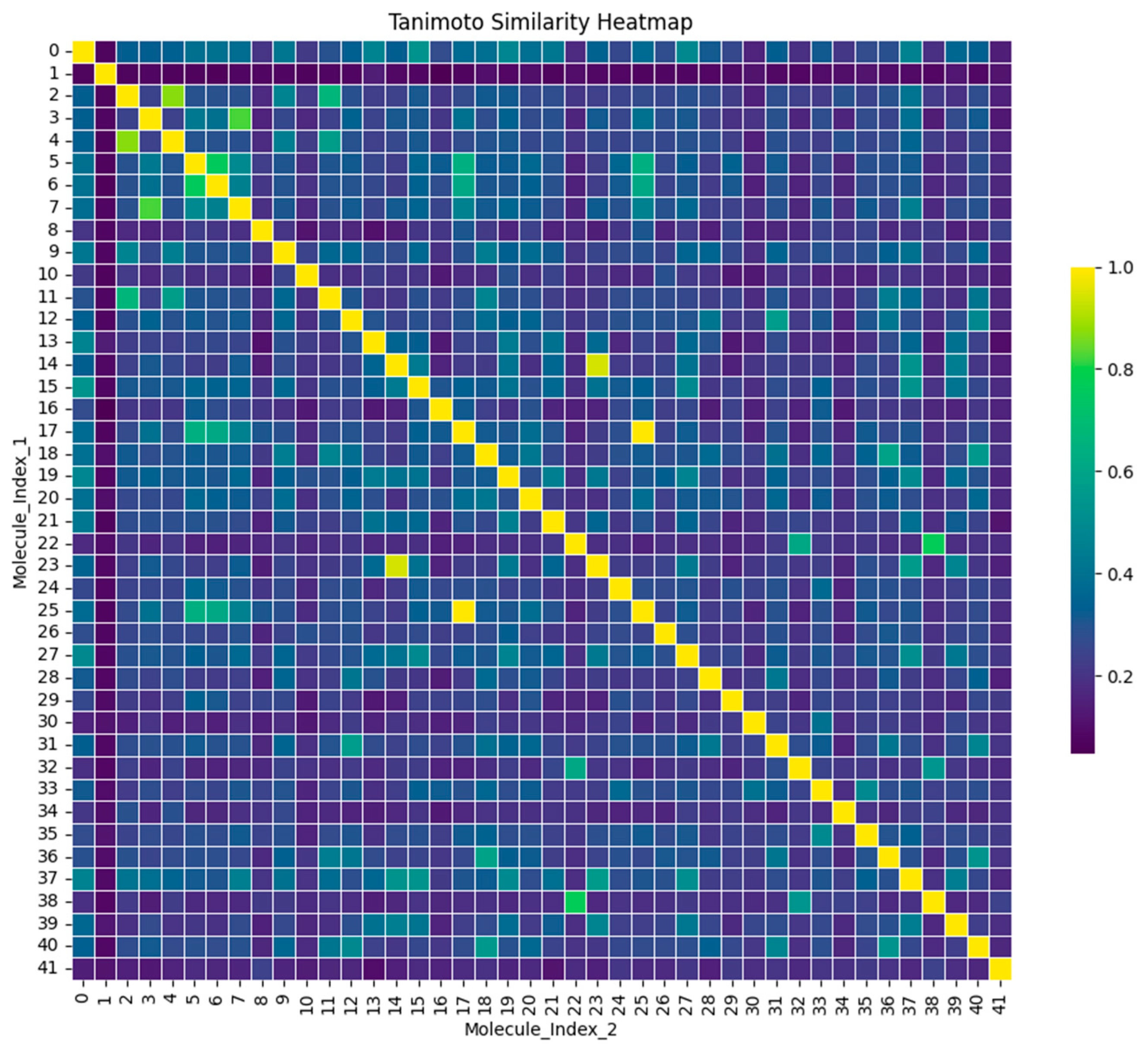
2.4. Tanimoto and Clustering
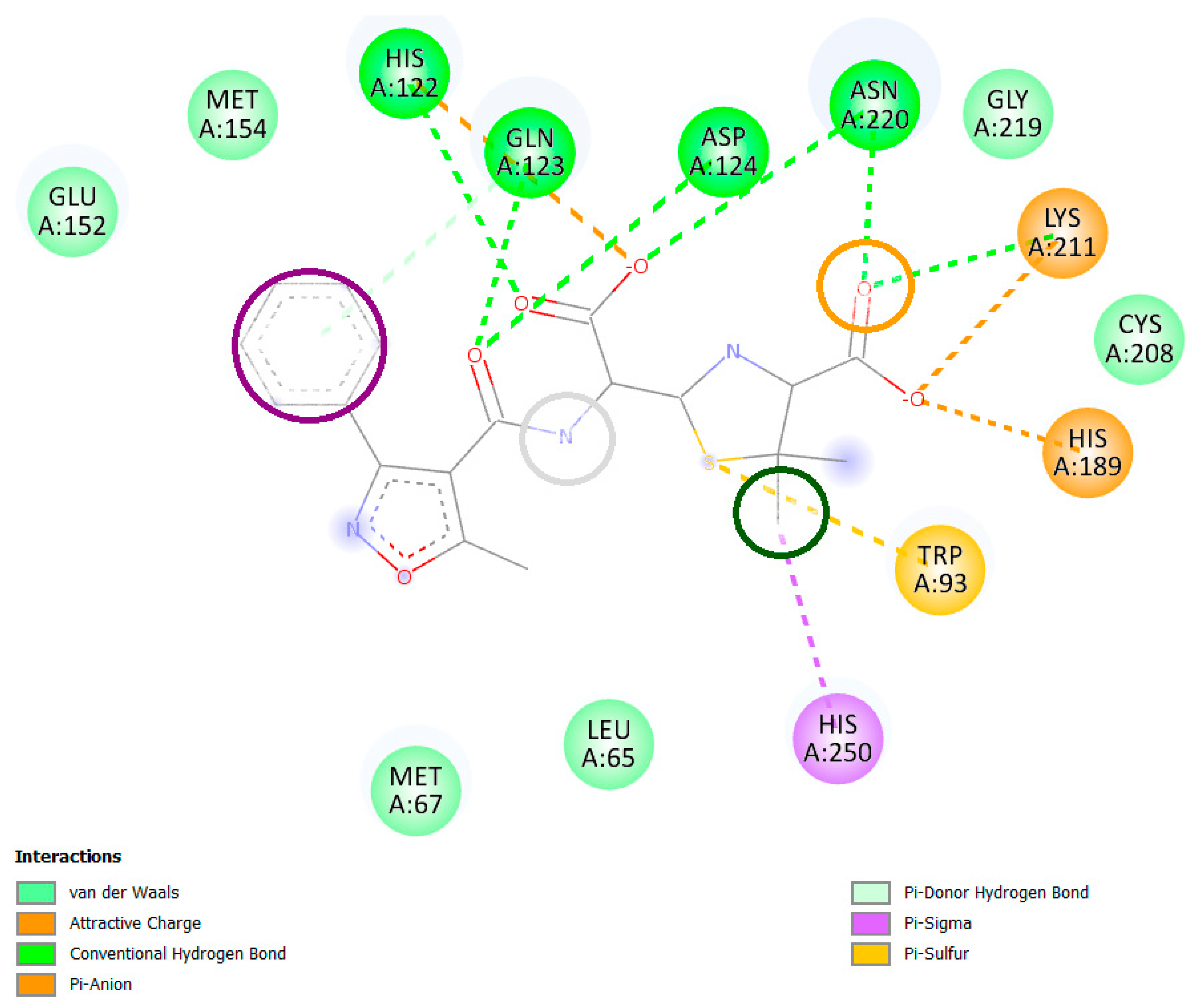
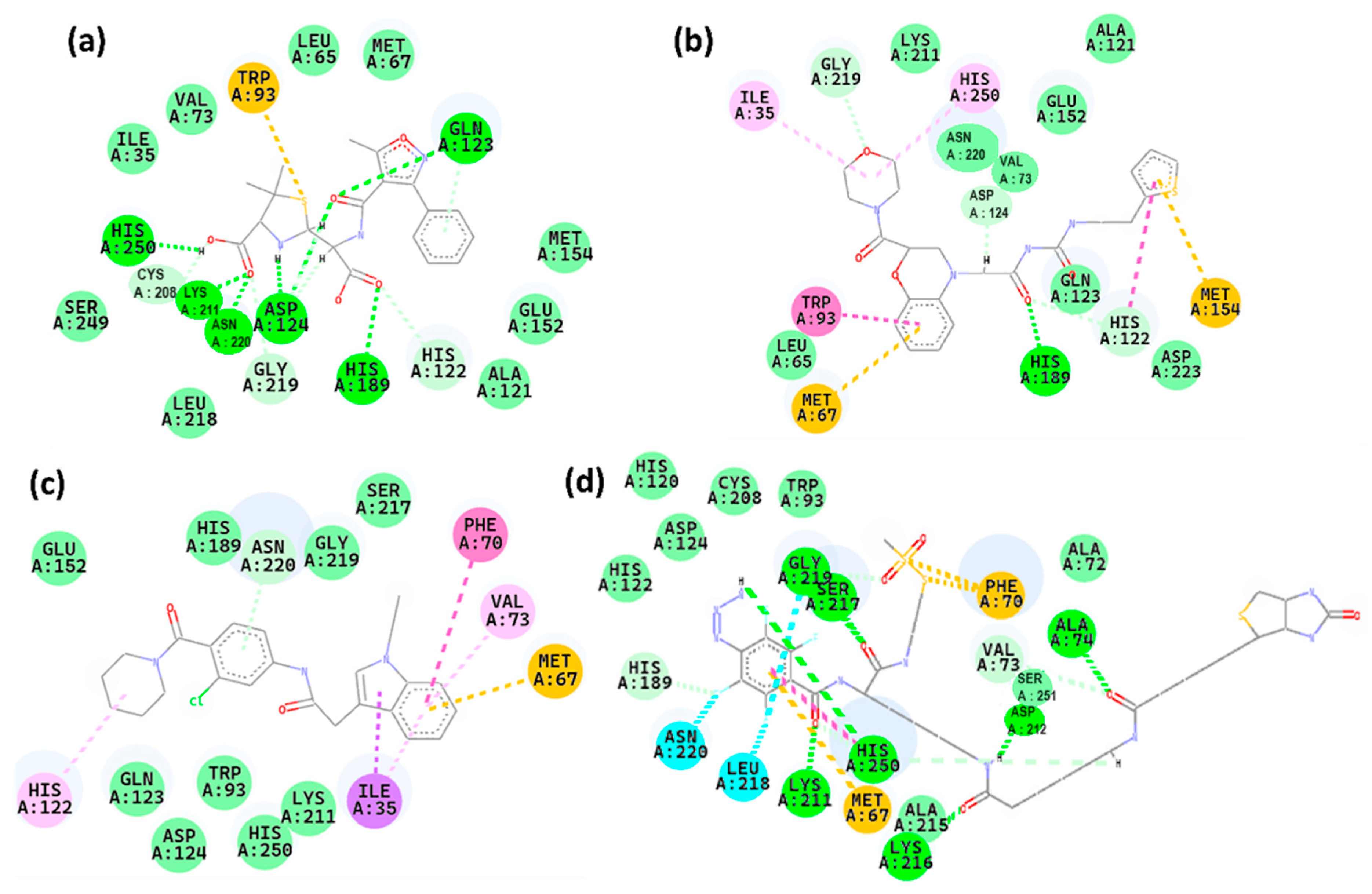
2.5. Interaction Analysis
2.6. ADMET Properties
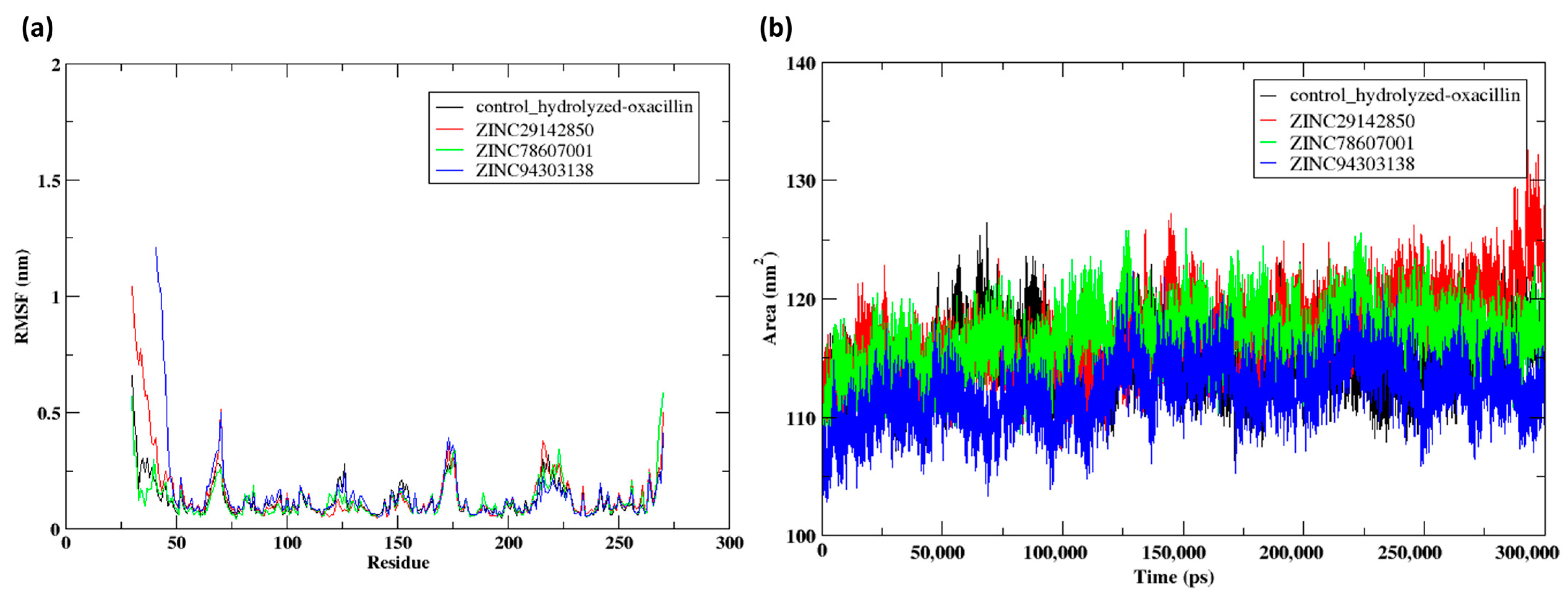
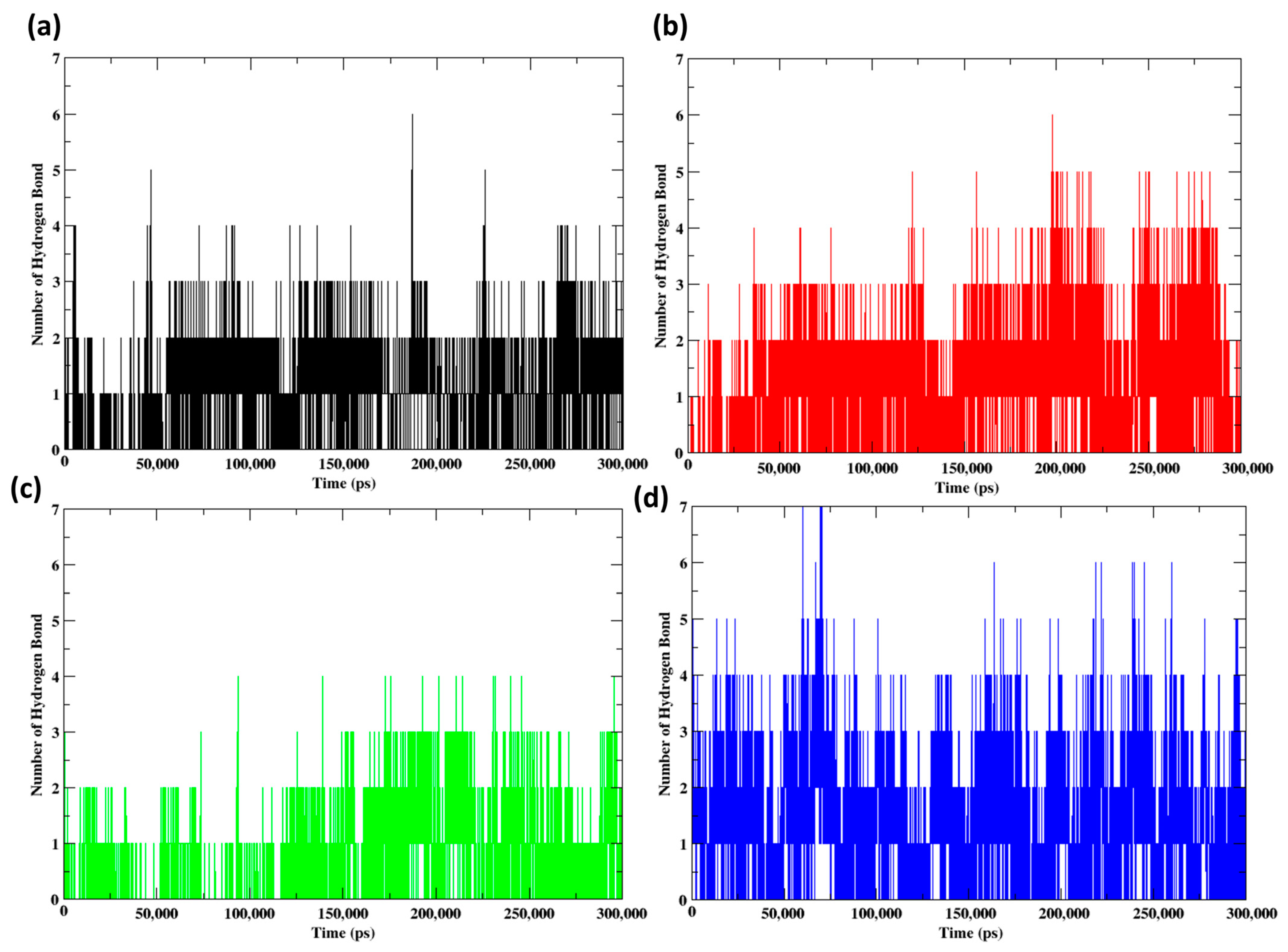
2.7. Molecular Dynamics Simulation (300 ns)
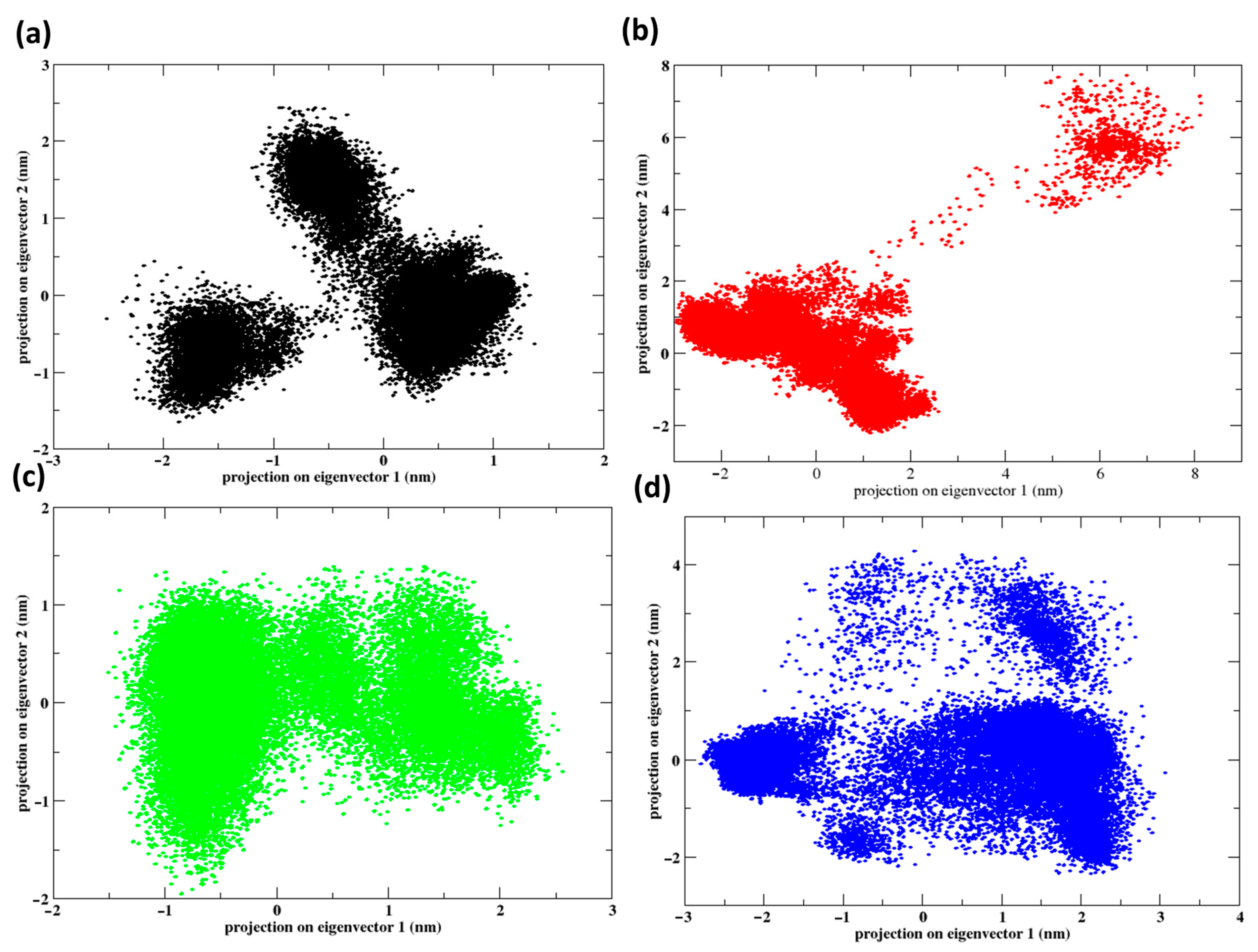
2.8. Principal Component Analysis
2.9. Free Energy Landscape
2.10. Binding Free Energies
3. Material and Methods
3.1. Protein Structure and Molecular Docking
3.2. Pharmacophore
3.3. Virtual Screening
3.4. Tanimoto Similarity and Clustering
3.5. ADMET Properties
3.6. Molecular Dynamics Simulation
3.7. PCA and FEL
3.8. MM/GBSA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- King, D.T.; Worrall, L.J.; Gruninger, R.; Strynadka, N.C. New Delhi metallo-β-lactamase: Structural insights into β-lactam recognition and inhibition. J. Am. Chem. Soc. 2012, 134, 11362–11365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Yang, K.W.; Zhou, Y.J.; LaCuran, A.E.; Oelschlaeger, P.; Crowder, M.W. Diaryl-substituted azolylthioacetamides: Inhibitor discovery of New Delhi Metallo-β-Lactamase-1 (NDM-1). ChemMedChem 2014, 9, 2445–2448. