
From the “One-Molecule, One-Target, One-Disease” Concept towards Looking for Multi-Target Therapeutics for Treating Non-Polio Enterovirus (NPEV) Infections




Abstract
:
1. Introduction
1.1. Pre-Proposal Context
1.2. The Need for Therapies
2. Results
- Part 1: NPEV-inhibitors evaluated in clinical trials.
2.1. Structural Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Publication Date | Type of Study * | Virus/Disease | Type of Infection (Time to Treatment Interval) † | Drug | Pharmaceutical Form | Patient Age | Ref. |
|---|---|---|---|---|---|---|---|
| April 1989 | DB PC phase II | RV-A9 | Inoculation (−28 h) | R61837 | Nasal spray | 18–50 years old | [24] |
| February 1993 | Rdm DB PC phase II | RV-A39 | Inoculation (−42 h) | WIN 54954 | Oral capsule | 18–50 years old | [25] |
| April 1992 | Rdm DB PC phase II | RV-A39 | Naturally occurred (+24 h) | Pirodavir (R77,975) | Nasal spray | 18–50 years old | [26] |
| February 1995 | Rdm DB PC phase II | RV colds | Inoculation (−6/12 h) | Pirodavir (R77,975) | Nasal spray | 18–65 years old | [27] |
| January 2000 | Rdm DB PC phase II | CVA21 | Inoculation (−14 h) | Pleconaril | Oral formulation | 18–37 years old | [28] |
| 15 January 2001 | Phase II ‡ | EV | Naturally occurred | Pleconaril | Suspension | 2–50 years old | [29] |
| 6 March 2002 | Rdm DB PC phase II | EV Sepsis | Naturally occurred (+36 h) | Pleconaril | Oral solution/suspension | >15 days | [30] |
| September 2002 | Clinical application | CVB | Naturally occurred (+48 h) | Pleconaril | / | <27 days | [31] |
| April 2003 | Rdm DB PC phase II | Suspected EV meningitis | Naturally occurred (+10 days) | Pleconaril | / | <12 months | [32] |
| November 2005 | Rdm DB PC phase III | RV common colds | Naturally occurred (+24 h) | Pleconaril | Oral tablets | >18 years old | [33] |
| July 2006 | Rdm DB PC phase II | EV meningitis | Naturally occurred (+10 days) | Pleconaril | Oral formulation | >14 years old | [34] |
| 31 October 2006 | Rdm DB PC phase II | RV asthma & common cold | Naturally occurred | Pleconaril | Nasal spray | 6–65 years old | [35] |
| 28 May 2008 | Rdm DB PC phase II | RV | Inoculation (+1/2 h) | Vapendavir | Oral capsules | NS | [36] |
| 3 August 2010 | Rdm DB PC phase II | RV | Naturally occurred | Vapendavir | / | 18–70 years old | [37] |
| 31 December 2014 | Rdm DB PC phase II | RV | Naturally occurred | Vapendavir | Oral capsules | >18 years old | [38] |
| 13 February 2015 | Rdm DB PC phase II | Asthma | Naturally occurred (same day) | Vapendavir | / | 18–75 years old | [39] |
| March 2016 | Rdm DB PC phase II | EV suspected sepsis | Naturally occurred | Pleconaril | Liquid/suspension | <27 days | [40] |
| 12 January 2017 | Rdm DB PC phase II | RV Upper Respiratory | Naturally occurred (+3/5 days) | Vapendavir | Oral tablets | 12–75 years old | [41] |
| 10 November 2023 | Rdm PC phase II | RV & EV COPD | Naturally occurred (+48 h) | Vapendavir | Oral tablets | 40–75 years old | [33] |
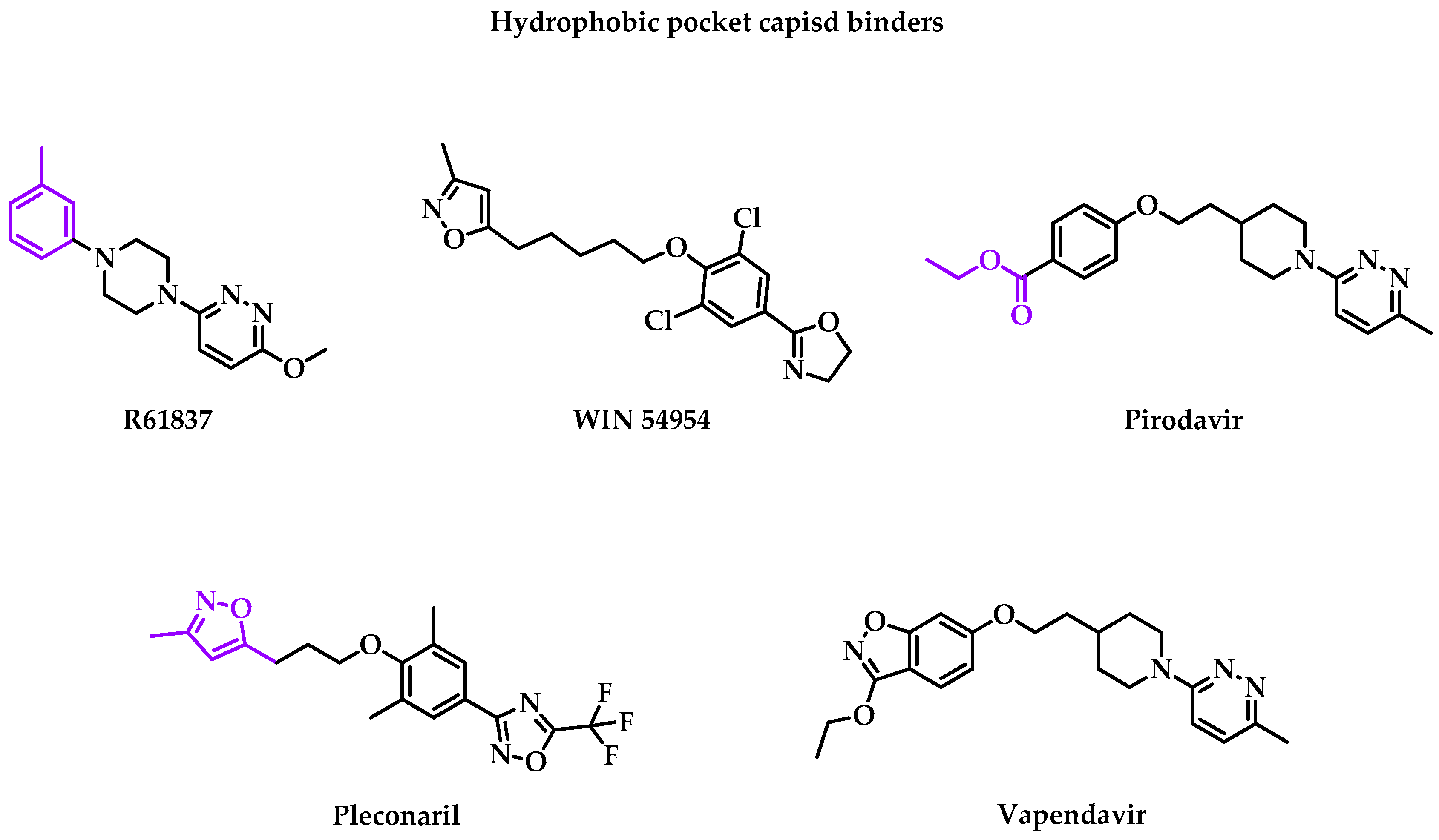
- R61837
- WIN 54954
- Pirodavir
- Pleconaril
- Vapendavir
2.2. Viral Non-Structural Protein
| Publication Date | Type of Study * | Virus/Disease | Type of Infection (Time to Treatment Interval) † | Drug | Target | Pharmaceutical Form | Patient Age | Ref. |
|---|---|---|---|---|---|---|---|---|
| 20 July 1981 | PC phase II | RV-A9 | Inoculation (−24 h) | Enviroxime | 3A/3AB protein | Nasal spray | 18–50 years old | [48] |
| July 1982 | Rdm DB PC phase II | RV-A39 | Inoculation (−24 h) | Enviroxime | 3A/3AB protein | Nasal spray/oral | 18–36 years old | [49] |
| December 1982 | Rdm DB PC phase II | RV | Inoculation (−24 h) | Enviroxime | 3A/3AB protein | Nasal spray | NS | [50] |
| July 1983 | DB PC phase II | RV-A9 | Inoculation (−24 h) | Enviroxime | 3A/3AB protein | Nasal spray | 18–50 years old | [51] |
| January 1985 | Rdm DB PC phase III | RV common cold | Inoculation (−44 h) | Enviroxime | 3A/3AB protein | Nasal suspension | 8–65 years old | [52] |
| February 2002 | Rdm DB PC phase I | RV | / | Rupintrivir | 3C protein | Nasal spray | 18–50 years old | [53] |
| December 2003 | Rdm DB PC phase II | RV-A39, RV-A21 | Inoculation (−6/+24 h) | Rupintrivir | 3C protein | Nasal spray | 18–60 years old | [54] |
