Abstract
Cystic fibrosis (CF) is the most common fatal genetic disease among Caucasian people, with over 2000 mutations in the CFTR gene. Although highly effective modulators have been developed to rescue the mutant CFTR protein, unresolved inflammation and persistent infections still threaten the lives of patients. While the central role of arachidonic acid (AA) and its metabolites in the inflammatory response is widely recognized, less is known about their impact on immunomodulation and metabolic implications in CF. To this end, here we provided a comprehensive analysis of the AA metabolism in CF. In this context, CFTR dysfunction appeared to complexly disrupt normal lipid processing, worsening the chronic airway inflammation, and compromising the immune responses to bacterial infections. As such, potential strategies targeting AA and its inflammatory mediators are being investigated as a promising approach to balance the inflammatory response while mitigating disease progression. Thus, a deeper understanding of the AA pathway dysfunction in CF may open innovative avenues for designing more effective therapeutic interventions.
1. Introduction
Cystic fibrosis (CF) is the most common fatal genetic disease in Caucasians, with an incidence of 1 in 3200 births in the North European population. The basic genetic defect has been localized in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, with more than 2000 mutations identified so far [1]. The CFTR gene product is primarily a cAMP-activated chloride ion channel, which is highly expressed by epithelial cells but also on endothelial cells [2], polymorphonuclear neutrophils (PMN) [3], monocytes/macrophages [4], dendritic cells [5], platelets [6], and lymphocytes [7]. CFTR mutations alter the fluid and electrolyte composition of secretions and the homeostasis of organs such as lung, pancreas, and liver, and sustain an unrelenting inflammatory reaction. Although CF affects almost every single tissue and organ in the body, chronic airway infection and inflammation, leading to pulmonary insufficiency, is responsible for at least 80% of CF-related deaths. However, disease severity is highly dependent on genetic variability and environmental factors (recently reviewed in [8]).
Under physiological conditions, acute inflammation requires timely resolution to ensure the return to tissue homeostasis after the removal of the biological threat. During resolution, actively regulated by several mechanisms, including the production of specialized pro-resolving lipid mediators (SPM), pro-inflammatory mediators are catabolized, PMN recruitment decreases, and return to tissue homeostasis is promoted by macrophages [9]. Failure of effective resolution leading to persistent inflammation, tissue damage, and organ fibrosis has been documented also in CF (recently reviewed in [10,11,12]).
Although progress in multidisciplinary care and the introduction of modulators [13] have led to a substantial increase in the median life expectancy of people with CF, the current strategies may not completely restore several molecular and clinical defects [14,15]. Therefore, new pharmacological approaches to manage inflammation in CF are still required.
In addition to cytokines, chemoattractants, and SPM, oxylipins are key regulatory players in the inflammatory response and its resolution. Oxylipins are oxygenated products of polyunsaturated fatty acids (PUFAs), which derive from the catabolism of the essential PUFA linoleic acid (LA) and alpha-linolenic acid (ALA) [16]. Arachidonic acid (AA) is widely recognized as the key oxylipin precursor driving the inflammatory process [17]. Thus, an imbalance in AA metabolism may sustain CF inflammation. Indeed, alterations in AA precursors and metabolites have been documented in CF [18]. Therefore, a comprehensive understanding of the dysfunctional AA pathway in CF is relevant to gain further knowledge about CF lung inflammation.
This review aims to summarize how dysfunctions of AA metabolism, related to CFTR-loss-of-function, fuel CF lung disease. Furthermore, novel AA-based approaches to prevent the progression of CF disease will be discussed.
2. Alteration of the AA Pathway in CF, from Precursors to Metabolites
AA is an omega-6 fatty acid (20:4 n-6 with 20 carbon atoms and 4 double bonds, the last of which is in position 6 (omega) from the terminal carboxyl group) which is key in the inflammatory response. In the following sections, we will describe the mechanisms of altered PUFA, AA, its metabolites, and enzymes, and how they contribute to CF airway pathology.
2.1. n-3 and n-6 PUFA
AA is derived from linoleic acid (LA), the essential fatty acid, along with alpha-linolenic acid (ALA). Although humans cannot synthesize essential fatty acids but absorb them with the diet, the organism has developed a series of enzymes with desaturating and elongating activity capable of converting LA to the omega-6 AA, and ALA to omega-3 eicosapentaenoic acid (EPA), which is further metabolized to docosahexaenoic acid (DHA) (Figure 1). Once synthesized, AA, EPA, and DHA become membrane phospholipids, the concentration of which depends on nutritional status, metabolic processes, specific cell/tissue type, enzyme activity, and disease state [19,20].
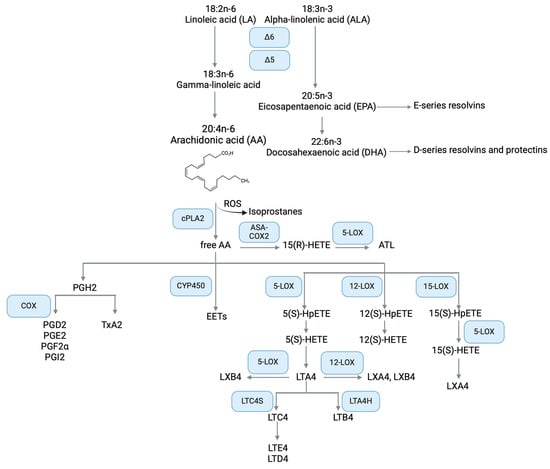
Figure 1.
Simplified overview of the omega-6 and omega-3 PUFAs metabolism, focusing on the AA metabolic pathway components with relevance in CF. ALA metabolism to EPA, DHA, and SPM is also depicted. Created with BioRender.com.
Since the 1970s [21], alterations in fatty acid metabolism have been widely reported in CF and recognized early in infants with CF and animal models of CF, indicating deficiencies owing to the genetic defect rather than secondary to pancreatic insufficiency or malnutrition [22,23]. Among fatty acids, cord-blood samples from CF newborns showed the absence or reduced levels of gamma-LA (a metabolic product of LA), alfa-LA (a precursor of DHA), and DHA, with an overall reduction in n-3 metabolites as compared to healthy newborns [22]. Similarly, the plasma of adults with CF showed LA and DHA deficiency [24], specifically due to defective essential fatty acid metabolism [24,25,26], consistently reproduced in mouse models (reviewed in [27]). Importantly, marked LA deficiency was associated with more severe CFTR mutations and phenotypes, indicating a crucial impact of this alteration on disease progression [27,28]. As discussed below, in addition to strikingly affecting the CF inflammatory response, this PUFA unbalance impaired plasma membrane fluidity [26], with pathobiological consequences that need to be elucidated. Importantly, n-3 and n-6 PUFA form highly specific interactions that affect their distribution in the cell membrane, and thus their biological effects depend on their cellular ratio rather than on their absolute levels [29]. Studies conducted on CFTR-expressing tissues in people with CF and rodents identified an elevated AA/DHA ratio in CF samples [30,31,32]. Corroborating the strict interdependence and the fine balance between bioactive lipids, DHA supplementation induced both a significant increase in DHA and a decrease in AA levels in the lungs and pancreas of treated mice, as well as in CF in vitro systems and in patients [32,33,34]. Thus, LA and ALA may share an unbalanced metabolism followed by dysregulated levels of both AA and DHA in CF. Indeed, most of the enzymes involved in the AA/EPA/DHA pathways are mutual and compete for metabolism (Figure 1). In line with this, a large body of evidence points to alteration in desaturase enzymes as responsible for the reduced LA and DHA levels found in CF [35]. Moreover, reduced LA correlated with boosted expression and activity of the Δ5- and ∆6-desaturase [35], suggesting that alteration in LA metabolism in CF is due to overreactive desaturases driven by the CFTR defect [36]. Supplementation with DHA reversed desaturase hyperactivity and restored intrinsic alteration of the PUFA unbalance in CF in vitro [33].
However, even if it is clear that DHA is reduced in CF, mechanisms leading to such reduction are less well understood. Since Δ6-desaturase activity is shared, it is surprising that LA levels are decreased in CF tissues, but DHA levels, the product of EPA metabolized by the same enzyme, are not. A mechanistic explanation of this apparent paradox was obtained in CF epithelial cell lines, where the conversion rate of EPA to DHA is reduced while the conversion of LA to AA is increased [35]. On the other hand, recent studies have reported that different isoforms of Δ6-desaturase and Δ5-desaturase may exist, so AA and EPA/DHA may be differentially metabolized within the cell [37].
2.2. Arachidonic Acid
As mentioned above, AA concentration is higher in CF tissues due to the enhanced metabolism of LA to AA. This increase is independent of persistent P. aeruginosa infection or chronic inflammation but is rather CF-specific [38], suggesting a CFTR-dependent alteration. Indeed, increased phospholipid-bound AA levels and decreased DHA levels were only found in the lung, pancreas, and ileum of CFTR −/− mice compared to healthy controls [18]. Similarly, the AA/DHA ratio was elevated in canonical CFTR-expressing tissues such as mucosal and submucosal nasal and rectal specimens in patients with CF [30]. Mechanistically, in addition to the increased conversion of LA to AA [35], the increase in free AA could also be due to enhanced release from the plasma membrane by the activity of the membrane-releasing enzyme cytosolic phospholipase A2 (cPLA2) [39,40]. Hyperactivation of cPLA2 may contribute to the establishment of the inflammatory environment in the CF airways even in the absence of infection. Indeed, mutant CFTR loses its inhibitory activity against cPLA2 by disrupting the Annexin A1/CFTR/cPLA2 complex at the plasma membrane. The hyperactivated cPLA2 drives the excessive AA release from the plasma membranes of CF cells [41]. In addition to its role in regulating cPLA2 activation, annexin-1 is an anti-inflammatory protein downregulated in the nasal epithelial cells of patients with CF and the lung and pancreas of CFTR KO mice [42]. Therefore, reduced annexin-1 expression driven by the basic CFTR genetic defect sustains chronic inflammation [43] and disrupts the cPLA2-CFTR assembly at the plasma membrane, finally triggering cPLA2 hyperactivation and AA release.