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, X.; Yu, T.; Liu, Y.; Zhang, X.; Yao, Y.; Feng, X.; Liu, H.; Yu, D.; Ma, L. Discovery of thiosemicarbazone derivatives as effective New Delhi metallo-β-lactamase-1 (NDM-1) inhibitors against NDM-1 producing clinical isolates. Acta Pharm. Sin. B 2021, 11, 203–221. [Google Scholar] [CrossRef]
- Thomas, P.W.; Zheng, M.; Wu, S.; Guo, H.; Liu, D.; Xu, D.; Fast, W. Characterization of purified New Delhi metallo-β-lactamase-1. Biochemistry 2011, 50, 10102–10113. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Yu, Y.; Chen, H.; Cao, X.; Lao, X.; Fang, Y.; Shi, Y.; Chen, J.; Zheng, H. Inhibitor discovery of full-length New Delhi metallo-β-lactamase-1 (NDM-1). PLoS ONE 2013, 8, e62955. [Google Scholar] [CrossRef]
- Walsh, T.R.; Weeks, J.; Livermore, D.M.; Toleman, M.A. Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: An environmental point prevalence study. Lancet Infect. Dis. 2011, 11, 355–362. [Google Scholar] [CrossRef]
- Fomda, B.A.; Khan, A.; Zahoor, D. NDM-1 (New Delhi metallo beta lactamase-1) producing Gram-negative bacilli: Emergence & clinical implications. Indian J. Med. Res. 2014, 140, 672–678. [Google Scholar]