| July 2005 | Phase I | RV | / | V-7404 | 3C protein | Oral formulation | 18–55 years old | [55] |
| 17 September 2021 | Rdm DB PC phase I | EV | / | V-7404 | 3C protein | Oral solution | 18–45 years old | [56] |
2.2.1. 3A/3AB Proteases
- Enviroxime
2.2.2. 3C Protease
- Rupintrivir
- V-7404
2.3. Targeting Human-Coded Protein Promoting Enterovirus Replication
| Publication Date | Type of Study * | Virus/Disease | Type of Infection (Time to Treatment Interval) † | Drug | Target | Pharmaceutical Form | Patient Age | Ref. |
|---|---|---|---|---|---|---|---|---|
| January 1987 | Rdm DB PC phase II | RV-A39 | Inoculation (−1 day) | Atropine Methonitrate | Anticholinergic | Nasal spray | >18 years old | [69] |
| 23 July 2020 | Rdm DB PC phase III | EV, RV | Inoculation | CUR-N399 | PI4KIII β | Oral capsule | >18 years old | [70] |
| September 2010 | Rdm DB PC phase II | RV-A39 | Inoculation (−3 h) | Oxymetazoline | α-Adrenoreceptor | Intranasal liquid | 18–65 years old | [71] |
2.3.1. Cholinergic Receptors
2.3.2. Phosphatidylinositol 4-Kinase Beta (PI4KIII β)
2.3.3. α-Adrenoceptor
2.4. Unknown or Other Mechanisms of Action
2.4.1. Anti-Rhinovirus
| Publication Date | Type of Study * | Virus/Disease | Type of Infection (Time to Treatment Interval) † | Drug | Pharmaceutical Form | Patient Age | Ref. |
|---|---|---|---|---|---|---|---|
| July 1976 | Rdm DB PC phase II | RV-B3 | Inoculation (−1 day) | SKF 40491, GL R9-338, RP 19326 | Solution spray/suspension drop | 18–50 years old | [79] |
| October 1984 | Rdm DB PC phase II | RV-A9 | Inoculation (−1 day) | Ro 09-0415 | Oral capsule | 18–50 years old | [80] |
| May 1985 | Rdm PC phase II | RV-EL, RV-1B | Inoculation (−1 day) | 44 081 R.P. | Nasal spray | 18–50 years old | [81] |
| December 1987 | DB PC phase II | RV-A2 | Inoculation | Ro 09-0410 | Nasal spray | >18 years old | [82] |
| July 1990 | Phase II | RV-A2 | Inoculation | Ro 09-0410 | DMSO solution | 18–50 years old | [83] |
| 13 May 2022 | Rdm DB PC phase I | Asthma with RV | Inoculation | GSK3923868 | Inhalation powder | 18–65 years old | [84] |
| 26 May 2022 | Rdm DB PC phase I | COPD with RV | Inoculation | GSK3923868 | Inhalation powder | 18–65 years old | [85] |
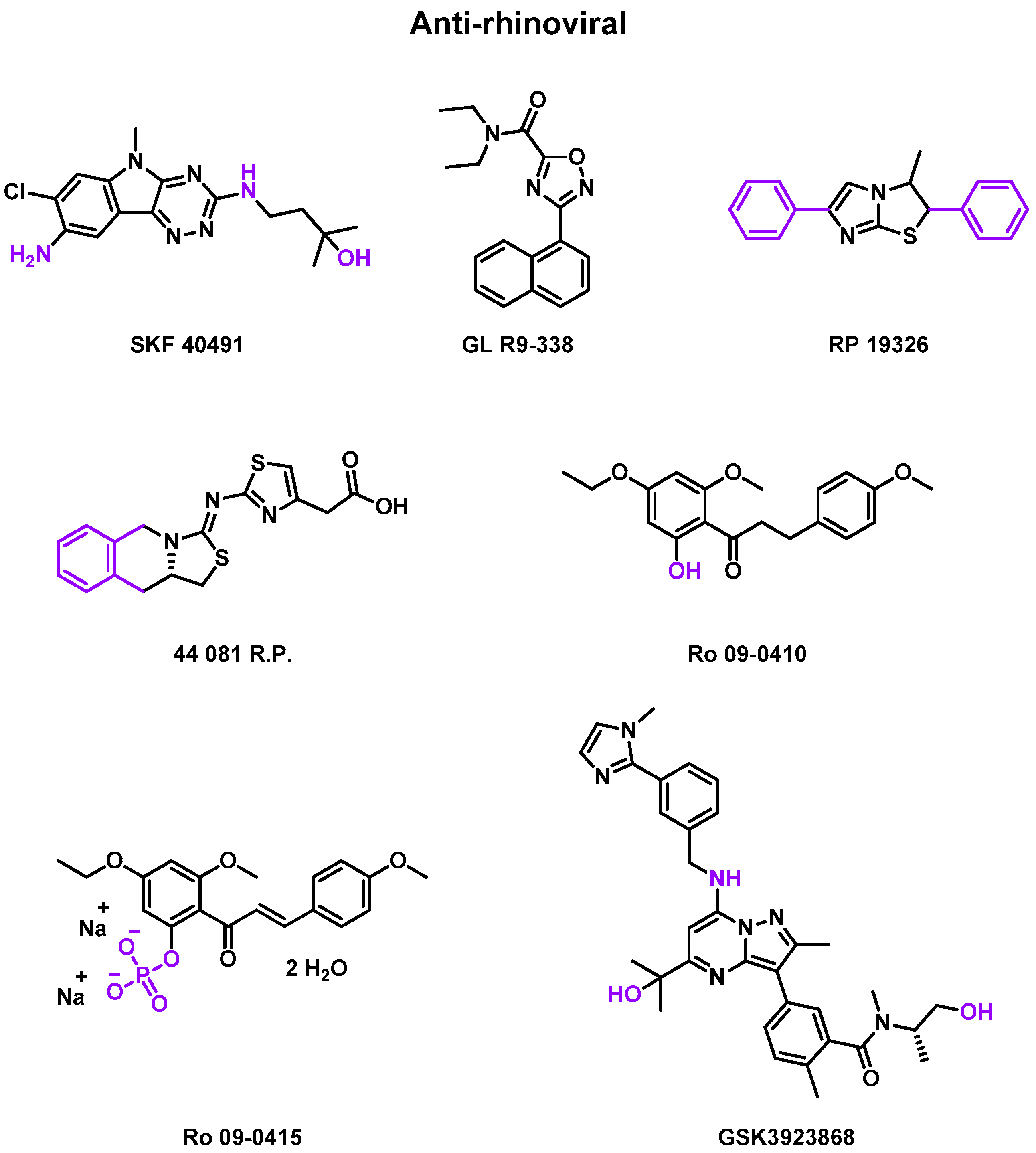
- Rhône Poulenc (RP) studies
- Roche (Ro) studies
- GlaxoSmithKline (GSK) studies
2.4.2. Broad Spectrum Antiviral
| Publication Date | Type of Study * | Virus/Disease | Type of Infection (Time to Treatment Interval) † | Drug | Pharmaceutical Form | Patient Age | Ref. |
|---|---|---|---|---|---|---|---|
| March 1973 | DB PC phase II | RV-A9, RV-A13 | Inoculation (−1 day) | Isoprinosine | Oral tablet | 18–50 years old | [87] |
| April 1974 | DB PC phase II | RV-A32, RV-B44 | Inoculation | Isoprinosine | Oral tablet | 25–48 years old | [88] |
| March 1977 | Rdm DB PC phase II | RV-A21 | Inoculation | Isoprinosine | Oral tablet | >18 years old | [89] |
| 18 July 2018 | Rdm DB PC phase III | EV, RV | Naturally occurred (+40 h) | Nitazoxanide | Oral tablets | >12 years old | [90] |
| 13 May 2020 | Rdm DB PC phase III | EV, RV | Naturally occurred (+72 h) | Nitazoxanide | Oral tablets | >12 years old | [91] |
| May 1970 | Rdm DB PC phase II | RV-A9 | Inoculation (−1 day) | UK2054 | Oral formulation | NS | [92] |
| December 1973 | DB PC phase II | RV-A24 | Inoculation (−1 day) | DIQA | Oral capsule | 21–42 years old | [93] |
| 14 January 2017 | Rdm DB PC phase II | Upper respiratory | Naturally occurred | ARM-1 | Oral spray | 18–43 years old | [94] |
- Isoprinosine (Inosiplex®)
- Nitazoxanide
- Others
2.4.3. Other Drug Repurposing
| Publication Date | Type of Study * | Virus/Disease | Type of Infection (Time to Treatment Interval) † | Drug | Type of Drug | Pharmaceutical Form | Patient Age | Ref. |
|---|---|---|---|---|---|---|---|---|
| January 1990 | Rdm DB PC phase II | RV-A9, RV-B14 | Inoculation (−1 day) | Nedocromil sodium | Mast cell stabilizer | Nasal spray | 18–50 years old | [98] |
| 15 April 2000 | Rdm DB PC phase II | RV-A16 | Inoculation (−1 day) | Clarithromycin | Antibiotic | Oral capsule | >18 years old | [100] |
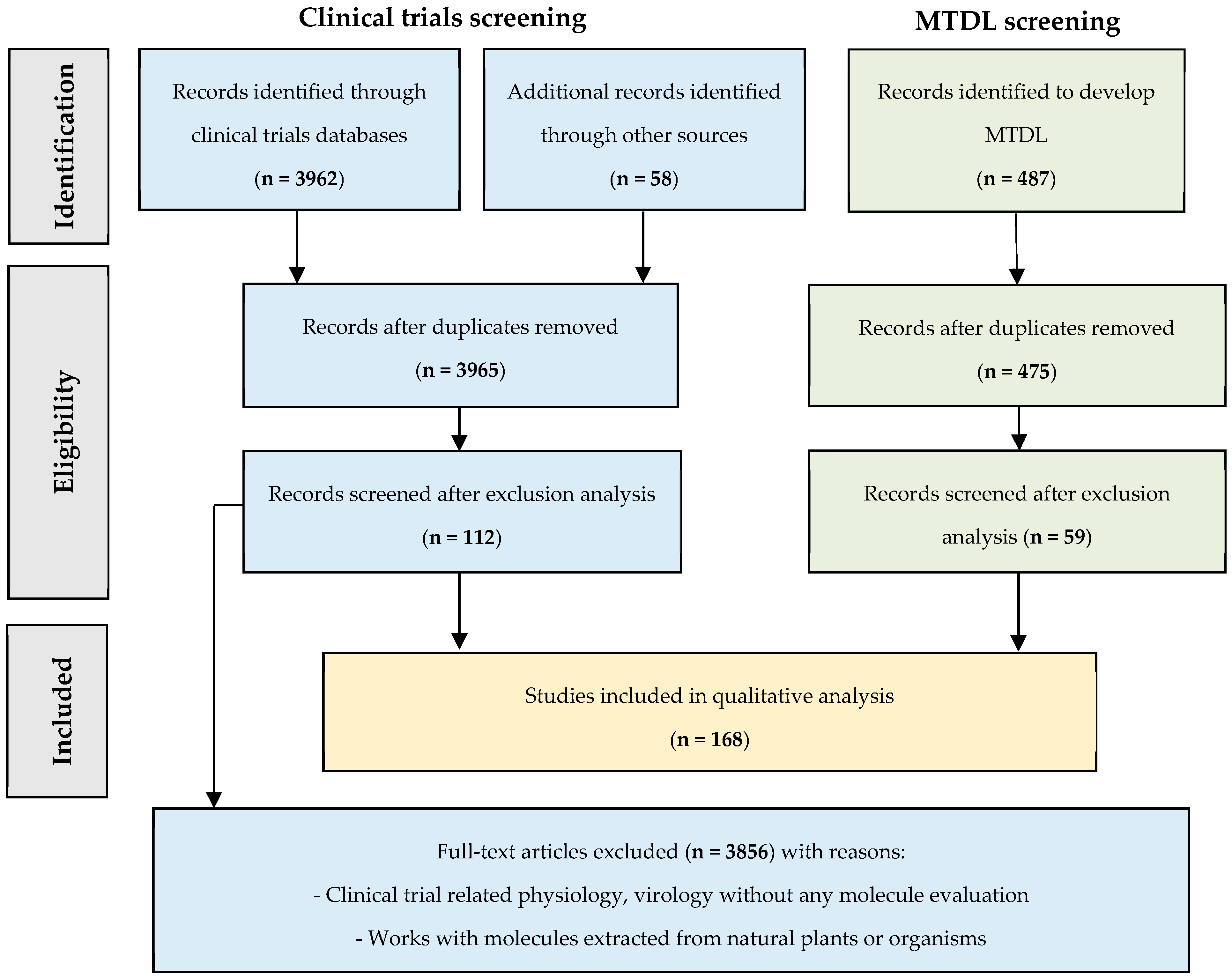
- Part 2: Filters to obtain an effective broad-spectrum anti-enterovirus drug.