In addition to altering eicosanoid levels (see the following paragraph), the increased AA concentration modifies plasma membrane composition and fluidity, the mobility of membrane-bound proteins, and ion channel activity [44]. Notably, AA, applied to the cytoplasmic side of excised membrane patches of baby hamster kidney cell lines, inhibited CFTR-dependent chloride flux by electrostatic interaction with cytoplasmic amino acid side chains of the CFTR channel pore [45,46]. Thus, the aberrant AA concentration in the CF airway may further contribute to aggravating the CFTR channel dysfunction. Along these lines, lipidomic analysis of CF bronchial epithelial cell lines showed lipid modifications in the cell membrane due to the altered balance between phospholipid types [47]. Compared to the wild type, CF cells showed a higher accumulation of ceramide and glycosylated sphingolipids, which are known to be involved in inflammatory processes [48]. Hence, these findings in CF models suggest that alterations in the AA compartment may impair both chloride efflux and plasma membrane dynamics, adding further levels of complexity to the understanding of the role of AA in CF.
2.3. Metabolites
Among PUFAs, the biological functions of AA are closely related to inflammation since it is the substrate of both a class of potent inflammatory lipids, the eicosanoids, and lipoxins, a family of SPM [49]. Thus, a balanced AA metabolism is crucial to maintain a homeostatic regulation of the inflammatory response. For this reason, the biological synthesis and degradation of AA-derived oxylipins must be tightly regulated. AA metabolism initiates with the release of membrane-bound AA into the cytoplasm by the action of cPLA2, which catalyzes the hydrolysis of the acyl bond between the second fatty acid tail and the glycerol molecule of membrane phospholipids. cPLA2 is activated by inflammatory stimuli and its activity is the rate-limiting step in the AA pathway [50]. Once free, AA is the substrate of three families of oxylipin-generating enzymes: cyclooxygenases (COX), responsible for the synthesis of prostanoids; lipoxygenases (LOX) that catalyze the biosynthesis of leukotrienes and lipoxins; and CYP450 epoxygenases form the epoxyeicosatrienoic acids (EET). In addition, AA can be non-enzymatically oxidized by reactive oxygen species to isoprostanes (recently reviewed in [19]) (Figure 1).
Lung inflammation and parenchymal injury are early detectable in CF, with accumulation of membrane bioactive lipids and AA metabolites. Indeed, in children with CF, eicosanoid levels (prostaglandin G2 and isoprostanes) in bronchoalveolar lavage fluids (BAL) were high and correlated with markers of airway inflammation such as PMN elastase and myeloperoxidase, and with lung damage [51]. Therefore, the alteration of AA metabolism deeply impacts CF inflammation and outcomes. In the following sections, we will examine the main pathways involved in AA-derived oxylipin production, presenting the most relevant changes occurring in CF.
2.3.1. The COX Pathway
AA can be transformed by COX-1 or COX-2 into prostaglandins (PG) or thromboxanes (Tx) (Figure 1). Both COX isoforms release an oxygenated partially cyclic lipid, However, COX-1 is constitutively expressed while COX-2 is inducible by inflammatory stimuli, hormones, and growth factors [19].
One of the key COX products is PGE2, a central bioactive lipid at the interface between the pro-inflammatory and pro-resolving response. PGE2 is released by immune cells during the onset of the acute inflammatory response, as it regulates vascular permeability, chemotaxis, and pro-inflammatory cytokine release. Secondly, PGE2 is also required to boost the release of lipoxins (LX), bioactive AA metabolites known for their pro-resolving and anti-inflammatory functions [52]. PGD2 and TxA2 could be also produced from the same PGE2 precursor (PGH2). PGD2 is the major COX-metabolite produced by activated macrophages and mast cells, while TxA2 is mainly released by activated platelets, monocytes, macrophages, PMN, and lung parenchyma during inflammation [19].
Marked staining for COX-2 has been found in the sinonasal tissue of people with CF, underlining its upregulation in the upper airways due to post-transcriptional mechanisms [53,54], also confirmed by in vitro assays on cell cultures [35]. COX-1 overexpression was also found in nasal CF polyps [54], even if its overexpression in other CF tissues is controversial [53]. As a consequence, patients with CF had excessive levels of COX-1 products such as PGE2 in saliva, urine, sputum, and BAL and PGD2 in sputum [55,56,57,58], suggesting their potential role in driving airway inflammation. Notably, PGE2 but not PGD2 levels correlated with the CF genotype and disease severity. Higher levels of urinary PGE2 and PGD2 metabolites were detected in patients with class I, II, and III mutations and with bronchiectasis as compared to control healthy volunteers [59,60]. Consistent with this, a longitudinal study on a cohort of children with CF showed that PGs correlated with structural lung damage and predicted the evolution of lung disease within two years [51].
TxA2 is also a key COX-derived product involved in CF lung chronic inflammation. Indeed, the concentration of its stable metabolite, TxB2, was consistently found to increase in the urines of patients with CF and correlated with pulmonary dysfunction [56,61]. As TxB2 excretion is considered an in vivo index of platelet activation [62], high TxB2 concentrations were correlated to sustained platelet activation that could be partially corrected by antioxidant supplementation [61].
2.3.2. The LOX Pathway
The involvement of AA metabolism in both pro-inflammatory and pro-resolving responses is well illustrated by the LOX pathway. There are several isoforms of this enzyme, namely 5-, 12-, and 15-LOX. Before being converted to leukotrienes or lipoxins, AA is oxidized to chiral-specific unstable intermediates denominated hydroperoxyl eicosatetraenoic acids: 5(S)-HpETE from 5-LOX, 12(S)-HpETE from 12-LOX, and 15(S)-HpETE from 15-LOX. The reduced products of HpETEs are hydroxy eicosatetraenoic acids (HETEs), which are secreted by epithelial cells and leukocytes during the inflammatory response (Figure 1). Overall, HETEs are associated with pro-inflammatory events, e.g., 5(S)-HETE and 15-(S)HETE are potent chemoattractants for PMN and stimulate their degranulation [63]. On the other hand, 15(S)-HETE inhibited PMN transepithelial migration, degranulation, and superoxide production [64]. Alterations in HETEs have been described in several chronic inflammatory diseases such as obesity, cardiovascular disease, and cancer [65], but little is known about HETE alterations in CF. Since 15(S)-HETE is the predominant LOX metabolite in the human lung [66], it may play a critical role in the regulation of the inflammatory response in CF. Thus, further investigations are needed in this field.
With 5-HpETE as an intermediate, 5-LOX converts AA to leukotriene A4 (LTA4). This bioactive lipid mediator is best known as a sparkplug of acute inflammation, acting as a chemoattractant for innate immune cells, but also for Th17 lymphocytes [67]. Apart from its physiological role, LTA4 could perpetuate the inflammatory state, leading to chronicity and further tissue damage in diseases such as asthma and atherosclerosis [68]. LTA4 is the precursor of two major classes of leukotrienes, cysteinyl LTs and LTB4. Specifically, LTA4 is converted by LTC4 synthase to LTC4 and finally to the stable metabolites LTD4 and LTE4. These metabolites are collectively termed cysteinyl leukotrienes (CysLT) (Figure 1). CysLT are contractile agonists of airway smooth muscles and stimulate mucus production by goblet cells, resulting in bronchoconstriction and mucus accumulation, key events in CF lung disease [69]. On the other hand, LTA4 hydrolase converts LTA4 into LTB4, a potent chemoattractant for leukocytes during immune responses (reviewed in [70]).
LT in CF airways significantly contribute to airway inflammation. Indeed, LTB4 and CysLT in the sputum of children with CF correlated with tumor necrosis factor-alpha (TNF-α) levels and parameters of airflow obstruction [71], pointing to key roles for these mediators in sustaining CF inflammation. Indeed, analysis of CF sputum and epithelial lining fluid revealed LTB4 as a major AA metabolite along with CysLT [57,72]. Elevated levels of LTB4 and CysLT are early detectable in the disease, as they were found at nanomolar concentrations in the sputum and urine of CF infants [73]. This evidence supports the hypothesis that the inflammatory response could be driven by altered lipid metabolism even in the absence of infection.
Actions of 12-LOX and 15-LOX, in cooperation with 5-LOX, can convert AA into lipoxins (LXs). Two main routes have been described for the generation of LXA4 and its positional isomer LXB4: the 5/15-LOX and the 5/12-LOX pathways (reviewed in [74]). The 5/15-LOX pathway can be either initiated by 5-LOX or 15-LOX in PMN, eosinophil or alveolar macrophages, to convert AA into intermediates that are transformed by the reciprocal LOX into both LXA4 and LXB4. Transcellular interactions between PMN and eosinophils or PMN and lung cells has also been described to generate LX through 5/15-LOX metabolism. Conversely, 5-LOX from PMN generates and transfers LTA4 as a precursor for platelets, which convert LTA4 into LXA4 and LXB4 through 12-LOX activity (Figure 1) [75,76]. The biological significance of this transcellular interaction is extremely important, as the physical proximity of inflammatory and resident cells at the site of injury promotes the initiation of specific signals toward resolution and homeostasis. Finally, in a third biosynthetic pathway involving transcellular interactions among PMN and endothelial or tumor cells [77,78], as well as hepatocytes and liver cells [79], COX-2 acetylated by aspirin produces 15(R)-HETE, which is converted by 5-LOX in 15-epimers of LXA4 and LXB4 (named aspirin-triggered lipoxins, ATL). As LX, ATL generate a range of anti-inflammatory actions, i.e., reduction in PMN migration and activation, and pro-resolving responses by supporting efferocytosis and active removal of pro-inflammatory mediators and debris [37,76,80,81].
LXA4 levels are significantly low in patients with CF, both adults and children. As compared to patients with other inflammatory lung diseases, people with CF had the lowest LXA4 concentration in airway fluids that contribute to maintain airway inflammation. Indeed, administration of a stable LXA4 analog to a chronically infected mouse model of CF ameliorated markers of inflammation and lung functions [82]. Mechanistically, this failure in LXA4 production could be ascribed to the downregulation of 15-LOX expression observed both in CF epithelial cells [83] and BAL from children with CF even in the absence of detectable infection [84]. On the other hand, CFTR loss-of-function deeply affects the activity of platelet 12-LOX, reducing considerably LXA4 and LXB4 generation during PMN/platelet coincubations [6]. In addition to the reduced LX production, CF macrophages showed lower expression of LXA4 receptor FPR2/ALX compared to cells from healthy volunteers. Importantly, restoration of normal levels of FPR2/ALX improved phagocytosis by CF macrophages, indicating the crucial role of LXA4 signaling in mechanism of CF inflammation [85]. Therefore, defects in LOX expression, activity, metabolites, and their receptors greatly contribute to impair resolution of inflammation in CF lung.
