
Development of a Hydrocortisone Orodispersible Thin Film Containing Its Succinate Prodrug

 , , ,
, , ,
Abstract

1. Introduction
2. Results
2.1. Stability-Indicating Liquid Chromatography HMS and HCT Assay
2.1.1. Validation of the HPLC Assay Method
2.1.2. Selectivity of the HMS and HCT Analytical Method
2.2. ODF Formulation Studies
2.2.1. Preparation of the Casting Gel and ODFs
- (1)
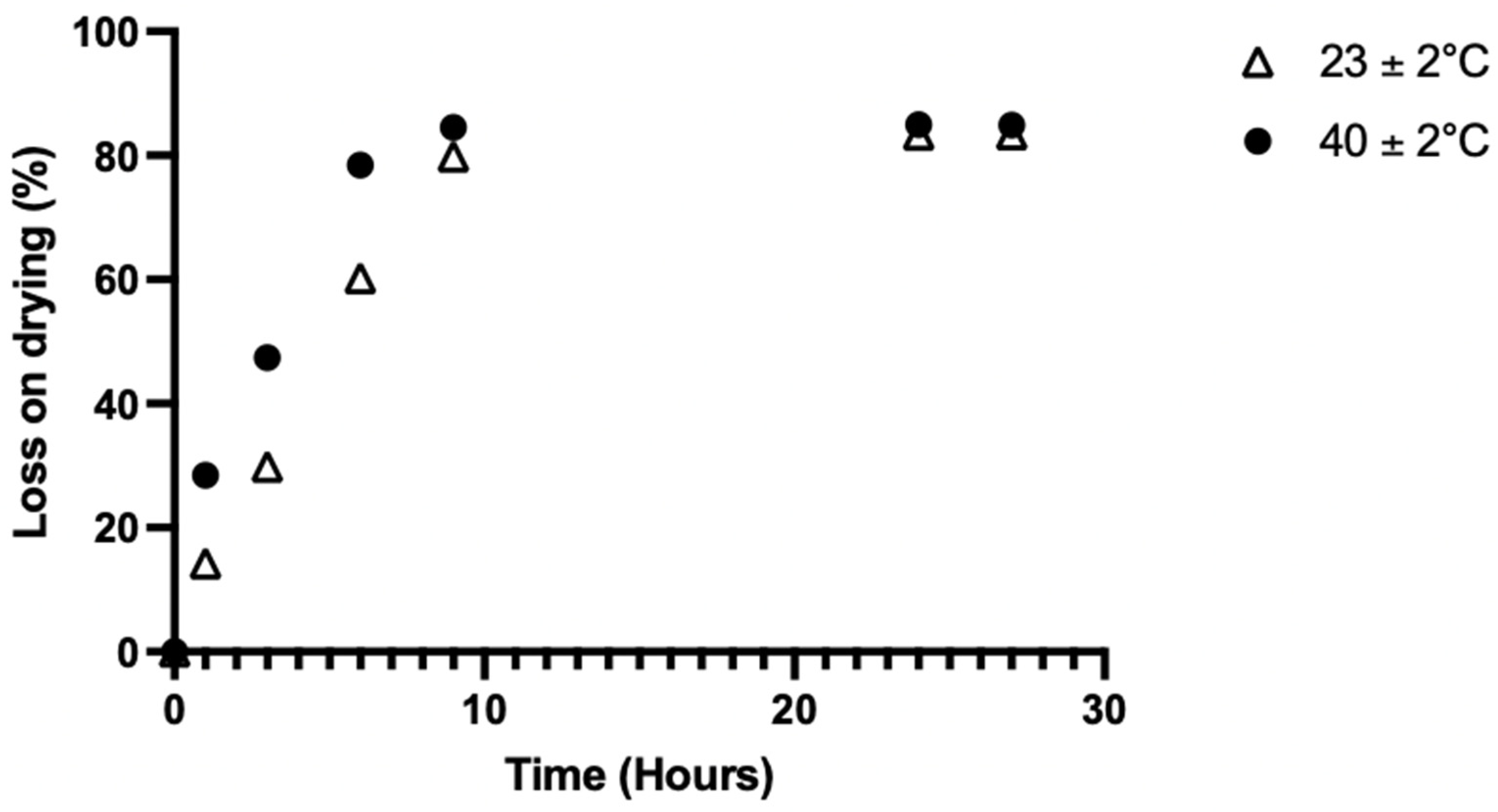
- Preparation of the casting gel: to obtain a perfectly homogenous gel, all the components except HPC and HMS were mixed in water. Once perfectly solubilized, the solution was heated to 40 °C, as HPC is insoluble over 38 °C. This temperature avoided the formation of small gelatinous masses and therefore improved the dispersion of the HPC. These steps were mandatory to avoid clots and aggregates in the casting gel, resulting in an ODF that would break at the slightest contact. To limit the exposure of the API to heating process, it was incorporated just before the HPC. The solution was then brought to room temperature under stirring, allowing the gel to form gradually.