2.5. Candidates’ Selection for MTDL Strategy
2.5.1. Combination of Two Compounds
| (A) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Synergistic Effect [µM2;%] ‡ | Virus | Pocapavir (V-073) | Vapendavir (BTA798) | Pleconaril | Disoxaril | Arildone | Enviroxime | HBB | MDL-860 | NITD008 | GPP3 | Itraconazole | Favipiravir | Suramin | Ref. | |
| AG-7404/ V-7404 | PV Sabin 1 | 580 | 463 | / | / | / | / | / | / | / | / | / | / | / | [115] | |
| PV Sabin 2 | 459 | 245 | / | / | / | / | / | / | / | / | / | / | / | |||
| PV Sabin 3 | 288 | 579 | / | / | / | / | / | / | / | / | / | / | / | |||
| MDL-860 | CVB1 Connecticut | / | / | 22.5 | / | / | / | / | / | / | / | / | / | / | [111,112] | |
| CVB3 Woodruff | / | / | 61.1 | / | / | / | / | / | / | / | / | / | / | |||
| Guanidine.HCl | CVB1 Connecticut | / | / | 58.4 | / | / | / | / | 47.5 | / | / | / | / | / | ||
| CVB3 Nancy | / | / | / | / | / | / | / | 1.1 | / | / | / | / | / | |||
| CVB3 Woodruff | / | / | 56.2 | / | / | / | / | 83.9 | / | / | / | / | / | |||
| Oxoglaucine | CVB1 Connecticut | / | / | 27.0 | / | / | / | / | 7.8 | / | / | / | / | / | ||
| CVB3 Nancy | / | / | / | / | / | / | / | 220.6 | / | / | / | / | / | |||
| CVB3 Woodruff | / | / | 18.6 | / | / | / | / | 151.0 | / | / | / | / | / | |||
| Enviroxime | CVB1 Connecticut | / | / | / | 82 | −39 a | / | 253 | / | / | / | / | / | / | ||
| PTU-23 | CVB1 Connecticut | / | / | / | 314 | 519 | 373 | 275 | / | / | / | / | / | / | ||
| HBB | CVB1 Connecticut | / | / | / | 689 | 852 | 253 | / | / | / | / | / | / | / | ||
| S-7 | CVB1 Connecticut | / | / | / | / | / | 399 | 368 | / | / | / | / | / | / | ||
| Compound 1 | CVB4 Edwards | / | / | 147 | / | / | / | / | / | / | / | / | / | / | [109] | |
| YZ-LY-0 | 0.125 µM | EV-A71 SK-EV006 | / | / | / | / | / | / | / | / | −400–−300 | 0–100 | / | / | / | [118] |
| 1 µM | / | / | / | / | / | / | / | / | −100–0 | 0–100 | / | / | / | |||
| Rupintrivir | EV-A71 FY573 | / | / | / | / | / | / | / | / | / | / | 450.64 | 438.07 | 4.96 | [116] | |
| Favipiravir | EV-A71 FY573 | / | / | / | / | / | / | / | / | / | / | −88.11 | / | 337.59 | ||
| Suramin | EV-A71 FY573 | / | / | / | / | / | / | / | / | / | / | −246.23 | 337.59 | / | ||
| GW5074 | EV-A71 FY573 | / | / | / | / | / | / | / | / | / | / | −167.68 | / | / | ||
| (B) | ||||||||||||||||
| ZIP Synergy Score [−30/30] † | Virus | Pleconaril | Rupintrivir | Ref. | ||||||||||||
| Rupintrivir | E1 Farouk (A549 cells) | 18.0 | / | [110] | ||||||||||||
| E1 Farouk (RPE cells) | 18.9 | / | ||||||||||||||
| Vemurafenib | E1 Farouk (A549 cells) | 13.6 | 15.6/16.00 | |||||||||||||
| E1 Farouk (RPE cells) | 20.2 | 17.2 | ||||||||||||||
| Pleconaril | E1 Farouk (A549 cells) | 18.0/18.3 | 18.0/18.3 | |||||||||||||
| E1 Farouk (RPE cells) | 18.9 | 18.9 | ||||||||||||||
| Cycloheximide | E1 Farouk (A549 cells) | / | 10.7 | |||||||||||||
| Remdesivir | E1 Farouk (A549 cells) | / | 8.23 | |||||||||||||
| Dalbavancin | E1 Farouk (A549 cells) | / | 8 | |||||||||||||
| Anisomycin | E1 Farouk (A549 cells) | / | 6.39 | |||||||||||||
| Emetine | E1 Farouk (A549 cells) | / | 4.1 | |||||||||||||
| Digoxin | E1 Farouk (A549 cells) | / | −5.6 | |||||||||||||
| Homoharringtonine | E1 Farouk (A549 cells) | / | −5.7 | |||||||||||||
| Halofuginone | E1 Farouk (A549 cells) | / | −7.5 | |||||||||||||
| Obatoclax | E1 Farouk (A549 cells) | / | −7.8 | |||||||||||||
| Gemcitabine | E1 Farouk (A549 cells) | / | −8.7 | |||||||||||||
| (C) | ||||||||||||||||
| Concentration of Ribavirin (µM) | 25 | 50 | 100 | 200 | 400 | Ref. | ||||||||||
| Combination Index with 0.4 µM of gemcitabine ° (CVB3 on Vero cells) | 0.28 | 0.23 | 0.15 | 0.14 | 0.14 | [120] | ||||||||||
2.5.2. Combination of Three Compounds
2.5.3. Other Criteria
2.6. Biological Parameters for MTDL Strategy
2.6.1. Enteroviruses of Interest
2.6.2. Biological Access to the Target
2.6.3. Conserved Residues of Targets—Antiviral Resistance
3. Discussion
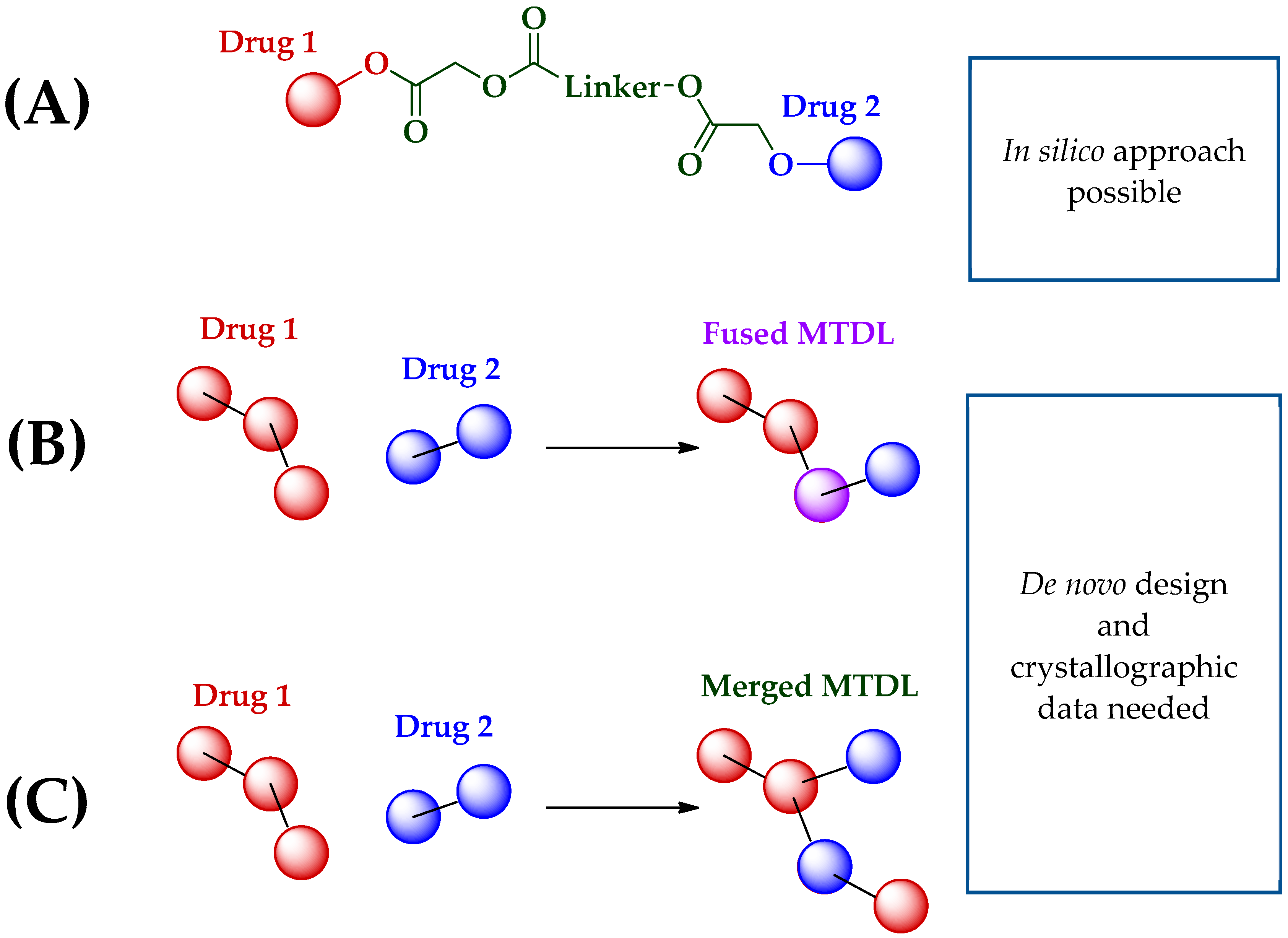
3.1. Multi-Target Directed Ligand (MTDL) Definition
3.2. MTLD Challenges and Alternatives
3.3. Benefit–Risk Balance of MTDL
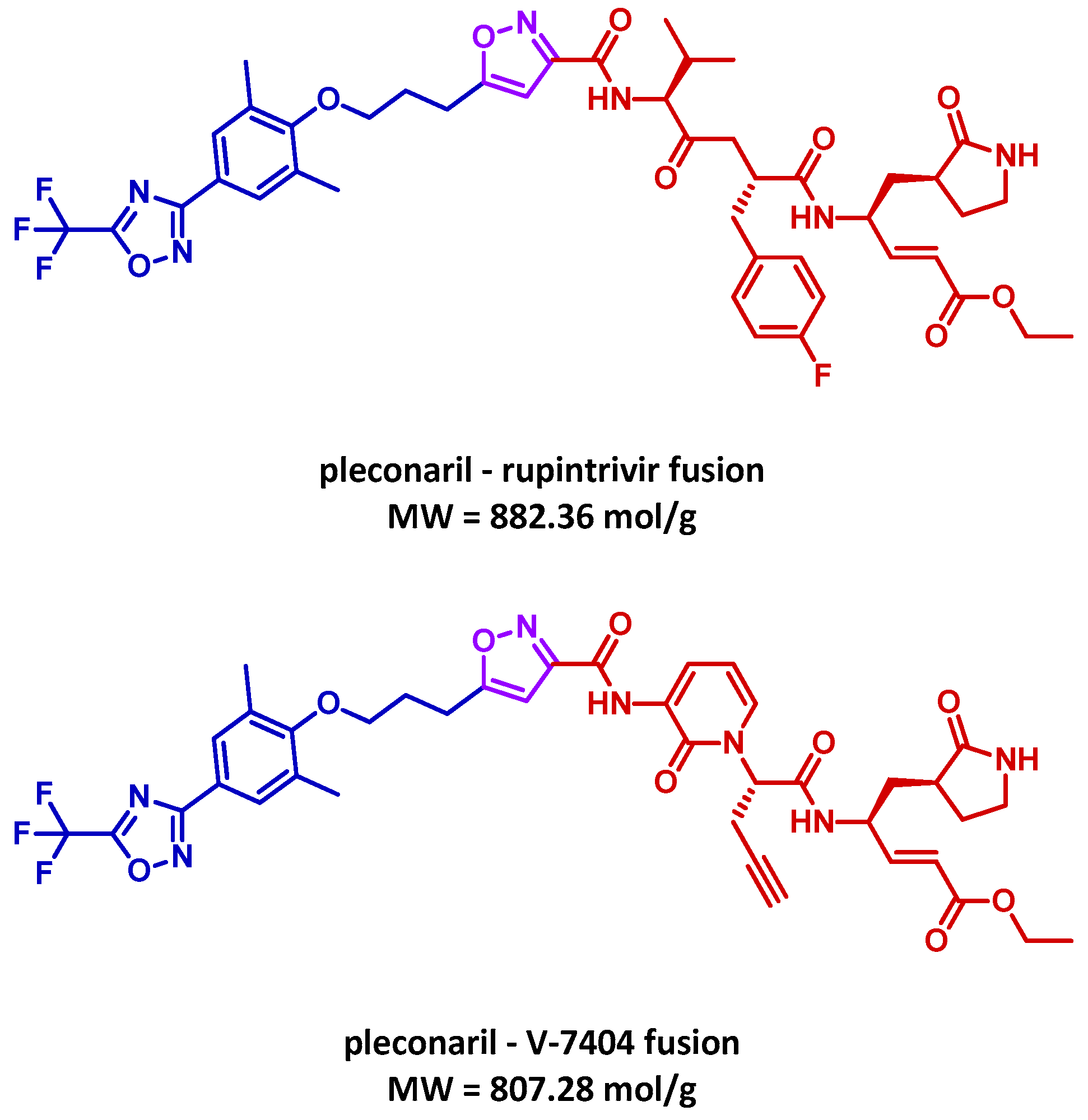
3.4. MTDL Proposal
4. Materials and Methods
4.1. Literature Survey
4.2. MTDL Candidate Proposal
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Peigue-Lafeuille, H. Picornaviridae. In Traité de Virologie Médicale, 1st ed.; Estem Edition, Ed.; SFM: Paris, France, 2003; pp. 389–405. [Google Scholar]
- Palmer, P.; Lebon, P. Entérovirus. In Les Virus Transmissibles de la Mère à L’enfant, 1st ed.; Médecine Sciences Edition, Ed.; La Librairie Médicale, Scientifique & Technique: Arcueil, France, 1999; pp. 305–318. [Google Scholar]
- Pons-Salort, M.; Parker, E.P.; Grassly, N.C. The Epidemiology of non-polio enteroviruses: Recent advances and outstanding questions. Curr. Opin. Infect. Dis. 2015, 28, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, H.; Poelman, R.; Knoester, M.; Van Leer-Buter, C.C.; Niesters, H.G.M. Enterovirus D68—The New polio? Front. Microbiol. 2018, 9, 2677. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Folkers, G.K.; Fauci, A.S. Acute flaccid myelitis: Something old and something new. mBio 2019, 10, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Zhang, Y.; Scheuermann, R.H. Epidemiology and sequence-based evolutionary analysis of circulating non-polio enteroviruses. Microorganisms 2020, 8, 1856. [Google Scholar] [CrossRef]