2.3.3. The CYP450 Pathway and Oxidative Stress
While the COX and LOX pathways are quite well studied in CF, less is known about the AA metabolites formed by CYP450. Once recognized by this class of enzymes, AA and its intermediates are epoxidized to EET, high-affinity ligands of PPAR-γ [86], and are generally involved in the anti-inflammatory response [87] (Figure 1). Thus, investigating the extent to which CYP450 and EETs are involved in non-resolving inflammation in CF may provide further information on lipid imbalance and potential therapeutic approaches.
Interestingly, a recent study reported an AA-oxylipin produced by P. aeruginosa [88], one of the most prevalent colonizing bacteria of CF airways [89]. Indeed, P. aeruginosa contains CYP168A1, a cytochrome P450 enzyme with high affinity for AA, which is readily found at high concentrations in the CF host environment. The main product of CYP168A1 catalysis is 19-HETE, a potent vasodilator and inhibitor of platelet aggregation [90], that facilitates blood vessel permeability and leukocyte transmigration. The reason beyond the release of 19-HETE by P. aeruginosa remains to be elucidated, but its ability to modulate the immune response by acting on host lipid mediator signaling may be fundamental for its persistent colonization, which remains a life-threatening condition in the compromised airways of people with CF.
The impact of oxidative stress on CF pathophysiology is receiving increasing attention. Accumulating evidence shows how the extent of pathological markers in CF lung parallels that of oxidative markers, during both chronic and acute exacerbations and disease progression [91]. PMNs are recognized as a major contributor to the exaggerated release of reactive oxygen species (ROS) during infections, due to failure of phagocytosis and continuous contact with bacteria in CF [92,93]. Furthermore, epithelial cells support the oxidative burst by continuously releasing ROS [94], even in the absence of infection. Worsening this scenario, patients with CF also showed impaired antioxidant detoxification mechanisms, such as extracellular glutathione (GSH) deficiency [95]. A contribution to lipid peroxidation also comes from pancreatic insufficiency, which could be responsible for the malabsorption of essential fat-soluble antioxidants [91]. Despite the generating mechanism, one immediate consequence of altered oxidant/antioxidant ratio is the ROS reaction with cellular membranes and lipoproteins [96]. Polyunsaturated fatty acid phospholipids are highly susceptible to oxidation with conversion to bioactive or toxic lipids called isoprostanes [97]. Because of their origin, they are considered markers of oxidative stress also in lung diseases [98]. In physiological conditions, isoprostanes are biologically active and are potent mediators of inflammation [99]. For instance, they can bind to TLR2 on macrophages and trigger the expression of COX-2 and interleukin 1-β, promoting inflammation, but they could also boost pathogen clearance and removal of apoptotic cells [100].
The presence of isoprostanes in the lungs of patients with CF can be detected at an early age, as found in BAL fluid from children aged 3–5 years [51]. The gold standard used as a reliable measure of lipid peroxidation is the isoprostane 8-iso-PGF-2α, whose levels positively correlate with markers of lung inflammation, disease severity [101], and exacerbations [102,103]. Remarkably, antibiotic therapy against P. aeruginosa infection does not alter levels of 8-iso-PGF2α, cys-LTs, and PGE2 in the airways, suggesting that, despite the decreased bacterial load and improved clinical status, there is persistent lipid peroxidation of AA [102,104]. In addition to lungs, elevated isoprostane levels have been found in the urine and plasma of people with CF compared to healthy controls [61,105], underlying a systemic circulation of oxidized lipids that could further affect peripheral tissues.
Moreover, also the LOX products HETEs can be non-enzymatically oxidized to form keto-phospholipids (KETEs) [106]. In a cohort including healthy subjects, people with CF, and people with other lung diseases, such as bronchiectasis and persistent bacterial bronchitis, CF samples showed significantly increased 15-KETE levels [106]. However, their role remains to be fully elucidated.
Figure 2 summarizes the alterations in AA precursors, metabolites, and enzymes found in CF, along with the reported molecular mechanisms leading to dysfunction.
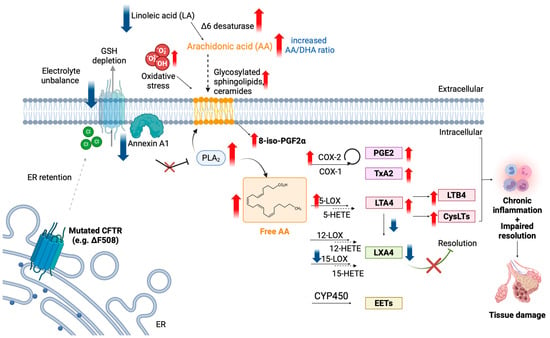
Figure 2.
Dysregulation of the AA pathway in CF. Blue arrows indicate the enzyme/lipid mediator found downregulated in CF, while red arrows indicate the upregulation. See text for details. Created with BioRender.com.
2.4. Role of CFTR Dysfunction in CF Abnormal Fatty Acid Metabolism
As previously highlighted, CFTR loss-of-function directly impacts AA production and metabolism (Figure 3). However, with the introduction of highly effective modulator therapy to rescue CFTR functions, it becomes crucial to determine whether this therapy modulates the unbalanced AA pathway. Although CFTR could control AA metabolism, bacterial antigens, pro-inflammatory cytokines, and dysfunctional immune cells may also significantly contribute to increase eicosanoid levels in the inflamed CF lung [107].
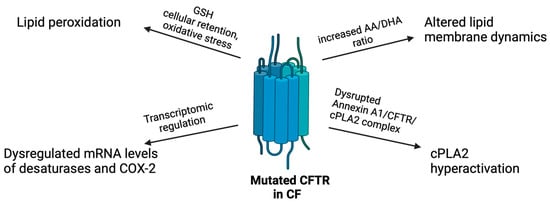
Figure 3.
Infographic of the direct impact of mutated CFTR in the AA metabolism in CF. See text for details. Created with BioRender.com.
As mentioned, the deficiency of plasma LA and DHA, together with the excessive release of AA, has been positively correlated with genotype severity in patients with CF [108]. The altered circulating AA/DHA ratio may reflect the alterations in membrane phospholipids [24]. CFTR is also localized at the cell membrane and, as a channel, can interact with phospholipids and affect their trafficking, incorporation, and turnover [109]. Thus, CFTR may contribute to the regulation of DHA and AA processing [32]. Indeed, in newborn pigs, CFTR loss caused blunted lung uptake of fatty acids, even if their circulating levels were high. Interestingly, AA was the only PUFA with an increased uptake, suggesting a specific mechanism driven by CFTR that directs AA from the bloodstream to the lung [110]. Consistent with this, the increased AA/DHA ratio occurs early in life due to changes in lipid membrane dynamics caused by CFTR loss-of-function. In line with the role of CFTR in regulating membrane composition, in vitro and in vivo CFTR knockout models highlighted the impact of CFTR on the organization of lipid rafts in the cell membrane, affecting both phospholipid and protein distribution. In human bronchial epithelial cells, CFTR loss-of-function increased the conversion of LA to AA and decreased DHA generation from precursors [36]. Similarly, FF508del CFTR enhanced the conversion of LA to AA compared to cells with wild-type CFTR [111]. Therefore, CFTR mutations directly affect AA metabolism by modulating membrane lipid dynamics. In line with this, CFTR inhibition caused membrane destabilization with disrupted formation of protein complexes, increased cPLA2 activity, and eicosanoid biosynthesis in airway epithelial cells [41], suggesting a direct relationship between CFTR, plasma membrane, and AA metabolism.
In addition to membrane dynamics, CFTR could also directly modify, mainly at the mRNA level, the expression of enzymes involved in AA metabolism, such as desaturases and COX-2 [54]. In addition to AA enzymes, a variety of changes in gene expression have been observed in cells with CFTR deletion [112], such as alterations in transcription factors, cytokines, and membrane receptors that are part of the inflammatory pathway. This reprogramming not only compensates for the absence of CFTR function but also suggests a direct role for CFTR as a transcriptional regulator perhaps through changes in protein–protein interactions and altered ion fluxes within the cell.
Importantly, CFTR dysfunction also causes the drastic reduction in antioxidant agents that act as scavengers of ROS, promoting lipid peroxidation and dysregulation of AA metabolism. Indeed, due to the impaired chloride efflux from CF cells, GSH transport in the extracellular environment is dampened (recently reviewed in [113]), thus contributing to enhance oxidative stress in CF.
2.5. Therapeutic Strategies to Modulate AA Metabolism in CF
The advent of CFTR modulators has revolutionized CF care. These medications enhance CFTR protein expression and activity, leading to significant improvements in lung function, pulmonary exacerbations, and overall quality of life [114,115]. Ivacaftor, lumacaftor, tezacaftor, and elexacaftor are among the most widely used modulators, However, knowledge on their impacts on CF chronic inflammation, as well as their influence on AA metabolism, remains incomplete.
Lipidomic characterization of CF cell lines treated with elexacaftor/tezacaftor/ivacaftor demonstrated that modulators could alter membrane and intracellular lipidome [116]. Indeed, an analysis from the GOAL study evidenced significantly reduced AA and PGE2 levels but not the rescue of LA and DHA deficiency in individuals with CF under ivacaftor [117], suggesting that CFTR correction could partially mitigate some aspects of the aberrant AA-derived inflammatory responses. Mechanistic studies reported that CFTR modulators lower AA levels and increase DHA levels in CF cell lines, but not in WT cells, thus confirming a direct impact of CFTR on lipid metabolism [118].