- Salari-Jazi, A.; Mahnam, K.; Sadeghi, P.; Damavandi, M.S.; Faghri, J. Discovery of potential inhibitors against New Delhi metallo-β-lactamase-1 from natural compounds: In silico-based methods. Sci. Rep. 2021, 11, 2390. [Google Scholar] [CrossRef] [PubMed]
- Spyrakis, F.; Santucci, M.; Maso, L.; Cross, S.; Gianquinto, E.; Sannio, F.; Verdirosa, F.; De Luca, F.; Docquier, J.-D.; Cendron, L. Virtual screening identifies broad-spectrum β-lactamase inhibitors with activity on clinically relevant serine-and metallo-carbapenemases. Sci. Rep. 2020, 10, 12763. [Google Scholar] [CrossRef]
- Linciano, P.; Cendron, L.; Gianquinto, E.; Spyrakis, F.; Tondi, D. Ten years with New Delhi metallo-β-lactamase-1 (NDM-1): From structural insights to inhibitor design. ACS Infect. Dis. 2018, 5, 9–34. [Google Scholar] [CrossRef]
- Li, X.; Zhao, D.; Li, W.; Sun, J.; Zhang, X. Enzyme inhibitors: The best strategy to tackle superbug NDM-1 and its variants. Int. J. Mol. Sci. 2021, 23, 197. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. Classification of beta-lactamases: Groups 1, 2a, 2b, and 2b’. Antimicrob. Agents Chemother. 1989, 33, 264–270. [Google Scholar] [CrossRef]