- Haston, J.C.; Dixon, T.C. Nonpolio enterovirus infections in neonates. Pediatr. Ann. 2015, 44, e103–e107. [Google Scholar] [CrossRef]
- Sun, L.; Tijsma, A.; Mirabelli, C.; Baggen, J.; Wahedi, M.; Franco, D.; De Palma, A.; Leyssen, P.; Verbeken, E.; van Kuppeveld, F.J.M.; et al. Intra-Host emergence of an enterovirus A71 variant with enhanced PSGL1 usage and neurovirulence. Emerg. Microbes. Infect. 2019, 8, 1076–1085. [Google Scholar] [CrossRef]
- Chong, P.; Liu, C.C.; Chow, Y.H.; Chou, A.H.; Klein, M. Review of enterovirus 71 vaccines. Clin. Infect. Dis. 2015, 60, 797–803. [Google Scholar] [CrossRef]
- Higgins, P.G. Enteroviral conjunctivitis and its neurological complications. Arch. Virol. 1982, 73, 91–101. [Google Scholar] [CrossRef]
- Lugo, D.; Krogstad, P. Enteroviruses in the early 21st century: New manifestations and challenges. Curr. Opin. Pediatr. 2016, 28, 107–113. [Google Scholar] [CrossRef]
- Disease Outbreak News (7th of June 2023) from World Health Organization Site. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON474 (accessed on 4 July 2024).
- Messacar, K.; Abzug, M.J.; Dominguez, S.R. The Emergence of enterovirus-D68. Microbiol. Spectr. 2016, 4, 105–119. [Google Scholar] [CrossRef]
- Benschop, K.S.; Albert, J.; Anton, A.; Andrés, C.; Aranzamendi, M.; Armannsdóttir, B.; Bailly, J.L.; Baldanti, F.; Baldvinsdóttir, G.E.; Beard, S.; et al. Re-Emergence of enterovirus D68 in Europe after easing the COVID-19 lockdown, September 2021. Euro. Surveill. 2021, 26, 2100998. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Yip, C.C.Y.; Yuen, K.Y. Rhinovirus—From Bench to bedside. J. Formos. Med. Assoc. 2017, 116, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Ljubin-Sternak, S.; Meštrović, T.; Ivković-Jureković, I.; Kolarić, B.; Slović, A.; Forčić, D.; Tot, T.; Mijač, M.; Vraneš, J. The Emerging role of rhinoviruses in lower respiratory tract infections in children—Clinical and molecular epidemiological study from Croatia, 2017–2019. Front. Microbiol. 2019, 10, 2737. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli, C.; Scheers, E.; Neyts, J. Novel therapeutic approaches to simultaneously target rhinovirus infection and asthma/COPD pathogenesis. F1000Research 2017, 6, 1860. [Google Scholar] [CrossRef] [PubMed]
- Grozdanov, P.; Joffret, M.L.; Stoyanova, A.; Polston, P.; Achouri, E.; Nikolova, I.; Delpeyroux, F.; Galabov, A.S. Genome analysis of coxsackievirus B1 isolates during the consecutive alternating administration course of triple antiviral combination in newborn mice. Antivir. Chem. Chemother. 2020, 28, 2040206620906061. [Google Scholar] [CrossRef]
- Lanko, K.; Shi, C.; Patil, S.; Delang, L.; Matthijnssens, J.; Mirabelli, C.; Neyts, J. Assessing in vitro resistance development in enterovirus A71 in the context of combination antiviral treatment. ACS. Infect. Dis. 2021, 7, 2801–2806. [Google Scholar] [CrossRef]
- Shyr, Z.A.; Cheng, Y.S.; Lo, D.C.; Zheng, W. Drug combination therapy for emerging viral diseases. Drug Discov. Today 2021, 26, 2367–2376. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Baggen, J.; Thibaut, H.J.; Strating, J.R.P.M.; van Kuppeveld, F.J.M. The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Microbiol. 2018, 16, 391. [Google Scholar] [CrossRef]
- Anasir, M.I.; Zarif, F.; Poh, C.L. Antivirals blocking entry of enteroviruses and therapeutic potential. J. Biomed. Sci. 2021, 28, 10. [Google Scholar] [CrossRef]
- Al-Nakib, W.; Higgins, P.G.; Barrow, G.I.; Tyrrell, D.A.; Andries, K.; Vanden Bussche, G.; Taylor, N.; Janssen, P.A. Suppression of colds in human volunteers challenged with rhinovirus by a new synthetic drug (R61837). Antimicrob. Agents Chemother. 1989, 33, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.B.; Dutko, F.J.; Goldstein, N.H.; Lockwood, G.; Hayden, F.G. Efficacy of oral WIN 54954 for prophylaxis of experimental rhinovirus infection. Antimicrob. Agents Chemother. 1993, 37, 297–300. [Google Scholar] [CrossRef]
- Hayden, F.G.; Hipskind, G.J.; Woerner, D.H.; Eisen, G.F.; Janssens, M.; Janssen, P.A.; Andries, K. Intranasal pirodavir (R77,975) treatment of rhinovirus colds. Antimicrob. Agents Chemother. 1995, 39, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Andries, K.; Janssen, P.A. Safety and efficacy of intranasal pirodavir (R77975) in experimental rhinovirus infection. Antimicrob. Agents Chemother. 1992, 36, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Schiff, G.M.; Sherwood, J.R. Clinical activity of pleconaril in an experimentally induced coxsackievirus A21 respiratory infection. J. Infect. Dis. 2000, 181, 20–26. [Google Scholar] [CrossRef]
- Rotbart, H.A.; Webster, A.D.; Pleconaril Treatment Registry Group. Treatment of potentially life-threatening enterovirus infections with pleconaril. Clin. Infect. Dis. 2001, 32, 228–235. [Google Scholar] [CrossRef]
- National Institute of Allergy and Infectious Diseases (NIAID). Pleconaril Enteroviral Sepsis Syndrome. A Double-Blind, Placebo-Controlled, Virologic Efficacy Trial of Pleconaril in the Treatment of Neonates with Enteroviral Sepsis Syndrome. National Library of Medecine, National Center for Biotechnology Information. Available online: https://clinicaltrials.gov/study/NCT00031512 (accessed on 4 July 2024).