Dietary supplementation with omega-3 fatty acids, such as EPA and DHA, has been proposed as an encouraging therapeutic approach to reduce inflammation and balance the dysregulated AA/DHA ratio. These fatty acids can compete with AA for incorporation into cell membranes and enzymatic conversion, resulting in a shift toward the production of anti-inflammatory and pro-resolutive lipid mediators. Indeed, both adults and children with CF showed restoration of the lipid profile of red blood cell membranes, with reduced AA/EPA and AA/DHA ratios [119,120]. In line with this, DHA supplementation resulted in increased levels of DHA and its anti-inflammatory metabolites, such as 17-hydroxy-docosahexaenoic acid (17OH-DHA), along with a reduction in LTB4, 15-HETE, and PGE2 levels in the airways of adults with CF [119], confirming that DHA modifies lipid metabolism. However, DHA supplementation did not improve pulmonary function parameters such as forced vital capacity (FVC) and forced expiratory volume in one second (FEV1), likely because of the short duration of the study. Supporting this hypothesis, inflammatory mediators and DHA-derived metabolites returned to baseline after washout, suggesting the need for continuous DHA supplementation to maintain anti-inflammatory effects. However, even if long-term DHA (48 weeks) in patients with CF altered the fatty acid balance by increasing omega-3 levels and decreasing omega-6 levels, including AA, pulmonary and inflammatory markers (interleukin release, FEV1, or pulmonary exacerbations) were not affected [121]. Similarly, a meta-analysis of five randomized controlled trials with 106 participants evidenced low benefits for DHA supplementation in CF also because of the low quality of evidence across outcomes [122]. A recent randomized, double-blind, placebo-controlled study reported significant improvements in FVC and FEV1 in children with CF taking DHA [123]. In contrast, no significant differences were found in sputum and serum levels of inflammatory biomarkers, such as PMN elastase, resolvin D1, IL-8, and TNF-α, suggesting that the anti-inflammatory effects of DHA may be limited. Overall, additional studies are needed to understand optimal dosage, timing, need for concomitant administration of pancreatic enzymes, and categories of patients with patients with CF that may benefit from DHA supplementation.
Notably, since LA levels are decreased in patients with CF, LA supplementation has been proposed to correct this defect. However, evidence suggests that this supplementation may even exacerbate inflammatory responses. In fact, increased LA intake leads to higher levels of AA and its pro-inflammatory metabolites, exacerbating lung inflammation and CF lung disease [124].
As part of anti-inflammatory treatments to reduce abnormal fatty acid metabolism in CF, drugs that target specific enzymes involved in AA metabolism have been also used. For example, COX inhibitors such as non-steroidal anti-inflammatory drugs (NSAIDs) can reduce the synthesis of inflammatory PG. Among them, ibuprofen (COX-1 and COX-2 inhibitor) is recommended to slow disease progression in people with CF older than 6 years and with mild lung disease, also based on a 4-year trial that showed slower progression in lung disease [125]. However, the risk of side effects (mainly GI bleeding) has limited the use of this drug, even if benefits prevailed over the risks. Interestingly, in vitro and in vivo studies showed that ibuprofen may act as a CFTR corrector to restore CFTR trafficking in bronchial epithelial cells [126] and has anti-microbial actions against CF-related bacterial species [127,128]. Therefore, given this triple function, the risk–benefit ratio of ibuprofen administration to people with CF should be carefully evaluated. In contrast, selective COX-2 inhibitors are not recommended because of their cardiovascular side effects [129].
In addition to NSAIDs, other popular anti-inflammatory drugs such as steroids can interfere with AA metabolism. Indeed, steroids interfere with cPLA2, thus reducing both pro-inflammatory LT and PG production [130]. Despite that, their systemic long-term use in CF is discouraged due to the severe side effects. Furthermore, clinical results with aerosol steroids, used to minimize the harmful effects of systemic administration, were modest benefits [131].
Along these lines, drugs interfering with the LOX arm of the AA metabolism have been tested in CF to control inflammation. As mentioned, LT are elevated in CF and, in particular, LTB4 exerts potent pro-inflammatory actions by stimulating specific membrane receptors named BLT1 and BLT2 [132]. The potential therapeutic effects of the BLT1 antagonist have been tested in pediatric and adult patients with CF, but the trial has been interrupted due to serious pulmonary adverse events [133]. Another tested strategy in CF to reduce the excessive LTB4 aimed to reduce its production by inhibiting the LTA4 hydrolase (Figure 1). Acebilustat is a direct and selective LTA4 hydrolase inhibitor, that was found to be safe, well tolerated, and able to reduce inflammatory biomarkers (total leukocytes, PMN, total DNA and elastase in sputum) [134]. Despite these promising results in patients with CF treated with 15 days in phase I trial, in a 48-week phase II study, acebilustat failed to improve pulmonary function or to significantly reduce exacerbations, although a trend toward diminished exacerbations in people with CF at early stage of lung disease (FEV1 > 75) indicated a potential effect [135].
CysLT also activates two receptors, CysLT1 and CysLT2 [136]. Montelukast, a CysLT1 receptor antagonist, has shown some efficacy in reducing eosinophilic inflammation, improving clinical symptoms such as exercise tolerance, cough, and wheezing in people with CF, and lowering several inflammatory markers such as IL-8 and myeloperoxidase [137,138,139,140]. Similarly, Zafirlukast provided some benefits including improvement in chest radiograph appearance and physical examination, but not in respiratory function [141]. However, more powered studies are needed to evaluate their safety and efficacy.
Finally, as lipid peroxidation plays a crucial role in determining the distinctive lipid unbalance in CF, antioxidant administration could be proposed as a strategy to reverse the AA dysfunction. Indeed, administration of inhaled GSH to patients with CF significantly reduced PGE2 levels, along with increased the levels of CD4+ and CD8+ T cells. However, markers of oxidative stress such as 8-isoprostane were not changed [142], suggesting that the primary benefit of GSH inhalation in people with CF may be through modulation of the immune response rather than direct antioxidant effects.
A summary of the strategies tested to restore a balanced AA in CF in clinical trials is presented in Table 1.

Table 1.
Therapeutic strategies proposed in CF clinical trials. For each therapeutic approach the cohort of patients with CF enrolled, the clinical outcome, and the respective reference are reported.
3. Conclusions
Despite substantial advancements in care, chronic airway inflammation in CF persists as a vexing issue, characterized by a disproportionate, persistent, malfunctional activation of immune cells, mainly PMN, compared to the actual need for bacterial clearance. Concurrently, increasing evidence indicates an imbalance of AA metabolites in CF. Thus, there is an urgent need to investigate how alterations in the AA pathway could sustain chronic inflammation and how tailored therapeutic approaches could be developed to overcome the AA-dependent clinical complications in CF.
The AA pathway, from its membrane-bound phospholipids to its metabolites, is a key player in immune regulation, as it orchestrates the different phases of the inflammatory response toward resolution in a tissue- and disease-specific manner [143], modulating the interactions between immune, resident, and endothelial cells. In line with this, a direct cause–effect relationship in CF has been established for the excessive airway concentration of LTB4 that induces further PMN adherence, superoxide production, degranulation, and neutrophil extracellular traps (NET) formation in the lung environment [72], thus sustaining PMN-mediated lung damage. Therefore, AA dysfunctions are a leading cause of CF pathology, suggesting that strategies to restore AA metabolism could be beneficial to hamper chronic inflammation and promote resolution. To this end, the integration of CFTR modulators, omega-3 fatty acids, enzyme inhibitors, receptor antagonists, and antioxidants holds some promise in preclinical studies for improving clinical outcomes through the restoration of physiological AA metabolism. However, failure of the BLT1 trial, likely due to the impairment of the beneficial physiological functions of LTB4, suggested that proper modulation of the AA pathway, despite inhibition of some enzymes or mediators, might be a better-tailored approach to preserve its physiology while dampening pathology. Importantly, inconsistent improvements in trials with DHA or acebilustat also suggested that drug timing, dosage, and target patients need to be carefully tailored to achieve durable drug efficacy. Thus, while some encouraging efficacies have been found in preclinical models, consistent evidence of improvement in patients remains elusive, highlighting the fact that future research is still warranted to restore a proper AA metabolism for people with CF.
Author Contributions
Conceptualization, S.D. and D.M.; writing—original draft preparation, S.D.; writing—review and editing, D.M.; supervision, D.M.; funding acquisition, D.M. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Italian Cystic Fibrosis Foundation (FFC) (FFC#11/2022 to D.M.). S. D’O. was a former FFC fellow. We are grateful to the FFC supporting group of Delegazione FFC Ricerca di Cuneo Alba for adopting the FFC-funded projects.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cystic Fibrosis Mutation Database: Statistics. Available online: http://www.genet.sickkids.on.ca/StatisticsPage.html (accessed on 8 July 2024).