- (2)
- Gel formation and degassing: The solution had to be left to stand for several hours (~10 h) until the HPC was completely dispersed. It was ready for spreading when the gel was visually completely homogeneous and the air bubbles were completely removed. The absence of air bubbles was essential for obtaining a complete ODF with a reproducible and accurate API content. Degassing at +4 °C showed no effect versus ambient temperature as it took several hours to remove all the trapped air. Use of an ultrasonic bath for 5 min helped to raise air bubbles in the upper half of the recipient, saving a few hours in this stage.
- (3)
- Casting process: the use of greaseproof paper was not satisfactory, as it absorbed too much water, resulting in an uneven thickness and discontinuities in the film. In addition, it did not allow for a good drying. A glass plate allowed for the formation of a smooth film, but demolding was impossible. Polyolefin liner allowed for the formation of a smooth film with very easy demolding, but we did not manage to stretch the liner enough, resulting in irregularities in the thickness of the film. Food grade silicone was the best tested surface, giving a smooth film that was easy to demold. This surface has been selected for the rest of study.
2.2.2. ODF Formula Optimization
2.3. ODF and Casting Gel Characterization Assay
2.3.1. ODF Disintegration Time
2.3.2. Casting Gel Content Uniformity
2.3.3. Casting Gel Viscosity
2.3.4. ODF Mass and Thickness Uniformity
2.3.5. ODF Content Uniformity
2.3.6. Casting Gel and Dissolved ODF pH Determination
2.3.7. ODFs Residual Water Content
2.3.8. ODF Dissolution Test
2.3.9. ODFs Stability Study
3. Discussion
4. Materials and Methods
4.1. Drugs and Chemicals
4.2. Validation of Stability-Indicating Liquid Chromatography HMS and HCT Assay
4.2.1. Equipment and Analytical Conditions
4.2.2. Validation of the HPLC Assay Method
4.2.3. Selectivity of the HMS and HCT Analytical Method
4.3. ODF Formulation Studies
4.3.1. Optimizing the Preparation of the Casting Solution and ODFs
- (1)
- The homogeneity of the casting solution has been improved through formulation testing by comparing the dissolution/content of HCT or the HMS in the casting solution. The order of inclusion of the components and management of the solution temperature (23 °C or 40 °C) were also investigated and evaluated by visual inspection.
- (2)
- Air bubbles were removed with or without five minutes’ sonication (Branson 2510, VWR, Rosny, France) and then left at +4 °C or 23 °C for 10 h and evaluated by visual inspection.
- (3)
- The gel was cast on the casting surface on an area of 5 cm × 20 cm and at a thickness of 1700 µm. The casting surface was evaluated on four different materials: food-grade greaseproof paper, glass, polyolefin liner, and food-grade silicone. Drying was carried out at 23 ± 2 °C for 24 h. This study was performed three times on each surface material.