- Bauer, S.; Gottesman, G.; Sirota, L.; Litmanovitz, I.; Ashkenazi, S.; Levi, I. Severe coxsackie virus B infection in preterm newborns treated with pleconaril. Eur. J. Pediatr. 2002, 161, 491–493. [Google Scholar] [CrossRef]
- Abzug, M.J.; Cloud, G.; Bradley, J.; Sánchez, P.J.; Romero, J.; Powell, D.; Lepow, M.; Mani, C.; Capparelli, E.V.; Blount, S.; et al. Double blind placebo-controlled trial of pleconaril in infants with enterovirus meningitis. Pediatr. Infect. Dis. J. 2003, 22, 335–341. [Google Scholar] [CrossRef]
- Pevear, D.C.; Hayden, F.G.; Demenczuk, T.M.; Barone, L.R.; McKinlay, M.A.; Collett, M.S. Relationship of pleconaril susceptibility and clinical outcomes in treatment of common colds caused by rhinoviruses. Antimicrob. Agents Chemother. 2005, 49, 4492–4499. [Google Scholar] [CrossRef]
- Desmond, R.A.; Accortt, N.A.; Talley, L.; Villano, S.A.; Soong, S.J.; Whitley, R.J. Enteroviral meningitis: Natural history and outcome of pleconaril therapy. Antimicrob. Agents Chemother. 2006, 50, 2409–2414. [Google Scholar] [CrossRef]
- Merck Sharp & Dohme LLC. Effects of Pleconaril Nasal Spray on Common Cold Symptoms and Asthma Exacerbations Following Rhinovirus Exposure (Study P04295). A Placebo-Controlled Study of the Effects of Pleconaril Nasal Spray on Common Cold Symptoms and Asthma Exacerbations Following Rhinovirus Exposure. National Library of Medecine, National Center for Biotechnology Information. Available online: https://ClinicalTrials.gov/show/NCT00394914 (accessed on 4 July 2024).
- Biota Scientific Management Pty Ltd. A Phase II, Double-Blind Placebo-Controlled Study to Determine the Prophylactic Efficacy of Oral BTA798 in an Experimental Rhinovirus Challenge Model. EU Clinical Trials Register. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2008-001714-24 (accessed on 4 July 2024).
- Biota Scientific Management Pty Ltd. A Phase 2 Study of BTA798 in Asthmatic Adults with Symptomatic Human Rhinovirus Infection (RHINO). A Phase 2 Multicenter, Randomized, Double-Blind, Placebo-Controlled Study of BTA798 in Asthmatic Adults with Symptomatic Human Rhinovirus Infection. National Library of Medecine, National Center for Biotechnology Information. Available online: https://ClinicalTrials.gov/show/NCT01175226 (accessed on 4 July 2024).
- Biota Pharmaceuticals, Inc. A Phase 2, Multicenter, Randomized, Double-Blind, Placebo-Controlled Dose-Ranging Study of Vapendavir in Moderate to Severe Asthmatic Adults with Symptomatic Human Rhinovirus Infection. EU Clinical Trials Register. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2014-001785-95 (accessed on 4 July 2024).
- Biota Pharmaceuticals, Inc. A Phase 2, Multicenter, Randomized, Double-Blind, Placebo-Controlled Dose-Ranging Study of Vapendavir in Moderate to Severe Asthmatic Adults with Symptomatic Human Rhinovirus Infection. World Health Organisation, International Clinical Trias Registery Platform. Available online: https://trialsearch.who.int/Trial2.aspx?TrialID=NCT02367313 (accessed on 4 July 2024).
- Abzug, M.J.; Michaels, M.G.; Wald, E.; Jacobs, R.F.; Romero, J.R.; Sánchez, P.J.; Wilson, G.; Krogstad, P.; Storch, G.A.; Lawrence, R.; et al. A Randomized, double-blind, placebo-controlled trial of pleconaril for the treatment of neonates with enterovirus sepsis. J. Pediatric. Infect. Dis. Soc. 2016, 5, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Vaxart, H. A Study of Vapendavir Treatment of Hematopoietic Stem Cell Transplant Subjects with Symptomatic Rhinovirus Infection. A Phase 2, Multicenter, Randomized, Double-Blind, Placebo-Controlled Study of Vapendavir Treatment of Hematopoietic Stem Cell Transplant Subjects with Symptomatic Rhinovirus Infection. National Library of Medecine, National Center for Biotechnology Information. Available online: https://ClinicalTrials.gov/show/NCT03024177 (accessed on 4 July 2024).
- Woods, M.G.; Diana, G.D.; Rogge, M.C.; Otto, M.J.; Dutko, F.J.; McKinlay, M.A. In vitro and in vivo activities of WIN 54954, a new broad-spectrum antipicornavirus drug. Antimicrob. Agents Chemother. 1989, 33, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Dewindt, B.; Snoeks, J.; Willebrords, R.; van Eemeren, K.; Stokbroekx, R.; Janssen, P.A. In vitro activity of pirodavir (R 77975), a substituted phenoxy-pyridazinamine with broad-spectrum antipicornaviral activity. Antimicrob. Agents Chemother. 1992, 36, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Pevear, D.C.; Tull, T.M.; Seipel, M.E.; Groarke, J.M. Activity of pleconaril against enteroviruses. Antimicrob. Agents Chemother. 1999, 43, 2109–2115. [Google Scholar] [CrossRef] [PubMed]
- Aradottir, E.; Alonso, E.M.; Shulman, S.T. Severe neonatal enteroviral hepatitis treated with pleconaril. Pediatr. Infect. Dis. J. 2001, 20, 457–459. [Google Scholar] [CrossRef]
- Watson, K.G.; Brown, R.N.; Cameron, R.; Chalmers, D.K.; Hamilton, S.; Jin, B.; Krippner, G.Y.; Luttick, A.; McConnell, D.B.; Reece, P.A.; et al. An orally bioavailable oxime ether capsid binder with potent activity against human rhinovirus. J. Med. Chem. 2003, 46, 3181–3184. [Google Scholar] [CrossRef]
- Altesa Biosciences, Inc. RCT of Vapendavir in Patients with COPD and Human Rhinovirus/Enterovirus Upper Respiratory Infection. A Trial in Participants with Chronic Obstructive Pulmonary Disease (COPD) to Evaluate the Impact of Vapendavir on the Development of Lower Respiratory Tract Symptoms Following Rhinovirus Challenge. National Library of Medecine, National Center for Biotechnology Information. 2017. Available online: https://ClinicalTrials.gov/show/NCT06149494 (accessed on 4 July 2024).
- Phillpotts, R.J.; Jones, R.W.; Delong, D.C.; Reed, S.E.; Wallace, J.; Tyrrell, D.A. The activity of enviroxime against rhinovirus infection in man. Lancet 1981, 1, 1342–1344. [Google Scholar] [CrossRef]
- Hayden, F.G.; Gwaltney, J.M., Jr. Prophylactic activity of intranasal enviroxime against experimentally induced rhinovirus type 39 infection. Antimicrob. Agents Chemother. 1982, 21, 892–897. [Google Scholar] [CrossRef]
- Miller, F.D.; Monto, A.S.; DeLong, D.C.; Exelby, A.; Bryan, E.R.; Srivastava, S. Controlled trial of enviroxime against natural rhinovirus infections in a community. Antimicrob. Agents Chemother. 1985, 27, 102–106. [Google Scholar] [CrossRef]
- Levandowski, R.A.; Pachucki, C.T.; Rubenis, M.; Jackson, G.G. Topical enviroxime against rhinovirus infection. Antimicrob. Agents Chemother. 1982, 22, 1004–1007. [Google Scholar] [CrossRef]
- Phillpotts, R.J.; Wallace, J.; Tyrrell, D.A.; Tagart, V.B. Therapeutic activity of enviroxime against rhinovirus infection in volunteers. Antimicrob. Agents Chemother. 1983, 23, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Hsyu, P.H.; Pithavala, Y.K.; Gersten, M.; Penning, C.A.; Kerr, B.M. Pharmacokinetics and safety of an antirhinoviral agent, ruprintrivir, in healthy volunteers. Antimicrob. Agents Chemother. 2002, 46, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Turner, R.B.; Gwaltney, J.M.; Chi-Burris, K.; Gersten, M.; Hsyu, P.; Patick, A.K.; Smith, G.J., 3rd; Zalman, L.S. Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob. Agents Chemother. 2003, 47, 3907–3916. [Google Scholar] [CrossRef] [PubMed]
- Patick, A.K.; Brothers, M.A.; Maldonado, F.; Binford, S.; Maldonado, O.; Fuhrman, S.; Petersen, A.; Smith, G.J., 3rd; Zalman, L.S.; Burns-Naas, L.A.; et al. In vitro antiviral activity and single-dose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 2005, 49, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Kankam, M.K.; Burns, J.M.; Collett, M.S.; Corrado, M.L.; Hincks, J.R. A Phase 1 study of the safety, tolerability, and pharmacokinetics of single and multiple oral doses of V-7404 in healthy adult volunteers. Antimicrob. Agents Chemother. 2021, 65, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Lei, X.; Zhang, Z.; Ma, Y.; Qi, J.; Wu, C.; Xiao, Y.; Li, L.; He, B.; Wang, J. Enterovirus 3A facilitates viral replication by promoting phosphatidylinositol 4-kinase IIIβ-ACBD3 Interaction. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J. Virol. 2011, 85, 2364–2372. [Google Scholar] [CrossRef]
- Qin, Y.; Lin, L.; Chen, Y.; Wu, S.; Si, X.; Wu, H.; Zhai, X.; Wang, Y.; Tong, L.; Pan, B.; et al. Curcumin inhibits the replication of enterovirus 71 in vitro. Acta. Pharm. Sin. B 2014, 4, 284–294. [Google Scholar] [CrossRef]
- Lu, G.; Qi, J.; Chen, Z.; Xu, X.; Gao, F.; Lin, D.; Qian, W.; Liu, H.; Jiang, H.; Yan, J. Enterovirus 71 and Coxsackievirus A16 3C Proteases: Binding to Rupintrivir and Their Substrates and Anti-Hand, Foot, and Mouth Disease Virus Drug Design. J. Virol. 2011, 85, 10319–10331. [Google Scholar] [CrossRef]
- Tang, F.; Xia, H.; Wang, P.; Yang, J.; Zhao, T.; Zhang, Q.; Hu, Y.; Zhou, X. The identification and characterization of nucleic acid chaperone activity of human enterovirus 71 nonstructural protein 3AB. Virology 2014, 464–465, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Enviroxime PubChem Page. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/enviroxime (accessed on 4 July 2024).