- Tousson, A.; Van Tine, B.A.; Naren, A.P.; Shaw, G.M.; Schwiebert, L.M. Characterization of CFTR Expression and Chloride Channel Activity in Human Endothelia. Am. J. Physiol. 1998, 275, C1555–C1564. [Google Scholar] [CrossRef]
- Painter, R.G.; Valentine, V.G.; Lanson, N.A.; Leidal, K.; Zhang, Q.; Lombard, G.; Thompson, C.; Viswanathan, A.; Nauseef, W.M.; Wang, G.; et al. CFTR Expression in Human Neutrophils and the Phagolysosomal Chlorination Defect in Cystic Fibrosis. Biochemistry 2006, 45, 10260–10269. [Google Scholar] [CrossRef] [PubMed]
- Porto, P.D.; Cifani, N.; Guarnieri, S.; Domenico, E.G.D.; Mariggiò, M.A.; Spadaro, F.; Guglietta, S.; Anile, M.; Venuta, F.; Quattrucci, S.; et al. Dysfunctional CFTR Alters the Bactericidal Activity of Human Macrophages against Pseudomonas Aeruginosa. PLoS ONE 2011, 6, e19970. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tertilt, C.; Krause, A.; Quadri, L.E.; Crystal, R.G.; Worgall, S. Influence of the Cystic Fibrosis Transmembrane Conductance Regulator on Expression of Lipid Metabolism-Related Genes in Dendritic Cells. Respir. Res. 2009, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Mattoscio, D.; Evangelista, V.; De Cristofaro, R.; Recchiuti, A.; Pandolfi, A.; Di Silvestre, S.; Manarini, S.; Martelli, N.; Rocca, B.; Petrucci, G.; et al. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Expression in Human Platelets: Impact on Mediators and Mechanisms of the Inflammatory Response. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3970–3980. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.V.; Nghiem, P.T.; Gardner, P.; Martens, C.L. Human Lymphocytes Transcribe the Cystic Fibrosis Transmembrane Conductance Regulator Gene and Exhibit CF-Defective cAMP-Regulated Chloride Current. J. Biol. Chem. 1992, 267, 3242–3248. [Google Scholar] [CrossRef]
- Grasemann, H.; Ratjen, F. Cystic Fibrosis. N. Engl. J. Med. 2023, 389, 1693–1707. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-Resolving Lipid Mediators Are Leads for Resolution Physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Recchiuti, A.; Mattoscio, D.; Isopi, E. Roles, Actions, and Therapeutic Potential of Specialized Pro-Resolving Lipid Mediators for the Treatment of Inflammation in Cystic Fibrosis. Front. Pharmacol. 2019, 10, 252. [Google Scholar] [CrossRef]
- Briottet, M.; Shum, M.; Urbach, V. The Role of Specialized Pro-Resolving Mediators in Cystic Fibrosis Airways Disease. Front. Pharmacol. 2020, 11, 1290. [Google Scholar] [CrossRef]
- Shum, M.; London, C.M.; Briottet, M.; Sy, K.A.; Baillif, V.; Philippe, R.; Zare, A.; Ghorbani-Dalini, S.; Remus, N.; Tarze, A.; et al. CF Patients’ Airway Epithelium and Sex Contribute to Biosynthesis Defects of Pro-Resolving Lipids. Front. Immunol. 2022, 13, 915261. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Caverly, L.J.; Riquelme, S.A.; Hisert, K.B. The Impact of Highly Effective Modulator Therapy on Cystic Fibrosis Microbiology and Inflammation. Clin. Chest Med. 2022, 43, 647–665. [Google Scholar] [CrossRef] [PubMed]
- Hisert, K.B.; Birket, S.E.; Clancy, J.P.; Downey, D.G.; Engelhardt, J.F.; Fajac, I.; Gray, R.D.; Lachowicz-Scroggins, M.E.; Mayer-Hamblett, N.; Thibodeau, P.; et al. Understanding and Addressing the Needs of People with Cystic Fibrosis in the Era of CFTR Modulator Therapy. Lancet Respir. Med. 2023, 11, 916–931. [Google Scholar] [CrossRef]
- Calder, P.C. Omega-3 Fatty Acids and Inflammatory Processes: From Molecules to Man. Biochem. Soc. Trans. 2017, 45, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, B. Arachidonic Acid Metabolism: Role in Inflammation. Z. Rheumatol. 1991, 50 (Suppl. S1), 3–6. [Google Scholar]
- Strandvik, B. Fatty Acid Metabolism in Cystic Fibrosis. Prostaglandins Leukot. Essent. Fat. Acids 2010, 83, 121–129. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism Pathways of Arachidonic Acids: Mechanisms and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef]
- Harayama, T.; Riezman, H. Understanding the Diversity of Membrane Lipid Composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Chase, H.P.; Dupont, J. Abnormal Levels of Prostaglandins and Fatty Acids in Blood of Children with Cystic Fibrosis. Lancet 1978, 2, 236–238. [Google Scholar] [CrossRef]
- Lloyd-Still, J.; Johnson, S.; Holman, R. Essential Fatty Acid Status and Fluidity of Plasma Phospholipids in Cystic Fibrosis Infants. Am. J. Clin. Nutr. 1991, 54, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Uc, A.; Strandvik, B.; Yao, J.; Liu, X.; Yi, Y.; Sun, X.; Welti, R.; Engelhardt, J.F.; Norris, A.W. The Fatty Acid Imbalance of Cystic Fibrosis Exists at Birth Independent of Feeding in Pig and Ferret Models. Clin. Sci. 2022, 136, 1773–1791. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; Mischler, E.H.; Engle, M.J.; Jeannette Brown, D.; Lau, S.-M. Fatty Acid Abnormalities in Cystic Fibrosis. Pediatr. Res. 1985, 19, 104–109. [Google Scholar] [CrossRef]
- Van Biervliet, S.; Vanbillemont, G.; Van Biervliet, J.-P.; Declercq, D.; Robberecht, E.; Christophe, A. Relation between Fatty Acid Composition and Clinical Status or Genotype in Cystic Fibrosis Patients. Ann. Nutr. Metab. 2007, 51, 541–549. [Google Scholar] [CrossRef]
- Roulet, M.; Frascarolo, P.; Rappaz, I.; Pilet, M. Essential Fatty Acid Deficiency in Well Nourished Young Cystic Fibrosis Patients. Eur. J. Pediatr. 1997, 156, 952–956. [Google Scholar] [CrossRef]
- Seegmiller, A. Abnormal Unsaturated Fatty Acid Metabolism in Cystic Fibrosis: Biochemical Mechanisms and Clinical Implications. IJMS 2014, 15, 16083–16099. [Google Scholar] [CrossRef]
- Strandvik, B.; O’Neal, W.K.; Ali, M.A.; Hammar, U. Low Linoleic and High Docosahexaenoic Acids in a Severe Phenotype of Transgenic Cystic Fibrosis Mice. Exp. Biol. Med. 2018, 243, 496–503. [Google Scholar] [CrossRef]
- Rubin, D.; Laposata, M. Cellular Interactions between N-6 and n-3 Fatty Acids: A Mass Analysis of Fatty Acid Elongation/Desaturation, Distribution among Complex Lipids, and Conversion to Eicosanoids. J. Lipid Res. 1992, 33, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.D.; Blanco, P.G.; Zaman, M.M.; Shea, J.C.; Ollero, M.; Hopper, I.K.; Weed, D.A.; Gelrud, A.; Regan, M.M.; Laposata, M.; et al. Association of Cystic Fibrosis with Abnormalities in Fatty Acid Metabolism. N. Engl. J. Med. 2004, 350, 560–569. [Google Scholar] [CrossRef]
- Shrestha, N.; Rout-Pitt, N.; McCarron, A.; Jackson, C.A.; Bulmer, A.C.; McAinch, A.J.; Donnelley, M.; Parsons, D.W.; Hryciw, D.H. Changes in Essential Fatty Acids and Ileal Genes Associated with Metabolizing Enzymes and Fatty Acid Transporters in Rodent Models of Cystic Fibrosis. IJMS 2023, 24, 7194. [Google Scholar] [CrossRef]
- Freedman, S.D.; Katz, M.H.; Parker, E.M.; Laposata, M.; Urman, M.Y.; Alvarez, J.G. A Membrane Lipid Imbalance Plays a Role in the Phenotypic Expression of Cystic Fibrosis in Cftr−/− Mice. Proc. Natl. Acad. Sci. USA 1999, 96, 13995–14000. [Google Scholar] [CrossRef]
- Njoroge, S.W.; Laposata, M.; Katrangi, W.; Seegmiller, A.C. DHA and EPA Reverse Cystic Fibrosis-Related FA Abnormalities by Suppressing FA Desaturase Expression and Activity. J. Lipid Res. 2012, 53, 257–265. [Google Scholar] [CrossRef]
- Van Biervliet, S.; Devos, M.; Delhaye, T.; Van Biervliet, J.P.; Robberecht, E.; Christophe, A. Oral DHA Supplementation in ΔF508 Homozygous Cystic Fibrosis Patients. Prostaglandins Leukot. Essent. Fat. Acids 2008, 78, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Njoroge, S.W.; Seegmiller, A.C.; Katrangi, W.; Laposata, M. Increased Δ5- and Δ6-Desaturase, Cyclooxygenase-2, and Lipoxygenase-5 Expression and Activity Are Associated with Fatty Acid and Eicosanoid Changes in Cystic Fibrosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2011, 1811, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.; Al-Turkmani, M.R.; Savaille, J.E.; Alturkmani, R.; Katrangi, W.; Cluette-Brown, J.E.; Zaman, M.M.; Laposata, M.; Freedman, S.D. Cell Culture Models Demonstrate That CFTR Dysfunction Leads to Defective Fatty Acid Composition and Metabolism. J. Lipid Res. 2008, 49, 1692–1700. [Google Scholar] [CrossRef]
- Das, U.N. Essential Fatty Acids and Their Metabolites in the Pathobiology of Inflammation and Its Resolution. Biomolecules 2021, 11, 1873. [Google Scholar] [CrossRef] [PubMed]