- (4)
- Drying was assessed by casting 1000 mg of gel onto a food-grade silicone surface and evaluated under three temperature conditions: 23 ± 2 °C, 40 ± 2 °C, and 60 ± 2 °C in the proofer (n = 3 per condition). The weight was measured at different time points using an analytical balance (Mettler Toledo AG204, Viroflay, France) and compared to the initial value for calculation of the water loss on drying (%) using this formula, Equation (2):
4.3.2. Formula Optimization
4.4. ODF Characterization Assay
4.4.1. ODF Disintegration Time
4.4.2. Drug Content Uniformity of the Casting Gel
4.4.3. Casting Gel Viscosity
4.4.4. ODF Mass and Thickness Uniformity
4.4.5. ODF Content Uniformity
4.4.6. Casting Gel and Dissolved ODF pH Determination
4.4.7. ODF Residual Water Content
4.4.8. ODF Dissolution Assay
4.4.9. ODF Drug Stability Study
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Claahsen-van der Grinten, H.L.; Speiser, P.W.; Ahmed, S.F.; Arlt, W.; Auchus, R.J.; Falhammar, H.; Flück, C.E.; Guasti, L.; Huebner, A.; Kortmann, B.B.M.; et al. Congenital Adrenal Hyperplasia-Current Insights in Pathophysiology, Diagnostics, and Management. Endocr. Rev. 2022, 43, 91–159. [Google Scholar] [CrossRef] [PubMed]
- Auer, M.K.; Nordenström, A.; Lajic, S.; Reisch, N. Congenital Adrenal Hyperplasia. Lancet Lond. Engl. 2023, 401, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Seth, A. Congenital Adrenal Hyperplasia: Issues in Diagnosis and Treatment in Children. Indian J. Pediatr. 2014, 81, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Coope, H.; Parviainen, L.; Withe, M.; Porter, J.; Ross, R.J. Hydrocortisone Granules in Capsules for Opening (Alkindi) as Replacement Therapy in Pediatric Patients with Adrenal Insufficiency. Expert Opin. Orphan Drugs 2021, 9, 67–76. [Google Scholar] [CrossRef]
- Nguyen, D.; Secretan, P.-H.; Auvity, S.; Vidal, F.; Postaire, M.; Cisternino, S.; Schlatter, J. Assessment of Practices for Suspended Oral Drugs by Tablet Crushing in Pediatric Units. Eur. J. Pharm. Biopharm. Off. J. Arbeitsgemeinschaft Pharm. Verfahrenstechnik EV 2020, 157, 175–182. [Google Scholar] [CrossRef]
- Neumann, U.; Burau, D.; Spielmann, S.; Whitaker, M.J.; Ross, R.J.; Kloft, C.; Blankenstein, O. Quality of Compounded Hydrocortisone Capsules Used in the Treatment of Children. Eur. J. Endocrinol. 2017, 177, 239–242. [Google Scholar] [CrossRef]
- Orlu, M.; Ranmal, S.R.; Sheng, Y.; Tuleu, C.; Seddon, P. Acceptability of Orodispersible Films for Delivery of Medicines to Infants and Preschool Children. Drug Deliv. 2017, 24, 1243–1248. [Google Scholar] [CrossRef]
- Klingmann, V.; Pohly, C.E.; Meissner, T.; Mayatepek, E.; Möltner, A.; Flunkert, K.; Breitkreutz, J.; Bosse, H.M. Acceptability of an Orodispersible Film Compared to Syrup in Neonates and Infants: A Randomized Controlled Trial. Eur. J. Pharm. Biopharm. Off. J. Arbeitsgemeinschaft Pharm. Verfahrenstechnik EV 2020, 151, 239–245. [Google Scholar] [CrossRef]
- European Directorate for the Quality of Medicines and Healthcare—European Directorate for the Quality of Medicines & HealthCare. Available online: https://www.edqm.eu/en/ (accessed on 26 August 2024).
- Ferlak, J.; Guzenda, W.; Osmałek, T. Orodispersible Films—Current State of the Art, Limitations, Advances and Future Perspectives. Pharmaceutics 2023, 15, 361. [Google Scholar] [CrossRef]
- Visser, J.C.; Woerdenbag, H.J.; Crediet, S.; Gerrits, E.; Lesschen, M.A.; Hinrichs, W.L.J.; Breitkreutz, J.; Frijlink, H.W. Orodispersible Films in Individualized Pharmacotherapy: The Development of a Formulation for Pharmacy Preparations. Int. J. Pharm. 2015, 478, 155–163. [Google Scholar] [CrossRef]
- Cilurzo, F.; Cupone, I.E.; Minghetti, P.; Selmin, F.; Montanari, L. Fast Dissolving Films Made of Maltodextrins. Eur. J. Pharm. Biopharm. Off. J. Arbeitsgemeinschaft Pharm. Verfahrenstechnik EV 2008, 70, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Sevinç özakar, R.; Özakar, E. Current Overview of Oral Thin Films. Turk. J. Pharm. Sci. 2021, 18, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Khan, Q.-U.-A.; Siddique, M.I.; Sarfraz, M.; Rehman, K.; Sohail, M.F.; Katas, H. Oral Dispersible Films from Product Development to End-User Acceptability: A Review. Crit. Rev. Ther. Drug Carr. Syst. 2022, 39, 33–64. [Google Scholar] [CrossRef] [PubMed]