- Sun, L.; Meijer, A.; Froeyen, M.; Zhang, L.; Thibaut, H.J.; Baggen, J.; George, S.; Vernachio, J.; van Kuppeveld, F.J.M.; Leyssen, P.; et al. Antiviral activity of broad-spectrum and enterovirus-specific inhibitors against clinical isolates of enterovirus D68. Antimicrob. Agents Chemother. 2015, 59, 7782–7785. [Google Scholar] [CrossRef] [PubMed]
- Chau, D.H.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2007, 12, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Amineva, S.P.; Aminev, A.G.; Palmenberg, A.C.; Gern, J.E. Rhinovirus 3C protease precursors 3CD and 3CD’ localize to the nuclei of infected cells. J. Gen. Virol. 2004, 85 Pt 10, 2969–2979. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Prins, T.J.; Zhou, R.; Webber, S.E.; Marakovits, J.T.; Fuhrman, S.A.; Patick, A.K.; Matthews, D.A.; Lee, C.A.; Ford, C.E.; et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as L-glutamine replacements. J. Med. Chem. 1999, 42, 1213–1224. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Prins, T.J.; Zhou, R.; Johnson, T.O.; Hua, Y.; Luu, H.T.; Sakata, S.K.; Brown, E.L.; Maldonado, F.C.; Tuntland, T.; et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 8. Pharmacological optimization of orally bioavailable 2-pyridone-containing peptidomimetics. J. Med. Chem. 2003, 46, 4572–4585. [Google Scholar] [CrossRef]
- Gaffey, M.J.; Gwaltney, J.M., Jr.; Dressler, W.E.; Sorrentino, J.V.; Hayden, F.G. Intranasally administered atropine methonitrate treatment of experimental rhinovirus colds. Am. Rev. Respir. Dis. 1987, 135, 241–244. [Google Scholar]
- Curovir, A.B. A Randomised, Double-Blind, Single-Centre, Placebo-Controlled, First-in-Human Clinical Trial Evaluating the Safety, Tolerability and Pharmacokinetics of Single and Multiple Ascending Doses of CUR-N399 in Healthy Volunteers. National Library of Medecine, National Center for Biotechnology Information. Available online: https://ClinicalTrials.gov/show/NCT05016687 (accessed on 4 July 2024).
- Winther, B.; Buchert, D.; Turner, R.B.; Hendley, J.O.; Tschaikin, M. Decreased rhinovirus shedding after intranasal oxymetazoline application in adults with induced colds compared with intranasal saline. Am. J. Rhinol. Allergy 2010, 24, 374–377. [Google Scholar] [CrossRef]
- Carlson, A.B.; Kraus, G.P. Physiology, Cholinergic Receptors. In StatPearls, 1st ed.; StatPearls Publishing: Saint Petersburg, FL, USA, 2024. [Google Scholar]
- Pierce, R.J.; Allen, C.J.; Campbell, A.H. A comparative study of atropine methonitrate, salbutamol, and their combination in airways obstruction. Thorax 1979, 34, 45–50. [Google Scholar] [CrossRef]
- Melia, C.E.; van der Schaar, H.M.; Lyoo, H.; Limpens, R.W.A.L.; Feng, Q.; Wahedi, M.; Overheul, G.J.; van Rij, R.P.; Snijder, E.J.; Koster, A.J.; et al. Escaping Host Factor PI4KB Inhibition: Enterovirus Genomic RNA Replication in the Absence of Replication Organelles. Cell Rep. 2017, 21, 587–599. [Google Scholar] [CrossRef]
- Cheong, D.H.J.; Yogarajah, T.; Wong, Y.H.; Arbrandt, G.; Westman, J.; Chu, J.J.H. CUR-N399, a PI4KB inhibitor, for the treatment of Enterovirus A71 infection. Antivir. Res. 2023, 218, 105713. [Google Scholar] [CrossRef] [PubMed]
- Biazi, G.R.; Frasson, I.G.; Miksza, D.R.; de Morais, H.; de Fatima Silva, F.; Bertolini, G.L.; de Souza, H.M. Decreased hepatic response to glucagon, adrenergic agonists, and cAMP in glycogenolysis, gluconeogenesis, and glycolysis in tumor-bearing rats. J. Cell Biochem. 2018, 119, 7300–7309. [Google Scholar] [CrossRef] [PubMed]
- Durkee, C.A.; Covelo, A.; Lines, J.; Kofuji, P.; Aguilar, J.; Araque, A. Gi/o protein-coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia 2019, 67, 1076–1093. [Google Scholar] [CrossRef]
- Haenisch, B.; Walstab, J.; Herberhold, S.; Bootz, F.; Tschaikin, M.; Ramseger, R.; Bönisch, H. Alpha-adrenoceptor agonistic activity of oxymetazoline and xylometazoline. Fundam. Clin. Pharmacol. 2010, 24, 729–739. [Google Scholar] [CrossRef]
- Reed, S.E.; Craig, J.W.; Tyrrell, D.A. Four compounds active against rhinovirus: Comparison in vitro and in volunteers. J. Infect. Dis. 1976, 133 (Suppl. S2), A128–A1235. [Google Scholar] [CrossRef] [PubMed]
- Phillpotts, R.J.; Higgins, P.G.; Willman, J.S.; Tyrrell, D.A.; Lenox-Smith, I. Evaluation of the antirhinovirus chalcone Ro 09-0415 given orally to volunteers. J. Antimicrob. Chemother. 1984, 14, 403–409. [Google Scholar] [CrossRef]
- Zerial, A.; Werner, G.H.; Phillpotts, R.J.; Willmann, J.S.; Higgins, P.G.; Tyrrell, D.A. Studies on 44 081 R.P., a new antirhinovirus compound, in cell cultures and in volunteers. Antimicrob. Agents Chemother. 1985, 27, 846–850. [Google Scholar] [CrossRef]
- Al-Nakib, W.; Higgins, P.G.; Barrow, I.; Tyrrell, D.A.; Lenox-Smith, I.; Ishitsuka, H. Intranasal chalcone, Ro 09-0410, as prophylaxis against rhinovirus infection in human volunteers. J. Antimicrob. Chemother. 1987, 20, 887–892. [Google Scholar] [CrossRef]
- Yasin, S.R.; Al-Nakib, W.; Tyrrell, D.A. Pathogenicity for humans of human rhinovirus type 2 mutants resistant to or dependent on chalcone Ro 09-0410. Antimicrob. Agents Chemother. 1990, 34, 963–966. [Google Scholar] [CrossRef]
- GlaxoSmithKline Research & Development Limited. A Randomised, Double-Blind, Placebo Controlled, Repeat Dose Phase 1b Study to Assess the Efficacy, Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Inhaled GSK3923868 during Experimental Human Rhinovirus Infection in Participants with Mild Asthma. World Health Organisation, International Clinical Trias Registery Platform. Available online: https://trialsearch.who.int/Trial2.aspx?TrialID=ISRCTN15115094 (accessed on 4 July 2024).
- GlaxoSmithKline. A Randomized, Double-Blind, Placebo Controlled, Repeat Dose Phase 1b Study to Assess the Efficacy, Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Inhaled GSK3923868 during Experimental Human Rhinovirus Infection in Participants with Mild Asthma. World Health Organisation, International Clinical Trias Registery Platform. Available online: https://trialsearch.who.int/Trial2.aspx?TrialID=NCT05398198 (accessed on 4 July 2024).
- Ishitsuka, H.; Ninomiya, Y.T.; Ohsawa, C.; Fujiu, M.; Suhara, Y. Direct and specific inactivation of rhinovirus by chalcone Ro 09-0410. Antimicrob. Agents Chemother. 1982, 22, 617–621. [Google Scholar] [CrossRef]
- Soto, A.J.; Hall, T.S.; Reed, S.E. Trial of the antiviral action of isoprinosine against rhinovirus infection of volunteers. Antimicrob. Agents Chemother. 1973, 3, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Pachuta, D.M.; Togo, Y.; Hornick, R.B.; Schwartz, A.R.; Tominaga, S. Evaluation of isoprinosine in experimental human rhinovirus infection. Antimicrob. Agents Chemother. 1974, 5, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Waldman, R.H.; Gangulyj, R. Therapeutic efficacy of inosiplex (Isoprinosine) in rhinovirus infection. Ann. N. Y. Acad. Sci. 1977, 284, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Romark Laboratories, L.C. A Phase III, Randomized, Double-Blind, Placebo Controlled Trial to Evaluate the Efficacy and Safety of Nitazoxanide in the Treatment of Colds Due to Enterovirus/Rhinovirus Infection. National Library of Medecine, National Center for Biotechnology Information. Available online: https://ClinicalTrials.gov/show/NCT03605862 (accessed on 4 July 2024).
- Romark Laboratories, L.C. Phase 3, A Randomized, Double-Blind, Placebo-Controlled Trial to Evaluate Efficacy and Safety of Nitazoxanide in the Treatment of Colds Due to Enterovirus/Rhinovirus Infection. National Library of Medecine, National Center for Biotechnology Information. Available online: https://clinicaltrials.gov/study/NCT04489381 (accessed on 4 July 2024).
- Reed, S.E.; Bynoe, M.L. The antiviral activity of isoquinoline drugs for rhinoviruses in vitro and in vivo. J. Med. Microbiol. 1970, 3, 346–352. [Google Scholar] [CrossRef]
- Togo, Y.; Schwartz, A.R.; Hornick, R.B. Antiviral effect of 3, 4-dihydro-1-isoquinolineacetamide hydrochloride in experimental human rhinovirus infection. Antimicrob. Agents Chemother. 1973, 4, 612–616. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Esper, F.; Buchheit, K.; Arters, K.; Adkins, I.; Ghannoum, M.A.; Salata, R.A. Randomized, double-blind, placebo-controlled clinical trial to assess the safety and effectiveness of a novel dual-action oral topical formulation against upper respiratory infections. BMC Infect. Dis. 2017, 17, 74. [Google Scholar] [CrossRef]
- Chang, T.W.; Weinstein, L. Antiviral activity of isoprinosine in vitro and in vivo. Am. J. Med. Sci. 1973, 265, 143–146. [Google Scholar] [CrossRef]
- Chang, T.W.; Heel, R.C. Ribavirin and inosiplex: A review of their present status in viral diseases. Drugs 1981, 22, 111–128. [Google Scholar] [CrossRef]
- Cetylpyridinuium Chloride Page in Adis Insight Website. Available online: https://adisinsight.springer.com/drugs/800048807 (accessed on 4 July 2024).