- Gilljam, H.; Strandvik, B.; Ellin, Á.; Wiman, L.-. Gös. Increased Mole Fraction of Arachidonic Acid in Bronchial Phospholipids in Patients with Cystic Fibrosis. Scand. J. Clin. Lab. Investig. 1986, 46, 511–518. [Google Scholar] [CrossRef]
- Carlstedt-Duke, J.; Brönnegård, M.; Strandvik, B. Pathological Regulation of Arachidonic Acid Release in Cystic Fibrosis: The Putative Basic Defect. Proc. Natl. Acad. Sci. USA 1986, 83, 9202–9206. [Google Scholar] [CrossRef]
- Dif, F.; Wu, Y.-Z.; Burgel, P.-R.; Ollero, M.; Leduc, D.; Aarbiou, J.; Borot, F.; Garcia-Verdugo, I.; Martin, C.; Chignard, M.; et al. Critical Role of Cytosolic Phospholipase A2 in Bronchial Mucus Hypersecretion in CFTR-Deficient Mice. Eur. Respir. J. 2010, 36, 1120–1130. [Google Scholar] [CrossRef]
- Borot, F.; Vieu, D.-L.; Faure, G.; Fritsch, J.; Colas, J.; Moriceau, S.; Baudouin-Legros, M.; Brouillard, F.; Ayala-Sanmartin, J.; Touqui, L.; et al. Eicosanoid Release Is Increased by Membrane Destabilization and CFTR Inhibition in Calu-3 Cells. PLoS ONE 2009, 4, e7116. [Google Scholar] [CrossRef]
- Bensalem, N.; Ventura, A.P.; Vallee, B.; Lipecka, J.; Tondelier, D.; Davezac, N.; Dos Santos, A.; Perretti, M.; Fajac, A.; Sermet-Gaudelus, I.; et al. Down-Regulation of the Anti-Inflammatory Protein Annexin A1 in Cystic Fibrosis Knock-out Mice and Patients. Mol. Cell. Proteomics 2005, 4, 1591–1601. [Google Scholar] [CrossRef]
- Dalli, J.; Rosignoli, G.; Hayhoe, R.P.G.; Edelman, A.; Perretti, M. CFTR Inhibition Provokes an Inflammatory Response Associated with an Imbalance of the Annexin A1 Pathway. Am. J. Pathol. 2010, 177, 176–186. [Google Scholar] [CrossRef]
- Meves, H. Arachidonic Acid and Ion Channels: An Update. Br. J. Pharmacol. 2008, 155, 4–16. [Google Scholar] [CrossRef]
- Linsdell, P. Inhibition of Cystic Fibrosis Transmembrane Conductance Regulator Chloride Channel Currents by Arachidonic Acid. Can. J. Physiol. Pharmacol. 2000, 78, 490–499. [Google Scholar] [CrossRef]
- Zhou, J.-J.; Linsdell, P. Molecular Mechanism of Arachidonic Acid Inhibition of the CFTR Chloride Channel. Eur. J. Pharmacol. 2007, 563, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Dei Cas, M.; Zulueta, A.; Mingione, A.; Caretti, A.; Ghidoni, R.; Signorelli, P.; Paroni, R. An Innovative Lipidomic Workflow to Investigate the Lipid Profile in a Cystic Fibrosis Cell Line. Cells 2020, 9, 1197. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N. Specialized Pro-Resolving Mediator Network: An Update on Production and Actions. Essays Biochem. 2020, 64, 443–462. [Google Scholar] [CrossRef]
- Lin, L.L.; Lin, A.Y.; Knopf, J.L. Cytosolic Phospholipase A2 Is Coupled to Hormonally Regulated Release of Arachidonic Acid. Proc. Natl. Acad. Sci. USA 1992, 89, 6147–6151. [Google Scholar] [CrossRef]
- Horati, H.; Janssens, H.M.; Margaroli, C.; Veltman, M.; Stolarczyk, M.; Kilgore, M.B.; Chou, J.; Peng, L.; Tiddens, H.A.M.W.; Chandler, J.D.; et al. Airway Profile of Bioactive Lipids Predicts Early Progression of Lung Disease in Cystic Fibrosis. J. Cyst. Fibros. 2020, 19, 902–909. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of Inflammation: The Beginning Programs the End. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Owens, J.M.; Shroyer, K.R.; Kingdom, T.T. Expression of Cyclooxygenase and Lipoxygenase Enzymes in Sinonasal Mucosa of Patients with Cystic Fibrosis. Arch. Otolaryngol. Head Neck Surg. 2008, 134, 825–831. [Google Scholar] [CrossRef]
- Roca-Ferrer, J.; Pujols, L.; Gartner, S.; Moreno, A.; Pumarola, F.; Mullol, J.; Cobos, N.; Picado, C. Upregulation of COX-1 and COX-2 in Nasal Polyps in Cystic Fibrosis. Thorax 2006, 61, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Eiserich, J.P.; Cross, C.E.; Morrissey, B.M.; Hammock, B.D. Metabolomic Profiling of Regulatory Lipid Mediators in Sputum from Adult Cystic Fibrosis Patients. Free Radic. Biol. Med. 2012, 53, 160–171. [Google Scholar] [CrossRef]
- Strandvik, B.; Svensson, E.; Seyberth, H.W. Prostanoid Biosynthesis in Patients with Cystic Fibrosis. Prostaglandins Leukot. Essent. Fat. Acids 1996, 55, 419–425. [Google Scholar] [CrossRef]
- Zakrzewski, J.T.; Barnes, N.C.; Piper, P.J.; Costello, J.F. Detection of Sputum Eicosanoids in Cystic Fibrosis and in Normal Saliva by Bioassay and Radioimmunoassay. Br. J. Clin. Pharmacol. 1987, 23, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Rigas, B.; Korenberg, J.R.; Merrill, W.W.; Levine, L. Prostaglandins E2 and E2 Alpha Are Elevated in Saliva of Cystic Fibrosis Patients. Am. J. Gastroenterol. 1989, 84, 1408–1412. [Google Scholar] [PubMed]
- Jabr, S.; Gartner, S.; Milne, G.L.; Roca-Ferrer, J.; Casas, J.; Moreno, A.; Gelpí, E.; Picado, C. Quantification of Major Urinary Metabolites of PGE2 and PGD2 in Cystic Fibrosis: Correlation with Disease Severity. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Gartner, S.; Roca-Ferrer, J.; Fernandez-Alvarez, P.; Lima, I.; Rovira-Amigo, S.; García-Arumi, E.; Tizzano, E.F.; Picado, C. Elevated Prostaglandin E2 Synthesis Is Associated with Clinical and Radiological Disease Severity in Cystic Fibrosis. J. Clin. Med. 2024, 13, 2050. [Google Scholar] [CrossRef]
- Ciabattoni, G.; Davì, G.; Collura, M.; Iapichino, L.; Pardo, F.; Ganci, A.; Romagnoli, R.; Maclouf, J.; Patrono, C. In Vivo Lipid Peroxidation and Platelet Activation in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2000, 162, 1195–1201. [Google Scholar] [CrossRef]
- Patrono, C.; Rocca, B. Measurement of Thromboxane Biosynthesis in Health and Disease. Front. Pharmacol. 2019, 10, 1244. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Brash, A.R.; Tauber, A.I.; Oates, J.A.; Hubbard, W.C. Modulation of Human Neutrophil Function by Monohydroxy-Eicosatetraenoic Acids. Immunology 1980, 39, 491–501. [Google Scholar] [PubMed]
- Takata, S.; Papayianni, A.; Matsubara, M.; Jimenez, W.; Pronovost, P.H.; Brady, H.R. 15-Hydroxyeicosatetraenoic Acid Inhibits Neutrophil Migration across Cytokine-Activated Endothelium. Am. J. Pathol. 1994, 145, 541–549. [Google Scholar]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef]
- Hamberg, M.; Hedqvist, P.; Rådegran, K. Identification of 15-Hydroxy-5,8,11,13-Eicosatetraenoic Acid (15-HETE) as a Major Metabolite of Arachidonic Acid in Human Lung. Acta Physiol. Scand. 1980, 110, 219–221. [Google Scholar] [CrossRef]
- Dennis, E.A.; Norris, P.C. Eicosanoid Storm in Infection and Inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Zampolli, A. From Asthma to Atherosclerosis--5-Lipoxygenase, Leukotrienes, and Inflammation. N. Engl. J. Med. 2004, 350, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T.; Peters-Golden, M.; Panettieri, R.A.; Henderson, W.R. Roles of Cysteinyl Leukotrienes in Airway Inflammation, Smooth Muscle Function, and Remodeling. J. Allergy Clin. Immunol. 2003, 111, S18–S34; discussion S34–S36. [Google Scholar] [CrossRef]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and Leukotriene Pathways: Biochemistry, Biology, and Roles in Disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef]
- Greally, P.; Hussein, M.J.; Cook, A.J.; Sampson, A.P.; Piper, P.J.; Price, J.F. Sputum Tumour Necrosis Factor-Alpha and Leukotriene Concentrations in Cystic Fibrosis. Arch. Dis. Child. 1993, 68, 389–392. [Google Scholar] [CrossRef][Green Version]
- Konstan, M.W.; Walenga, R.W.; Hilliard, K.A.; Hilliard, J.B. Leukotriene B4 Markedly Elevated in the Epithelial Lining Fluid of Patients with Cystic Fibrosis. Am. Rev. Respir. Dis. 1993, 148, 896–901. [Google Scholar] [CrossRef]
- Sampson, A.; Spencer, D.; Green, C.; Piper, P.; Price, J. Leukotrienes in the Sputum and Urine of Cystic Fibrosis Children. Br. J. Clin. Pharmacol. 1990, 30, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Romano, M. Lipoxin and Aspirin-Triggered Lipoxins. Sci. World J. 2010, 10, 458217. [Google Scholar] [CrossRef]
- Romano, M.; Serhan, C.N. Lipoxin Generation by Permeabilized Human Platelets. Biochemistry 1992, 31, 8269–8277. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Chen, X.S.; Takahashi, Y.; Yamamoto, S.; Funk, C.D.; Serhan, C.N. Lipoxin Synthase Activity of Human Platelet 12-Lipoxygenase. Biochem. J. 1993, 296, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Claria, J.; Serhan, C.N. Aspirin Triggers Previously Undescribed Bioactive Eicosanoids by Human Endothelial Cell-Leukocyte Interactions. Proc. Natl. Acad. Sci. USA 1995, 92, 9475–9479. [Google Scholar] [CrossRef] [PubMed]