- Visser, J.C.; Woerdenbag, H.J.; Hanff, L.M.; Frijlink, H.W. Personalized Medicine in Pediatrics: The Clinical Potential of Orodispersible Films. AAPS PharmSciTech 2017, 18, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Umemura, K.; Tahara, K.; Takeuchi, H. Formulation Design of Hydroxypropyl Cellulose Films for Use as Orally Disintegrating Dosage Forms. J. Drug Deliv. Sci. Technol. 2018, 46, 93–100. [Google Scholar] [CrossRef]
- Speer, I.; Steiner, D.; Thabet, Y.; Breitkreutz, J.; Kwade, A. Comparative Study on Disintegration Methods for Oral Film Preparations. Eur. J. Pharm. Biopharm. 2018, 132, 50–61. [Google Scholar] [CrossRef]
- Kittipongpatana, O.S.; Trisopon, K.; Wattanaarsakit, P.; Kittipongpatana, N. Fabrication and Characterization of Orodispersible Composite Film from Hydroxypropylmethyl Cellulose-Crosslinked Carboxymethyl Rice Starch. Membranes 2022, 12, 594. [Google Scholar] [CrossRef]
- Steiner, D.; Tidau, M.; Finke, J.H. Embedding of Poorly Water-Soluble Drugs in Orodispersible Films—Comparison of Five Formulation Strategies. Pharmaceutics 2023, 15, 17. [Google Scholar] [CrossRef]
- Turner, M.A.; Duncan, J.; Shah, U.; Metsvaht, T.; Varendi, H.; Nellis, G.; Lutsar, I.; Vaconsin, P.; Storme, T.; Rieutord, A.; et al. European Study of Neonatal Exposure to Excipients: An Update. Int. J. Pharm. 2013, 457, 357–358. [Google Scholar] [CrossRef]
- Cupone, I.E.; Roselli, G.; Marra, F.; Riva, M.; Angeletti, S.; Dugo, L.; Spoto, S.; Fogolari, M.; Giori, A.M. Orodispersible Film Based on Maltodextrin: A Convenient and Suitable Method for Iron Supplementation. Pharmaceutics 2023, 15, 1575. [Google Scholar] [CrossRef]
- Johnson, T.N.; Whitaker, M.J.; Keevil, B.; Ross, R.J. Bioavailability of Oral Hydrocortisone Corrected for Binding Proteins and Measured by LC-MS/MS Using Serum Cortisol and Salivary Cortisone. J. Bioequivalence Bioavailab. 2018, 10, 001–003. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, E.; Baba, M. Pharmacokinetics and pharmacodynamics of hydrocortisone in asthmatic children. Arerugi Allergy 1993, 42, 1555–1562. [Google Scholar] [PubMed]
- Cupone, I.E.; Dellera, E.; Marra, F.; Giori, A.M. Development and Characterization of an Orodispersible Film for Vitamin D3 Supplementation. Molecules 2020, 25, 5851. [Google Scholar] [CrossRef] [PubMed]
- Chappe, J.; Osman, N.; Cisternino, S.; Fontan, J.E.; Schlatter, J. Stability of Hydrocortisone Preservative-Free Oral Solutions. J. Pediatr. Pharmacol. Ther. 2015, 20, 197–202. [Google Scholar] [CrossRef]
- Bobillot, M.; Delannoy, V.; Trouillard, A.; Kinowski, J.M.; Sanchez-Ballester, N.M.; Soulairol, I. Potentially Harmful Excipients: State of the Art for Oral Liquid Forms Used in Neonatology and Pediatrics Units. Pharmaceutics 2024, 16, 119. [Google Scholar] [CrossRef]
- Harley, K.G.; Berger, K.P.; Kogut, K.; Parra, K.; Lustig, R.H.; Greenspan, L.C.; Calafat, A.M.; Ye, X.; Eskenazi, B. Association of Phthalates, Parabens and Phenols Found in Personal Care Products with Pubertal Timing in Girls and Boys. Hum. Reprod. Oxf. Engl. 2019, 34, 109–117. [Google Scholar] [CrossRef]
- Ivanovska, V.; Rademaker, C.M.A.; van Dijk, L.; Mantel-Teeuwisse, A.K. Pediatric Drug Formulations: A Review of Challenges and Progress. Pediatrics 2014, 134, 361–372. [Google Scholar] [CrossRef]
- Klingmann, V.; Vallet, T.; Münch, J.; Stegemann, R.; Wolters, L.; Bosse, H.-M.; Ruiz, F. Dosage Forms Suitability in Pediatrics: Acceptability of Analgesics and Antipyretics in a German Hospital. Pharmaceutics 2022, 14, 337. [Google Scholar] [CrossRef]
- Vallet, T.; Bensouda, Y.; Saito, J.; Mathiesen, L.; Pokharkar, V.; Klingmann, V.; Peak, M.; Elhamdaoui, O.; Yamatani, A.; Ivanovic, I.; et al. Exploring Acceptability Drivers of Oral Antibiotics in Children: Findings from an International Observational Study. Pharmaceutics 2021, 13, 1721. [Google Scholar] [CrossRef]
- ICH Official Web Site: ICH. Available online: https://www.ich.org/page/quality-guidelines (accessed on 26 August 2024).