- Ambler, R.P. The structure of β-lactamases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1980, 289, 321–331. [Google Scholar] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, Y.; Tesar, C.; Mire, J.; Jedrzejczak, R.; Binkowski, A.; Babnigg, G.; Sacchettini, J.; Joachimiak, A. Structure of apo- and monometalated forms of NDM-1--a highly potent carbapenem-hydrolyzing metallo-β-lactamase. PLoS ONE 2011, 6, e24621. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, g.; Hao, Q. Crystal structure of NDM-1 reveals a common β-lactam hydrolysis mechanism. FASEB J. 2011, 25, 2574–2582. [Google Scholar] [CrossRef]
- King, D.; Strynadka, N. Crystal structure of New Delhi metallo-β-lactamase reveals molecular basis for antibiotic resistance. Protein Sci. 2011, 20, 1484–1491. [Google Scholar] [CrossRef]
- Chiou, J.; Leung, T.Y.-C.; Chen, S. Molecular mechanisms of substrate recognition and specificity of New Delhi metallo-β-lactamase. Antimicrob. Agents Chemother. 2014, 58, 5372–5378. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, J.; Niu, G.; Shui, W.; Sun, Y.; Zhou, H.; Zhang, Y.; Yang, C.; Lou, Z.; Rao, Z. A structural view of the antibiotic degradation enzyme NDM-1 from a superbug. Protein Cell 2011, 2, 384–394. [Google Scholar] [CrossRef]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef] [PubMed]
- Wallach, I.; Jaitly, N.; Nguyen, K.; Schapira, M.; Lilien, R. Normalizing molecular docking rankings using virtually generated decoys. J. Chem. Inf. Model. 2011, 51, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular Similarity in Medicinal Chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Y.; Gao, Y.; Niu, X. Discovery of the novel inhibitor against New Delhi Metallo-β-Lactamase based on virtual screening and molecular modelling. Int. J. Mol. Sci. 2020, 21, 3567. [Google Scholar] [CrossRef]
- Rehman, M.T.; AlAjmi, M.F.; Hussain, A.; Rather, G.M.; Khan, M.A. High-throughput virtual screening, molecular dynamics simulation, and enzyme kinetics identified ZINC84525623 as a potential inhibitor of NDM-1. Int. J. Mol. Sci. 2019, 20, 819. [Google Scholar] [CrossRef]