- Claria, J.; Lee, M.H.; Serhan, C.N. Aspirin-Triggered Lipoxins (15-Epi-LX) Are Generated by the Human Lung Adenocarcinoma Cell Line (A549)-Neutrophil Interactions and Are Potent Inhibitors of Cell Proliferation. Mol. Med. 1996, 2, 583. [Google Scholar] [CrossRef]
- Titos, E.; Chiang, N.; Serhan, C.N.; Romano, M.; Gaya, J.; Pueyo, G.; Clària, J. Hepatocytes Are a Rich Source of Novel Aspirin-Triggered 15-Epi-Lipoxin A(4). Am. J. Physiol. 1999, 277, C870–C877. [Google Scholar] [CrossRef]
- Serhan, C.N.; Sheppard, K.A. Lipoxin Formation during Human Neutrophil-Platelet Interactions. Evidence for the Transformation of Leukotriene A4 by Platelet 12-Lipoxygenase in Vitro. J. Clin. Investig. 1990, 85, 772–780. [Google Scholar] [CrossRef]
- Romano, M.; Cianci, E.; Simiele, F.; Recchiuti, A. Lipoxins and Aspirin-Triggered Lipoxins in Resolution of Inflammation. Eur. J. Pharmacol. 2015, 760, 49–63. [Google Scholar] [CrossRef]
- Karp, C.L.; Flick, L.M.; Park, K.W.; Softic, S.; Greer, T.M.; Keledjian, R.; Yang, R.; Uddin, J.; Guggino, W.B.; Atabani, S.F.; et al. Defective Lipoxin-Mediated Anti-Inflammatory Activity in the Cystic Fibrosis Airway. Nat. Immunol. 2004, 5, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Jeanson, L.; Guerrera, I.C.; Papon, J.-F.; Chhuon, C.; Zadigue, P.; Prulière-Escabasse, V.; Amselem, S.; Escudier, E.; Coste, A.; Edelman, A. Proteomic Analysis of Nasal Epithelial Cells from Cystic Fibrosis Patients. PLoS ONE 2014, 9, e108671. [Google Scholar] [CrossRef] [PubMed]
- Ringholz, F.C.; Buchanan, P.J.; Clarke, D.T.; Millar, R.G.; McDermott, M.; Linnane, B.; Harvey, B.J.; McNally, P.; Urbach, V. Reduced 15-Lipoxygenase 2 and Lipoxin A4/Leukotriene B4 Ratio in Children with Cystic Fibrosis. Eur. Respir. J. 2014, 44, 394–404. [Google Scholar] [CrossRef]
- Pierdomenico, A.M.; Patruno, S.; Codagnone, M.; Simiele, F.; Mari, V.C.; Plebani, R.; Recchiuti, A.; Romano, M. microRNA-181b Is Increased in Cystic Fibrosis Cells and Impairs Lipoxin A4 Receptor-Dependent Mechanisms of Inflammation Resolution and Antimicrobial Defense. Sci. Rep. 2017, 7, 13519. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.; Schmelzer, K.; Lee, T.-S.; Fang, X.; Zhu, Y.; Spector, A.A.; Gill, S.; Morisseau, C.; Hammock, B.D.; et al. The Antiinflammatory Effect of Laminar Flow: The Role of PPARgamma, Epoxyeicosatrienoic Acids, and Soluble Epoxide Hydrolase. Proc. Natl. Acad. Sci. USA 2005, 102, 16747–16752. [Google Scholar] [CrossRef] [PubMed]
- Campbell, W.B. New Role for Epoxyeicosatrienoic Acids as Anti-Inflammatory Mediators. Trends Pharmacol. Sci. 2000, 21, 125–127. [Google Scholar] [CrossRef]
- Tooker, B.C.; Kandel, S.E.; Work, H.M.; Lampe, J.N. Pseudomonas Aeruginosa Cytochrome P450 CYP168A1 Is a Fatty Acid Hydroxylase That Metabolizes Arachidonic Acid to the Vasodilator 19-HETE. J. Biol. Chem. 2022, 298, 101629. [Google Scholar] [CrossRef]
- Malhotra, S.; Hayes, D.; Wozniak, D.J. Cystic Fibrosis and Pseudomonas Aeruginosa: The Host-Microbe Interface. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef]
- Tunaru, S.; Chennupati, R.; Nüsing, R.M.; Offermanns, S. Arachidonic Acid Metabolite 19(S)-HETE Induces Vasorelaxation and Platelet Inhibition by Activating Prostacyclin (IP) Receptor. PLoS ONE 2016, 11, e0163633. [Google Scholar] [CrossRef]
- Galli, F.; Battistoni, A.; Gambari, R.; Pompella, A.; Bragonzi, A.; Pilolli, F.; Iuliano, L.; Piroddi, M.; Dechecchi, M.C.; Cabrini, G.; et al. Oxidative Stress and Antioxidant Therapy in Cystic Fibrosis. Biochim. Biophys. Acta 2012, 1822, 690–713. [Google Scholar] [CrossRef]
- Thomson, E.; Brennan, S.; Senthilmohan, R.; Gangell, C.L.; Chapman, A.L.; Sly, P.D.; Kettle, A.J.; Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF); Balding, E.; Berry, L.J.; et al. Identifying Peroxidases and Their Oxidants in the Early Pathology of Cystic Fibrosis. Free Radic. Biol. Med. 2010, 49, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Chan, T.; Osberg, I.; Senthilmohan, R.; Chapman, A.L.P.; Mocatta, T.J.; Wagener, J.S. Myeloperoxidase and Protein Oxidation in the Airways of Young Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2004, 170, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Leto, T.L. Oxidative Innate Immune Defenses by Nox/Duox Family NADPH Oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [CrossRef] [PubMed]
- Roum, J.H.; Buhl, R.; McElvaney, N.G.; Borok, Z.; Crystal, R.G. Systemic Deficiency of Glutathione in Cystic Fibrosis. J. Appl. Physiol. (1985) 1993, 75, 2419–2424. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.-L.; Binder, C.J.; Stöckl, J. Generation and Biological Activities of Oxidized Phospholipids. Antioxid. Redox Signal 2010, 12, 1009–1059. [Google Scholar] [CrossRef] [PubMed]
- Dias, I.H.K.; Milic, I.; Heiss, C.; Ademowo, O.S.; Polidori, M.C.; Devitt, A.; Griffiths, H.R. Inflammation, Lipid (Per)Oxidation, and Redox Regulation. Antioxid. Redox Signal 2020, 33, 166–190. [Google Scholar] [CrossRef]
- Janssen, L.J. Isoprostanes: An Overview and Putative Roles in Pulmonary Pathophysiology. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2001, 280, L1067–L1082. [Google Scholar] [CrossRef]
- Janssen, L.J.; Catalli, A.; Helli, P. The Pulmonary Biology of Isoprostanes. Antioxid. Redox Signal 2005, 7, 244–255. [Google Scholar] [CrossRef]
- Freigang, S. The Regulation of Inflammation by Oxidized Phospholipids. Eur. J. Immunol. 2016, 46, 1818–1825. [Google Scholar] [CrossRef]
- Galiniak, S.; Mołoń, M.; Rachel, M. Links between Disease Severity, Bacterial Infections and Oxidative Stress in Cystic Fibrosis. Antioxidants 2022, 11, 887. [Google Scholar] [CrossRef]
- Wood, L.G.; Fitzgerald, D.A.; Gibson, P.G.; Cooper, D.M.; Garg, M.L. Increased Plasma Fatty Acid Concentrations after Respiratory Exacerbations Are Associated with Elevated Oxidative Stress in Cystic Fibrosis Patients. Am. J. Clin. Nutr. 2002, 75, 668–675. [Google Scholar] [CrossRef][Green Version]
- Reid, D.W.; Misso, N.; Aggarwal, S.; Thompson, P.J.; Walters, E.H. Oxidative Stress and Lipid-Derived Inflammatory Mediators during Acute Exacerbations of Cystic Fibrosis. Respirology 2007, 12, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Taccetti, G.; Campana, S.; Neri, A.S.; Boni, V.; Festini, F. Antibiotic Therapy against Pseudomonas Aeruginosa in Cystic Fibrosis. J. Chemother. 2008, 20, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.E.; Quaggiotto, P.; Wood, L.; O’Loughlin, E.V.; Henty, R.L.; Garg, M.L. Elevated Plasma Levels of F2α Isoprostane in Cystic Fibrosis. Lipids 1999, 34, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Hammond, V.J.; Morgan, A.H.; Lauder, S.; Thomas, C.P.; Brown, S.; Freeman, B.A.; Lloyd, C.M.; Davies, J.; Bush, A.; Levonen, A.-L.; et al. Novel Keto-Phospholipids Are Generated by Monocytes and Macrophages, Detected in Cystic Fibrosis, and Activate Peroxisome Proliferator-Activated Receptor-γ. J. Biol. Chem. 2012, 287, 41651–41666. [Google Scholar] [CrossRef]
- Gartner, S.; Fernandez-Alvarez, P.; Lima, I.; Rovira, S.; Picado, C.; Garcia-Arumí, E.; Mir, I.D.; Torrent, A.; Moreno, A.; Iglesias, I.; et al. Inflammation in Cystic Fibrosis Patients: Study of the Eicosanoid Pathway and Severity Correlations. Eur. Respir. J. 2021, 58, PA2106. [Google Scholar] [CrossRef]
- Strandvik, B.; Gronowitz, E.; Enlund, F.; Martinsson, T.; Wahlström, J. Essential Fatty Acid Deficiency in Relation to Genotype in Patients with Cystic Fibrosis. J. Pediatr. 2001, 139, 650–655. [Google Scholar] [CrossRef]
- Stubbs, C.D.; Smith, A.D. The Modification of Mammalian Membrane Polyunsaturated Fatty Acid Composition in Relation to Membrane Fluidity and Function. Biochim. Biophys. Acta 1984, 779, 89–137. [Google Scholar] [CrossRef]
- Bae, H.; Kim, B.R.; Jung, S.; Le, J.; Heide, D.M. van der; Yu, W.; Park, S.H.; Hilkin, B.M.; Gansemer, N.D.; Powers, L.S.; et al. Arteriovenous Metabolomics in Pigs Reveals CFTR Regulation of Metabolism in Multiple Organs. Available online: https://www.jci.org/articles/view/174500/pdf (accessed on 27 June 2024).