- Guide Méthodologique des Etudes de Stabilité des Préparations—Edition 2013. Available online: https://www.gerpac.net/plateforme/course/view.php?id=8 (accessed on 26 August 2024).
- El-Setouhy, D.A.; Abd El-Malak, N.S. Formulation of a Novel Tianeptine Sodium Orodispersible Film. AAPS PharmSciTech 2010, 11, 1018–1025. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glycerol | 0.5% | 1.0% | 1.5% |
|---|---|---|---|
| Flexibility (/5) (mean ± SD) | 1.4 ± 0.5 | 3.2 ± 1.1 | 4.6 ± 0.5 |
| Stretch resistance (/5) (mean ± SD) | 1.4 ± 0.9 | 3.8 ± 1.3 | 4.2 ± 0.8 |
| Stickiness (/5) (mean ± SD) | 4.0 ± 0.7 | 3.6 ± 0.5 | 3.0 ± 1.0 |
| Total score (/15) (mean ± SD) | 7.2 ±1.5 | 10.6 ± 0.3 | 11.8 ± 0.3 |
| Ingredients | Composition (% w/v) |
|---|---|
| HMS | 0.30 (in equivalent HCT) |
| HPC | 4.0–6.0 |
| Glycerol | 1.5 |
| Povidone K25 | 5.0 |
| Surfactant | 0.20 |
| Sucralose | 0.25 |
| Cola flavor | 0.50 |
| Water | Up to 100 |
| Setofilm® | HPC 4% | HPC 5% | HPC 6% | |
|---|---|---|---|---|
| First break time (mean ± SD) (s) | 35 ± 10 | 62 ± 39 | 264 ± 68 * | 511 ± 101 ** |
| Time for complete disintegration (mean ± SD) (s) | 219 ± 149 | 541 ± 86 * | 1 620 ± 114 *** | 2 073 ± 234 *** |
| Ingredients | Range Tested (% w/v) |
|---|---|
| HMS | 0.30 (in equivalent HCT) |
| HPC | 2–6 |
| Glycerol | 0.5–3.0 |
| Povidone K25 | 5–10 |
| Surfactant | 0–0.4 |
| Sucralose | 0.25 |
| Cola flavor | 0.50 |
| Water | Up to 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boisseillier, C.; Demange-Labriet, L.; Kariyawasam, D.; Marchadour, P.; Fauqueur, A.-S.; Annereau, M.; Denis, L.; Cotteret, C.; Cisternino, S.; Schweitzer-Chaput, A. Development of a Hydrocortisone Orodispersible Thin Film Containing Its Succinate Prodrug. Pharmaceuticals 2025, 18, 86. https://doi.org/10.3390/ph18010086
Boisseillier C, Demange-Labriet L, Kariyawasam D, Marchadour P, Fauqueur A-S, Annereau M, Denis L, Cotteret C, Cisternino S, Schweitzer-Chaput A. Development of a Hydrocortisone Orodispersible Thin Film Containing Its Succinate Prodrug. Pharmaceuticals. 2025; 18(1):86. https://doi.org/10.3390/ph18010086
Chicago/Turabian StyleBoisseillier, Clément, Lucas Demange-Labriet, Dulanjalee Kariyawasam, Pauline Marchadour, Anne-Sophie Fauqueur, Maxime Annereau, Lucas Denis, Camille Cotteret, Salvatore Cisternino, and Arnaud Schweitzer-Chaput. 2025. "Development of a Hydrocortisone Orodispersible Thin Film Containing Its Succinate Prodrug" Pharmaceuticals 18, no. 1: 86. https://doi.org/10.3390/ph18010086
APA StyleBoisseillier, C., Demange-Labriet, L., Kariyawasam, D., Marchadour, P., Fauqueur, A.-S., Annereau, M., Denis, L., Cotteret, C., Cisternino, S., & Schweitzer-Chaput, A. (2025). Development of a Hydrocortisone Orodispersible Thin Film Containing Its Succinate Prodrug. Pharmaceuticals, 18(1), 86. https://doi.org/10.3390/ph18010086