- Li, X.; Wang, Q.; Zheng, J.; Guan, Y.; Liu, C.; Han, J.; Liu, S.; Liu, T.; Xiao, C.; Wang, X. PHT427 as an effective New Delhi metallo-β-lactamase-1 (NDM-1) inhibitor restored the susceptibility of meropenem against Enterobacteriaceae producing NDM-1. Front. Microbiol. 2023, 14, 1168052. [Google Scholar] [CrossRef]
- Bibi, Z.; Asghar, I.; Ashraf, N.M.; Zeb, I.; Rashid, U.; Hamid, A.; Ali, M.K.; Hatamleh, A.A.; Al-Dosary, M.A.; Ahmad, R. Prediction of Phytochemicals for Their Potential to Inhibit New Delhi Metallo β-Lactamase (NDM-1). Pharmaceuticals 2023, 16, 1404. [Google Scholar] [CrossRef]
- Salih, T.; Ali, P.G. Rational design of novel compounds to serve as potential NDM-1 inhibitors using molecular docking, molecular dynamics simulation, and physicochemical studies. Mol. Simul. 2023, 49, 1373–1387. [Google Scholar] [CrossRef]
- Muteeb, G.; Alsultan, A.; Farhan, M.; Aatif, M. Risedronate and methotrexate are high-affinity inhibitors of New Delhi Metallo-β-Lactamase-1 (NDM-1): A drug repurposing approach. Molecules 2022, 27, 1283. [Google Scholar] [CrossRef]
- Bank, R.P.D. RCSB PDB—4EYB: Crystal Structure of NDM-1 Bound to Hydrolyzed Oxacillin. Available online: https://www.rcsb.org/structure/4EYB (accessed on 25 April 2024).