- Barrow, G.I.; Higgins, P.G.; Al-Nakib, W.; Smith, A.P.; Wenham, R.B.; Tyrrell, D.A. The effect of intranasal nedocromil sodium on viral upper respiratory tract infections in human volunteers. Clin. Exp. Allergy 1990, 20, 45–51. [Google Scholar] [CrossRef]
- Gazdik, F.; Jahnova, E.; Horvathova, M.; Pijak, M.R.; Gazdikova, K. Antiasthmatic effects of nedocromil sodium. Bratisl. Lek. Listy 2003, 104, 222–226. [Google Scholar]
- Abisheganaden, J.A.; Avila, P.C.; Kishiyama, J.L.; Liu, J.; Yagi, S.; Schnurr, D.; Boushey, H.A. Effect of clarithromycin on experimental rhinovirus-16 colds: A randomized, double-blind, controlled trial. Am. J. Med. 2000, 108, 453–459. [Google Scholar] [CrossRef]
- Clarithromycin page in United Kingdom National Health Service. Available online: https://www.nhs.uk/medicines/clarithromycin/about-clarithromycin/ (accessed on 4 July 2024).
- Hao, W.; Bernard, K.; Patel, N.; Ulbrandt, N.; Feng, H.; Svabek, C.; Wilson, S.; Stracener, C.; Wang, K.; Suzich, J.; et al. Infection and propagation of human rhinovirus C in human airway epithelial cells. J. Virol. 2012, 86, 13524–13532. [Google Scholar] [CrossRef] [PubMed]
- Basta, H.A.; Sgro, J.Y.; Palmenberg, A.C. Modeling of the human rhinovirus C capsid suggests a novel topography with insights on receptor preference and immunogenicity. Virology 2014, 448, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, Y.; Zheng, M. Enterovirus A71 Antivirals: Past, Present, and Future. Acta. Pharm. Sin. B 2022, 12, 1542–1566. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Zhu, F.; Ma, X.; Cao, Z.W.; Li, Y.X.; Chen, Y.Z. Mechanisms of drug combinations: Interaction and network perspectives. Nat. Rev. Drug. Discov. 2009, 8, 11–128. [Google Scholar] [CrossRef]
- Van Waardenburg, R.C.; de Jong, L.A.; van Eijndhoven, M.A.; Verseyden, C.; Pluim, D.; Jansen, L.E.; Bjornsti, M.-A.; Schellens, J.H.M. Platinated DNA adducts enhance poisoning of DNA topoisomerase I by camptothecin. J. Biol. Chem. 2004, 29, 54502–54509. [Google Scholar] [CrossRef]
- Lai, G.H.; Zhang, Z.; Sirica, A.E. Celecoxib acts in a cyloooxygenase-2-independent manner and in synergy with emodin to suppress rat cholangiocarcinoma growth in vitro through a mechanism involving enhanced akt inactivation and increased activation of caspase-9 and -3. Mol. Cancer. Ther. 2003, 2, 262–271. [Google Scholar]
- Cottagnoud, P.; Cottagnoud, M.; Täuber, M.G. Vancomycin acts systematically with gentamicin against penicillin-resistant pneumococci by increasing the intracellular penetration of gentamicin. Antimicrob. Agents Chemother. 2003, 47, 144–147. [Google Scholar] [CrossRef]
- Ma, Y.; Abdelnabi, R.; Delang, L.; Froeyen, M.; Luyten, W.; Neyts, J.; Mirabelli, C. New class of early-stage enterovirus inhibitors with a novel mechanism of action. Antivir. Res. 2017, 147, 67–74. [Google Scholar] [CrossRef]
- Ianevski, A.; Zusinaite, E.; Tenson, T.; Oksenych, V.; Wang, W.; Afset, J.E.; Bjørås, M.; Kainov, D.E. Novel synergistic anti-enteroviral drug combinations. Viruses 2022, 14, 1866. [Google Scholar] [CrossRef]
- Stoyanova, A.; Galabov, A.S. Effects of double combinations of enterovirus replication inhibitors against coxsackie B viruses. Acta. Virol. 2021, 65, 411–419. [Google Scholar] [CrossRef]
- Nikolaeva-Glomb, L.; Galabov, A.S. Synergistic combinations against the replication in vitro of coxsackievirus B1. Antivir. Res. 2004, 62, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lonberg-Holm, K.; Gosser, L.B.; Kauer, J.C. Early alteration of poliovirus in infected cells and its specific inhibition. J. Gen. Virol. 1975, 27, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.C.; Wang, H.C.; Shih, S.R.; Teng, I.F.; Tseng, C.P.; Hsu, J.T. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J. Infect. Dis. 2011, 203, 1784–1790. [Google Scholar] [CrossRef] [PubMed]
- Rhoden, E.; Liu, H.M.; Wang-Chern, S.W.; Oberste, M.S. Anti-poliovirus potency of protease inhibitor AG-7404, and assessment of in vitro potency in combination with antiviral capsid inhibitor compounds. Antiviral. Res. 2013, 98, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, G.; Yuan, S.; Gao, Q.; Lan, K.; Altmeyer, R.; Zou, G. In Vitro assessment of combinations of enterovirus inhibitors against enterovirus 71. Antimicrob. Agents Chemother. 2016, 60, 5357–5367. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.W.; Yam, W.K.; Kooi, R.J.W.; Westman, J.; Arbrandt, G.; Chu, J.J.H. Novel capsid binder and PI4KIIIbeta inhibitors for EV-A71 replication inhibition. Sci. Rep. 2021, 11, 9719. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Jie, Q.; Shaw, N.; Li, L.; Rao, Z.; Yin, Z.; Lou, Z. Studies on inhibition of proliferation of enterovirus-71 by compound YZ-LY-0. Int. J. Biol. Sci. 2015, 11, 1337–1347. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, B.; Zhai, Y.; Sun, Y.; Rao, Z. Peptidyl aldehyde NK-1.8k suppresses enterovirus 71 and enterovirus 68 infection by targeting protease 3C. Antimicrob. Agents Chemother. 2015, 59, 2636–2646. [Google Scholar] [CrossRef]
- Kang, H.; Kim, C.; Kim, D.E.; Song, J.H.; Choi, M.; Choi, K.; Kang, M.; Lee, K.; Kim, H.S.; Shin, J.S.; et al. Synergistic antiviral activity of gemcitabine and ribavirin against enteroviruses. Antivir. Res. 2015, 124, 6. [Google Scholar] [CrossRef]
- Vassileva-Pencheva, R.; Galabov, A.S. Avoiding drug-resistance development by novel approach of combining anti-enteroviral substances against coxsackievirus B1 infection in mice. Antivir. Res. 2010, 85, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Vassileva-Pencheva, R.; Galabov, A.S. Effectiveness of the consecutive alternating administration course of a triple antiviral combination in coxsackievirus B3 infections in mice. Drug. Res. 2016, 66, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, A.; Nikolova, I.; Pürstinger, G.; Dobrikov, G.; Dimitrov, V.; Philipov, S.; Galabov, A.S. Anti-enteroviral triple combination of viral replication inhibitors: Activity against coxsackievirus B1 neuroinfection in mice. Antivir. Chem. Chemother. 2015, 24, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Galabov, A.S.; Nikolova, I.; Vassileva-Pencheva, R.; Stoyanova, A. Antiviral Combination Approach as a Perspective to Combat Enterovirus Infections. Prilozi (Makedon. Akad. Nauk. Umet. Odd. Med. Nauki.) 2015, 36, 91–99. [Google Scholar] [CrossRef]
- Stoyanova, A.; Nikolova, I.; Galabov, A.S. Effect of consecutive altenarting administration (CAA) of triple anti-enteroviral combination on coxsackievirus B1 neuroinfection in mice. Antivir. Res. 2015, 121, 138–144. [Google Scholar] [CrossRef]
- Stoyanova, A.; Galabov, A.S. Effect of consecutive alternating administration of a triple combination of anti-enteroviral compounds in mice infected with coxsackievirus B3. Pathog. Dis. 2020, 78, ftaa065. [Google Scholar] [CrossRef]
- Stoyanova, A.; Galabov, A.S. Consecutive alternating administration as an effective anti-coxsackievirus B3 in vivo treatment scheme. Arch. Virol. 2021, 166, 1869–1875. [Google Scholar] [CrossRef]
- Nikolaeva, L.; Galabov, A.S. Antiviral effect of combination of enviroxime and disoxaril on coxsackievirus B1 Infection. Acta. Virol. 2000, 44, 73–78. [Google Scholar]
- Ianevski, A.; Frøysa, I.T.; Lysvand, H.; Calitz, C.; Smura, T.; Schjelderup Nilsen, H.J.; Høyer, E.; Afset, J.E.; Sridhar, A.; Wolthers, K.C.; et al. The combination of pleconaril, rupintrivir, and remdesivir efficiently inhibits enterovirus infections in vitro, delaying the development of drug-resistant virus variants. Antivir. Res. 2024, 224, 105842. [Google Scholar] [CrossRef]
- Hayden, F.G.; Herrington, D.T.; Coats, T.L.; Kim, K.; Cooper, E.C.; Villano, S.A.; Liu, S.; Hudson, S.; Pevear, D.C.; Collett, M.; et al. Efficacy and safety of oral pleconaril for treatment of colds due to picornaviruses in adults: Results of 2 double-blind, randomized, placebo-controlled trials. Clin. Infect. Dis. 2003, 36, 1523–1532. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Baggen, J.; Mirabelli, C.; Neyts, J. Antiviral strategies against (non-polio) picornaviruses. In New Drug Development for Known and Emerging Viruses, 1st ed.; Wiley-VCH GmbH.: Hoboken, NJ, USA, 2022. [Google Scholar]
- Chang, Y.K.; Chen, K.H.; Chen, K.T. Hand, foot and mouth disease and herpangina caused by enterovirus A71 infections: A Review of enterovirus A71 molecular epidemiology, pathogenesis, and current vaccine development. Rev. Inst. Med. Trop. Sao Paulo. 2018, 60, e70. [Google Scholar] [CrossRef] [PubMed]
- Holm-Hansen, C.C.; Midgley, S.E.; Fischer, T.K. Global emergence of enterovirus D68: A Systematic review. Lancet Infect. Dis. 2016, 16, e64–e75. [Google Scholar] [CrossRef] [PubMed]
- Royston, L.; Tapparel, C. Rhinoviruses and respiratory enteroviruses: Not as simple as ABC. Viruses 2016, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Egorova, A.; Ekins, S.; Schmidtke, M.; Makarov, V. Back to the future: Advances in development of broad-spectrum capsid-binding inhibitors of enteroviruses. Eur. J. Med. Chem. 2019, 178, 606–622. [Google Scholar] [CrossRef] [PubMed]
- Limpens, R.W.; van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; van Kuppeveld, F.J.; Bárcena, M. The Transformation of enterovirus replication structures: A Three-dimensional study of single- and double-membrane compartments. mBio 2011, 2, 10–1128. [Google Scholar] [CrossRef]