- Bhura-Bandali, F.N.; Suh, M.; Man, S.F.P.; Clandinin, M.T. The ΔF508 Mutation in the Cystic Fibrosis Transmembrane Conductance Regulator Alters Control of Essential Fatty Acid Utilization in Epithelial Cells. J. Nutr. 2000, 130, 2870–2875. [Google Scholar] [CrossRef]
- Xu, Y.; Clark, J.C.; Aronow, B.J.; Dey, C.R.; Liu, C.; Wooldridge, J.L.; Whitsett, J.A. Transcriptional Adaptation to Cystic Fibrosis Transmembrane Conductance Regulator Deficiency. J. Biol. Chem. 2003, 278, 7674–7682. [Google Scholar] [CrossRef]
- Moliteo, E.; Sciacca, M.; Palmeri, A.; Papale, M.; Manti, S.; Parisi, G.F.; Leonardi, S. Cystic Fibrosis and Oxidative Stress: The Role of CFTR. Molecules 2022, 27, 5324. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Dohna, M.; Graeber, S.Y.; Sommerburg, O.; Renz, D.M.; Pallenberg, S.T.; Voskrebenzev, A.; Schütz, K.; Hansen, G.; Doellinger, F.; et al. Impact of Elexacaftor/Tezacaftor/Ivacaftor Therapy on Lung Clearance Index and Magnetic Resonance Imaging in Children with Cystic Fibrosis and One or Two F508del Alleles. Eur. Respir. J. 2024, 2400004. [Google Scholar] [CrossRef] [PubMed]
- Szabo, M.M.; Foushee, S.E.; McPheeters, C.M.; O’Hagan, A.R.; Ramirez, A.M.; O’Reilly, E.A. Impact of Elexacaftor/Tezacaftor/Ivacaftor on Respiratory Colonization in an Adult Cystic Fibrosis Clinic. Am. J. Med. Sci. 2024, 367, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Liessi, N.; Pesce, E.; Braccia, C.; Bertozzi, S.M.; Giraudo, A.; Bandiera, T.; Pedemonte, N.; Armirotti, A. Distinctive Lipid Signatures of Bronchial Epithelial Cells Associated with Cystic Fibrosis Drugs, Including Trikafta. JCI Insight 2020, 5, e138722. [Google Scholar] [CrossRef]
- O’Connor, M.G.; Seegmiller, A. The Effects of Ivacaftor on CF Fatty Acid Metabolism: An Analysis from the GOAL Study. J. Cyst. Fibros. 2017, 16, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Veltman, M.; De Sanctis, J.B.; Stolarczyk, M.; Klymiuk, N.; Bähr, A.; Brouwer, R.W.; Oole, E.; Shah, J.; Ozdian, T.; Liao, J.; et al. CFTR Correctors and Antioxidants Partially Normalize Lipid Imbalance but Not Abnormal Basal Inflammatory Cytokine Profile in CF Bronchial Epithelial Cells. Front. Physiol. 2021, 12, 619442. [Google Scholar] [CrossRef]
- Teopompi, E.; Risé, P.; Pisi, R.; Buccellati, C.; Aiello, M.; Pisi, G.; Tripodi, C.; Fainardi, V.; Clini, E.; Chetta, A.; et al. Arachidonic Acid and Docosahexaenoic Acid Metabolites in the Airways of Adults With Cystic Fibrosis: Effect of Docosahexaenoic Acid Supplementation. Front. Pharmacol. 2019, 10, 938. [Google Scholar] [CrossRef]
- Ayats-Vidal, R.; Bosque-García, M.; Cordobilla, B.; Asensio-De la Cruz, O.; García-González, M.; Castro-Marrero, J.; López-Rico, I.; Domingo, J.C. Changes of Erythrocyte Fatty Acids after Supplementation with Highly Concentrated Docosahexaenoic Acid (DHA) in Pediatric Cystic Fibrosis: A Randomized Double-Blind Controlled Trial. J. Clin. Med. 2023, 12, 3704. [Google Scholar] [CrossRef]
- López-Neyra, A.; Suárez, L.; Muñoz, M.; de Blas, A.; Ruiz de Valbuena, M.; Garriga, M.; Calvo, J.; Ribes, C.; Girón Moreno, R.; Máiz, L.; et al. Long-Term Docosahexaenoic Acid (DHA) Supplementation in Cystic Fibrosis Patients: A Randomized, Multi-Center, Double-Blind, Placebo-Controlled Trial. Prostaglandins Leukot. Essent. Fat. Acids 2020, 162, 102186. [Google Scholar] [CrossRef]
- Watson, H.; Stackhouse, C. Omega-3 Fatty Acid Supplementation for Cystic Fibrosis. Cochrane Database Syst. Rev. 2020, 2020, CD002201. [Google Scholar] [CrossRef]
- Ayats-Vidal, R.; Bosque-García, M.; Cordobilla, B.; Asensio-De la Cruz, O.; García-González, M.; Loureda-Pérez, S.; Fernández-López, E.; Robert-Barriocanal, E.; Valiente-Planas, A.; Domingo, J.C. Impact of 1-Year Supplementation with High-Rich Docosahexaenoic Acid (DHA) on Clinical Variables and Inflammatory Biomarkers in Pediatric Cystic Fibrosis: A Randomized Double-Blind Controlled Trial. Nutrients 2024, 16, 970. [Google Scholar] [CrossRef]
- Vij, N. Linoleic Acid Supplement in Cystic Fibrosis: Friend or Foe? Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L597–L598. [Google Scholar] [CrossRef]
- Konstan, M.W.; Byard, P.J.; Hoppel, C.L.; Davis, P.B. Effect of High-Dose Ibuprofen in Patients with Cystic Fibrosis. N. Engl. J. Med. 1995, 332, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Carlile, G.W.; Robert, R.; Goepp, J.; Matthes, E.; Liao, J.; Kus, B.; Macknight, S.D.; Rotin, D.; Hanrahan, J.W.; Thomas, D.Y. Ibuprofen Rescues Mutant Cystic Fibrosis Transmembrane Conductance Regulator Trafficking. J. Cyst. Fibros. 2015, 14, 16–25. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shah, P.N.; Shah, K.N.; Smolen, J.A.; Tagaev, J.A.; Torrealba, J.; Zhou, L.; Zhang, S.; Zhang, F.; Wagers, P.O.; Panzner, M.J.; et al. A Novel in Vitro Metric Predicts in Vivo Efficacy of Inhaled Silver-Based Antimicrobials in a Murine Pseudomonas Aeruginosa Pneumonia Model. Sci. Rep. 2018, 8, 6376. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ilanga, M.; Simbassa, S.B.; Chirra, B.; Shah, K.N.; Cannon, C.L. Synergistic Antimicrobial Effects of Ibuprofen Combined with Standard-of-Care Antibiotics against Cystic Fibrosis Pathogens. Biomedicines 2023, 11, 2936. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D.; FitzGerald, G.A. COX-2 Inhibitors and Cardiovascular Risk. J. Cardiovasc. Pharmacol. 2007, 50, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M. New Mechanisms for Effects of Anti-Inflammatory Glucocorticoids. Biofactors 1991, 3, 97–102. [Google Scholar]
- Dezateux, C.; Walters, S.; Balfour-Lynn, I. Inhaled Corticosteroids for Cystic Fibrosis. Cochrane Database Syst. Rev. 2000, 2000, CD001915. [Google Scholar] [CrossRef]
- Okuno, T.; Yokomizo, T.; Hori, T.; Miyano, M.; Shimizu, T. Leukotriene B4 Receptor and the Function of Its Helix 8*. J. Biol. Chem. 2005, 280, 32049–32052. [Google Scholar] [CrossRef]
- Schmitt-Grohé, S.; Zielen, S. Leukotriene Receptor Antagonists in Children with Cystic Fibrosis Lung Disease: Anti-Inflammatory and Clinical Effects. Paediatr. Drugs 2005, 7, 353–363. [Google Scholar] [CrossRef]
- Elborn, J.; Horsley, A.; MacGregor, G.; Bilton, D.; Grosswald, R.; Ahuja, S.; Springman, E. Phase I Studies of Acebilustat: Biomarker Response and Safety in Patients with Cystic Fibrosis. Clin. Transl. Sci. 2017, 10, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S.; Konstan, M.W.; Taylor-Cousar, J.L.; Fajac, I.; Horsley, A.; Sutharsan, S.; Aaron, S.D.; Daines, C.L.; Uluer, A.; Downey, D.G.; et al. Empire-CF Study: A Phase 2 Clinical Trial of Leukotriene A4 Hydrolase Inhibitor Acebilustat in Adult Subjects with Cystic Fibrosis. J. Cyst. Fibros. 2021, 20, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.F. The Cysteinyl Leukotriene Receptors. Prostaglandins Leukot. Essent. Fat. Acids 2003, 69, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Morice, A.H.; Kastelik, J.A.; Aziz, I. Montelukast Sodium in Cystic Fibrosis. Thorax 2001, 56, 244–245. [Google Scholar] [CrossRef][Green Version]
- Schmitt-Grohé, S.; Eickmeier, O.; Schubert, R.; Bez, C.; Zielen, S. Anti-Inflammatory Effects of Montelukast in Mild Cystic Fibrosis. Ann. Allergy Asthma Immunol. 2002, 89, 599–605. [Google Scholar] [CrossRef]
- Stelmach, I.; Korzeniewska, A.; Stelmach, W.; Majak, P.; Grzelewski, T.; Jerzynska, J. Effects of Montelukast Treatment on Clinical and Inflammatory Variables in Patients with Cystic Fibrosis. Ann. Allergy Asthma Immunol. 2005, 95, 372–380. [Google Scholar] [CrossRef]
- Schmitt-Grohé, S.; Eickmeier, O.; Naujoks, C.; Schubert, R.; Lentze, M.J.; Zielen, S.; Rietschel, E. Effects of Long-Term Treatment with Montelukast in Mild Cystic Fibrosis: (Long Term Treatment with Montelukast in Cystic Fibrosis). Respir. Med. 2007, 101, 684. [Google Scholar] [CrossRef][Green Version]
- Conway, S.P.; Etherington, C.; Peckham, D.G.; Whitehead, A. A Pilot Study of Zafirlukast as an Anti-Inflammatory Agent in the Treatment of Adults with Cystic Fibrosis. J. Cyst. Fibros. 2003, 2, 25–28. [Google Scholar] [CrossRef][Green Version]
- Hartl, D.; Starosta, V.; Maier, K.; Beck-Speier, I.; Rebhan, C.; Becker, B.F.; Latzin, P.; Fischer, R.; Ratjen, F.; Huber, R.M.; et al. Inhaled Glutathione Decreases PGE2 and Increases Lymphocytes in Cystic Fibrosis Lungs. Free. Radic. Biol. Med. 2005, 39, 463–472. [Google Scholar] [CrossRef]
- Schett, G.; Neurath, M.F. Resolution of Chronic Inflammatory Disease: Universal and Tissue-Specific Concepts. Nat. Commun. 2018, 9, 3261. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).