- Rivière, G.; Oueslati, S.; Gayral, M.; Créchet, J.B.; Nhiri, N.; Jacquet, E.; Cintrat, J.C.; Giraud, F.; Van Heijenoort, C.; Lescop, E.; et al. NMR Characterization of the Influence of Zinc (II) Ions on the Structural and Dynamic Behavior of the New Delhi Metallo-β-Lactamase-1 and on the Binding with Flavonols as Inhibitors. ACS Omega 2020, 5, 10466–10480. [Google Scholar] [CrossRef] [PubMed]
- Hornstein, M.; Sautjeau-Rostoker, C.; Péduzzi, J.; Vessières, A.; Hong, L.T.H.; Barthélémy, M.; Scavizzi, M.; Labia, R. Oxacillin-hydrolyzing β-lactamase involved in resistance to imipenem in Acinetobacter baumannii. FEMS Microbiol. Lett. 1997, 153, 333–339. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Koes, D.R.; Pabon, N.A.; Deng, X.; Phillips, M.A.; Camacho, C.J. A Teach-Discover-Treat application of ZincPharmer: An online interactive pharmacophore modeling and virtual screening tool. PLoS ONE 2015, 10, e0134697. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-source cheminformatics. Release 2014.03. 1. Zenodo 2014. Available online: https://www.semanticscholar.org/paper/RDKit%3A-Open-source-cheminformatics.-Release-Landrum/548846422a26d7173c176ba4858d1bfb26d9f7f2 (accessed on 25 April 2024).
- Miranda-Quintana, R.A.; Bajusz, D.; Rácz, A.; Héberger, K. Differential Consistency Analysis: Which Similarity Measures Can Be Applied in Drug Discovery? Mol. Inform. 2021, 40, 2060017. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Suyash, S.; Jha, A.; Maitra, P.; Punia, P.; Mishra, A. Differentiating Stable and Unstable Protein Using Convolution Neural Network and Molecular Dynamics Simulations. Comput. Biol. Chem. 2024, 110, 108081. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell Jr, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Harrach, M.F.; Drossel, B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014, 140, 174501. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Martoňák, R.; Laio, A.; Parrinello, M. Predicting crystal structures: The Parrinello-Rahman method revisited. Phys. Rev. Lett. 2003, 90, 075503. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, E.; Mereghetti, P.; Fantucci, P.; Grandori, R.; De Gioia, L. Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: The myoglobin case. J. Mol. Graph. Model. 2009, 27, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.no | Top BS (kcal/mol) | Compound ID | Normalized BS | S.no | Top BS (kcal/mol) | Compound ID | Normalized BS |
|---|---|---|---|---|---|---|---|
| 1 | −8.884 | ZINC14802618 | −7.86967 | 35 | −7.131 | ZINC29142850 | −6.72855 |
| 2 | −8.104 | ZINC94303138 | −7.79811 | 36 | −7.305 | ZINC14265783 | −6.72711 |
| 3 | −8.696 | ZINC71983666 | −7.71111 | 37 | −6.965 | ZINC72008621 | −6.70222 |
| 4 | −8.296 | ZINC02927577 | −7.53545 | 38 | −7.233 | ZINC11748858 | −6.69533 |
| 5 | −8.45 | ZINC71986759 | −7.50078 | 39 | −7.442 | ZINC04851607 | −6.69456 |
| 6 | −8.168 | ZINC71924775 | −7.43711 | 40 | −6.921 | ZINC40034395 | −6.63233 |
| 7 | −7.715 | ZINC71924780 | −7.40022 | 41 | −6.824 | ZINC78498764 | −6.62466 |
| 8 | −8.327 | ZINC02927521 | −7.39134 | 42 | −7.307 | control_hydrolyzed-oxacillin | −6.59467 |
| 9 | −7.827 | ZINC35548858 | −7.29667 | 43 | −6.939 | ZINC12384097 | −6.56033 |
| 10 | −7.927 | ZINC78607001 | −7.23967 | 44 | −7.142 | ZINC04851208 | −6.52722 |
| 11 | −7.496 | ZINC32932272 | −7.21522 | 45 | −6.751 | ZINC39357686 | −6.52045 |
| 12 | −8.214 | ZINC32932744 | −7.19656 | 46 | −7.175 | ZINC40868984 | −6.51555 |
| 13 | −7.915 | ZINC40133417 | −7.18156 | 47 | −6.987 | ZINC12741918 | −6.50422 |
| 14 | −7.529 | ZINC11418769 | −7.15845 | 48 | −6.894 | ZINC58344565 | −6.45766 |