- Wolff, G.; Melia, C.E.; Snijder, E.J.; Bárcena, M. Double-membrane vesicles as platforms for viral replication. Trends Microbiol. 2020, 28, 1022–1033. [Google Scholar] [CrossRef]
- Lennemann, N.J.; Evans, A.S.; Coyne, C.B. Imaging-based reporter systems to define CV-B-induced membrane remodeling in living cells. Viruses 2020, 12, 1074. [Google Scholar] [CrossRef]
- Belov, G.A.; Sztul, E. Rewiring of cellular membrane homeostasis by picornaviruses. J. Virol. 2014, 88, 9478–9489. [Google Scholar] [CrossRef]
- Ledford, R.M.; Patel, N.R.; Demenczuk, T.M.; Watanyar, A.; Herbetz, T.; Collet, M.S.; Pevear, D.C. VP1 Sequencing of all human rhinovirus serotypes: Insights into genus phylogeny and susceptibility to antiviral capsid-binding compounds. J. Virol. 2004, 78, 3663–3674. [Google Scholar] [CrossRef]
- Huang, Y.L.; Lin, T.M.; Wang, S.Y.; Wang, J.R. The Role of conserved arginine and proline residues in enterovirus VP1 protein. J. Microbiol. Immunol. Infect. 2022, 55, 590–597. [Google Scholar] [CrossRef]
- Oberste, M.S.; Peñaranda, S.; Maher, K.; Pallansch, M.A. Complete genome sequences of all members of the species human enterovirus A. J. Gen. Virol. 2004, 85 Pt 6, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.C.; Hamilton, S.; Krippner, G.Y.; Lin, B.; Luttick, A.; McConnell, D.B.; Nearn, R.; Parker, M.W.; Ryan, J.; Stanislawski, P.C.; et al. An Orally available 3-ethoxybenzisoxazole capsid binder with clinical activity against human rhinovirus. ACS Med. Chem. Lett. 2012, 3, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.L.; Yeo, H.; Ye, H.Q.; Liu, S.Q.; Shang, B.D.; Gong, P.; Alonso, S.; Shi, P.Y.; Zhang, B. Inhibition of enterovirus 71 by adenosine analog NITD008. J. Virol. 2014, 88, 11915–11923. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cao, L.; Zhai, Y.; Ma, J.; Nie, Q.; Li, T.; Yin, Z.; Sun, Y.; Shang, L. Inhibition of enterovirus 71 replication by an α-hydroxy-nitrile derivative NK-1.9k. Antivir. Res. 2017, 141, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Wang, K.; Zhao, K.; Hua, S.C.; Du, J. The Structure, function, and mechanisms of action of enterovirus non-structural protein 2C. Front. Microbiol. 2020, 11, 615965. [Google Scholar] [CrossRef]
- Visser, L.J.; Langereis, M.A.; Rabouw, H.H.; Wahedi, M.; Muntjewerff, E.M.; de Groot, R.J.; van Kuppeveld, F.J.M. Essential role of enterovirus 2A protease in counteracting stress granule formation and the induction of type I interferon. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef]
- Li, P.; Wu, S.; Xiao, T.; Li, Y.; Su, Z.; Wei, W.; Hao, F.; Hu, G.; Lin, F.; Chen, X.; et al. Design, synthesis, and evaluation of a novel macrocyclic anti-EV71 agent. Bioorg. Med. Chem. 2020, 28, 115551. [Google Scholar] [CrossRef]
- Deutschmann-Olek, K.M.; Yue, W.W.; Bezerra, G.A.; Skern, T. Defining substrate selection by rhinoviral 2A proteinase through its crystal structure with the inhibitor zVAM.fmk. Virology 2021, 562, 128–141. [Google Scholar] [CrossRef]
- El Kazzi, P.; Yaacoub, C.; Fajloun, Z.; Vanelle, P.; Decoly, E.; Coutard, B.; Barral, K. 2C Protein of enterovirus: Key protein of viral replication and antiviral target. Virologie 2023, 27, 35–49. [Google Scholar] [CrossRef]
- Doppalapudi, S.; Jain, A.; Khan, W.; Domb, A.J. Biodegradable Polymers—An Overview. Polym. Adv. Technol. 2014, 25, 427–435. [Google Scholar] [CrossRef]
- Chang, G.H.; Luo, Y.J.; Wu, X.Y.; Si, B.Y.; Lin, L.; Zhu, Q.Y. Monoclonal antibody induced with inactived EV71-Hn2 virus protects mice against lethal EV71-Hn2 virus infection. Virol. J. 2010, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, C.; Dai, W.; Wang, Y.; Liu, Z.; Zhang, X.; Wang, X.; Wang, H.; Gong, S.; Cong, Y.; et al. Functional and structural characterization of a two-MAb cocktail for delayed treatment of enterovirus D68 infections. Nat. Commun. 2021, 12, 2904. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, A.M.; Cleator, G.C.; Ras, A. External quality assessment of enterovirus detection and typing. European Union Concerted Action on Virus Meningitis and Encephalitis. Bull. World Health Organ. 1999, 77, 217–223. [Google Scholar] [PubMed]
- Tan, C.W.; Chan, Y.F.; Sim, K.M.; Tan, E.L.; Poh, C.L. Inhibition of enterovirus 71 (EV-71) infections by a novel antiviral peptide derived from EV-71 capsid protein VP1. PLoS ONE 2012, 7, e34589. [Google Scholar] [CrossRef] [PubMed]
- Pourianfar, H.R.; Poh, C.L.; Fecondo, J.; Grollo, L. In vitro evaluation of the antiviral activity of heparan sulfate mimetic compounds against Enterovirus 71. Virus Res. 2012, 169, 22–29. [Google Scholar] [CrossRef]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational design of multitarget-directed ligands: Strategies and emerging paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Liu, F.; Li, S.; Shi, D. Rational multitargeted drug design strategy from the perspective of a medecinal chemist. J. Med. Chem. 2021, 64, 10581–10605. [Google Scholar] [CrossRef]
- Chaves, S.; Resta, S.; Santos, M.A. Design, synthesis, and in vitro evaluation of hydroxybenzimidazole-donepezil analogues as multitarget-directed ligands for the treatment of Alzheimer’s disease. Molecules 2020, 25, 985. [Google Scholar] [CrossRef]
- Abdolmaleki, A.; Ghasemi, J.B. Dual-acting of hybrid compounds—A New dawn in the discovery of multi-target drugs: Lead generation approaches. Curr. Top. Med. Chem. 2017, 17, 1096–1114. [Google Scholar] [CrossRef]
- Csermely, P.; Agoston, V.; Pongor, S. The Efficacy of multi-target drugs: The Network approach might help drug design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef]
- Puls, L.N.; Eadens, M.; Messersmith, W. Current status of SRC inhibitors in solid tumor malignancies. Oncologist 2011, 16, 566–578. [Google Scholar] [CrossRef]
- Boran, A.D.; Iyengar, R. Systems approaches to polypharmacology and drug discovery. Curr. Opin. Drug. Discov. Devel. 2010, 13, 297–309. [Google Scholar] [PubMed]
- Ikram, N.; Mirza, M.U.; Vanmeert, M.; Froeyen, M.; Salo-Ahen, O.M.H.; Tahir, M.; Qazi, A.; Ahmad, S. Inhibition of oncogenic kinases: An in vitro validated computational approach identified potential multi-target anticancer compounds. Biomolecules 2019, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Gupta, S.; Chand Yadav, T.; Pruthi, V.; Kumar Varadwaj, P.; Goel, N. Extrapolation of phenolic compounds as multi-target agents against cancer and inflammation. J. Biomol. Struct. Dyn. 2019, 37, 2355–2369. [Google Scholar] [CrossRef] [PubMed]
- Sunil, D.; Kamath, P.R. Multi-target directed indole based hybrid molecules in cancer therapy: An Up-to-date evidence-based review. Curr. Top. Med. Chem. 2017, 17, 959–985. [Google Scholar] [CrossRef] [PubMed]
- Sbaraglini, M.L.; Talevi, A. Hybrid compounds as anti-infective agents. Curr. Top. Med. Chem. 2017, 17, 1080–1095. [Google Scholar] [CrossRef]
- Naik, B.; Gupta, N.; Ojha, R.; Singh, S.; Prajapati, V.K.; Prusty, D. High throughput virtual screening reveals SARS-CoV-2 multi-target binding natural compounds to lead instant therapy for COVID-19 treatment. Int. J. Biol. Macromol. 2020, 160, 1–17. [Google Scholar] [CrossRef]
- Tassini, S.; Sun, L.; Lanko, K.; Crespan, E.; Langron, E.; Falchi, F.; Kissova, M.; Armijos-Rivera, J.I.; Delang, L.; Mirabelli, C.; et al. Discovery of multi-target agents active as broad-spectrum antivirals and correctors of cystic fibrosis transmembrane conductance regulator (CFTR) for associated pulmonary diseases. J. Med. Chem. 2017, 60, 1400–1416. [Google Scholar] [CrossRef]
- Ambure, P.; Bhat, J.; Puzyn, T.; Roy, K. Identifying natural compounds as multi-target-directed ligands against Alzheimer’s disease: An in Silico approach. J. Biomol. Struct. Dyn. 2019, 37, 1282–1306. [Google Scholar] [CrossRef]
- Scotti, L.; Mendonca Junior, F.J.; Ishiki, H.M.; Ribeiro, F.F.; Singla, R.K.; Barbosa Filho, J.M.; Da Silva, M.S.; Scotti, M.T. Docking studies for multi-target drugs. Curr. Drug. Targets 2017, 18, 592–604. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roux, H.; Touret, F.; Rathelot, P.; Vanelle, P.; Roche, M. From the “One-Molecule, One-Target, One-Disease” Concept towards Looking for Multi-Target Therapeutics for Treating Non-Polio Enterovirus (NPEV) Infections. Pharmaceuticals 2024, 17, 1218. https://doi.org/10.3390/ph17091218
Roux H, Touret F, Rathelot P, Vanelle P, Roche M. From the “One-Molecule, One-Target, One-Disease” Concept towards Looking for Multi-Target Therapeutics for Treating Non-Polio Enterovirus (NPEV) Infections. Pharmaceuticals. 2024; 17(9):1218. https://doi.org/10.3390/ph17091218
Chicago/Turabian StyleRoux, Hugo, Franck Touret, Pascal Rathelot, Patrice Vanelle, and Manon Roche. 2024. "From the “One-Molecule, One-Target, One-Disease” Concept towards Looking for Multi-Target Therapeutics for Treating Non-Polio Enterovirus (NPEV) Infections" Pharmaceuticals 17, no. 9: 1218. https://doi.org/10.3390/ph17091218
APA StyleRoux, H., Touret, F., Rathelot, P., Vanelle, P., & Roche, M. (2024). From the “One-Molecule, One-Target, One-Disease” Concept towards Looking for Multi-Target Therapeutics for Treating Non-Polio Enterovirus (NPEV) Infections. Pharmaceuticals, 17(9), 1218. https://doi.org/10.3390/ph17091218