| 15 | −7.749 | ZINC38697967 | −7.12411 | 49 | −7.243 | ZINC04851257 | −6.45056 |
| 16 | −7.591 | ZINC05441336 | −7.11533 | 50 | −6.844 | ZINC39357685 | −6.43023 |
| 17 | −7.578 | ZINC05006123 | −7.03278 | 51 | −6.813 | ZINC32764688 | −6.42144 |
| 18 | −7.66 | ZINC65610187 | −7.03189 | 52 | −7.085 | ZINC78444367 | −6.41122 |
| 19 | −7.414 | ZINC57076878 | −7.03111 | 53 | −6.945 | ZINC13012826 | −6.40767 |
| 20 | −7.499 | ZINC24929111 | −7.016 | 54 | −6.649 | ZINC14481796 | −6.32767 |
| 21 | −7.591 | ZINC78899383 | −7.00111 | 55 | −7.135 | ZINC78556987 | −6.32434 |
| 22 | −7.381 | ZINC12753875 | −6.95856 | 56 | −6.918 | ZINC05393487 | −6.29611 |
| 23 | −7.495 | ZINC12360899 | −6.95567 | 57 | −7.049 | ZINC04851606 | −6.29289 |
| 24 | −7.378 | ZINC11486594 | −6.93356 | 58 | −6.935 | ZINC09743679 | −6.28045 |
| 25 | −7.708 | ZINC78717525 | −6.91089 | 59 | −6.642 | ZINC38698169 | −6.25534 |
| 26 | −7.488 | ZINC65610185 | −6.90978 | 60 | −6.914 | ZINC09743666 | −6.24689 |
| 27 | −7.4 | ZINC14519389 | −6.88178 | 61 | −6.907 | ZINC16818978 | −6.23034 |
| 28 | −7.156 | ZINC11486626 | −6.84944 | 62 | −7.147 | ZINC02333181 | −6.17489 |
| 29 | −7.123 | ZINC71881695 | −6.77878 | 63 | −6.804 | ZINC19526877 | −6.11789 |
| 30 | −7.115 | ZINC48302108 | −6.77722 | 64 | −6.42 | ZINC14500916 | −6.03978 |
| 31 | −7.605 | ZINC06606192 | −6.765 | 65 | −6.601 | ZINC15419422 | −5.98367 |
| 32 | −7.146 | ZINC31954796 | −6.75566 | 66 | −6.813 | ZINC06606196 | −5.88189 |
| 33 | −7.155 | ZINC04851213 | −6.74545 | 67 | −6.292 | ZINC05253733 | −5.68456 |
| 34 | −7.097 | ZINC01028321 | −6.736 |
| Compounds | Conventional Hydrogen Bonds | Carbon-Hydrogen Bonds | Pi-Cation | Pi-Alkyl | Pi-Pi T-Shaped Interaction | Pi-Sigma Bonds |
|---|---|---|---|---|---|---|
| Control | Gln123, His250, Lys211, Asn220, Asp124, His189 | His122, Gly219, | Trp93 | |||
| Z1 | His 189 | His122, Gly219, Asp124 | Met67, Met154 | Ile35, His250 | Trp96 | |
| Z2 | Asn220 | Met67 | His122, Val73 | Phe70 | Ile35 | |
| Z3 | Gly219, Ser217, His250, Lys211, Lys216, Asp212, Ala74 | His189, Val73 | Met67, Phe70 |
| Molecule | ZINC29142850 (ZN1) | ZINC78607001 (ZN2) | ZINC94303138 (ZN3) |
|---|---|---|---|
| MW | 458.53 | 423.94 | 839.94 |
| Rotatable bonds | 10 | 7 | 28 |
| H-bond acceptors | 5 | 2 | 14 |
| H-bond donors | 2 | 1 | 6 |
| MR | 125.96 | 125.94 | 203.12 |
| TPSA | 128.45 | 54.34 | 300.4 |
| iLOGP | 1.49 | 3.69 | 0 |
| ESOL class | Soluble | Moderately soluble | Moderately soluble |
| Lipinski violations | 0 | 0 | 3 |
| PAINS alerts | 0 | 0 | 1 |
| Predicted toxicity class | 4 | 4 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkhatabi, H.A.; Alatyb, H.N. Pharmacophore-Based Study: An In Silico Perspective for the Identification of Potential New Delhi Metallo-β-lactamase-1 (NDM-1) Inhibitors. Pharmaceuticals 2024, 17, 1183. https://doi.org/10.3390/ph17091183
Alkhatabi HA, Alatyb HN. Pharmacophore-Based Study: An In Silico Perspective for the Identification of Potential New Delhi Metallo-β-lactamase-1 (NDM-1) Inhibitors. Pharmaceuticals. 2024; 17(9):1183. https://doi.org/10.3390/ph17091183
Chicago/Turabian StyleAlkhatabi, Heba Ahmed, and Hisham N. Alatyb. 2024. "Pharmacophore-Based Study: An In Silico Perspective for the Identification of Potential New Delhi Metallo-β-lactamase-1 (NDM-1) Inhibitors" Pharmaceuticals 17, no. 9: 1183. https://doi.org/10.3390/ph17091183
APA StyleAlkhatabi, H. A., & Alatyb, H. N. (2024). Pharmacophore-Based Study: An In Silico Perspective for the Identification of Potential New Delhi Metallo-β-lactamase-1 (NDM-1) Inhibitors. Pharmaceuticals, 17(9), 1183. https://doi.org/10.3390/ph17091183