
Diverse Roles of Antibodies in Antibody–Drug Conjugates

Abstract
:
{kind=link}
{kind=link}
1. Introduction
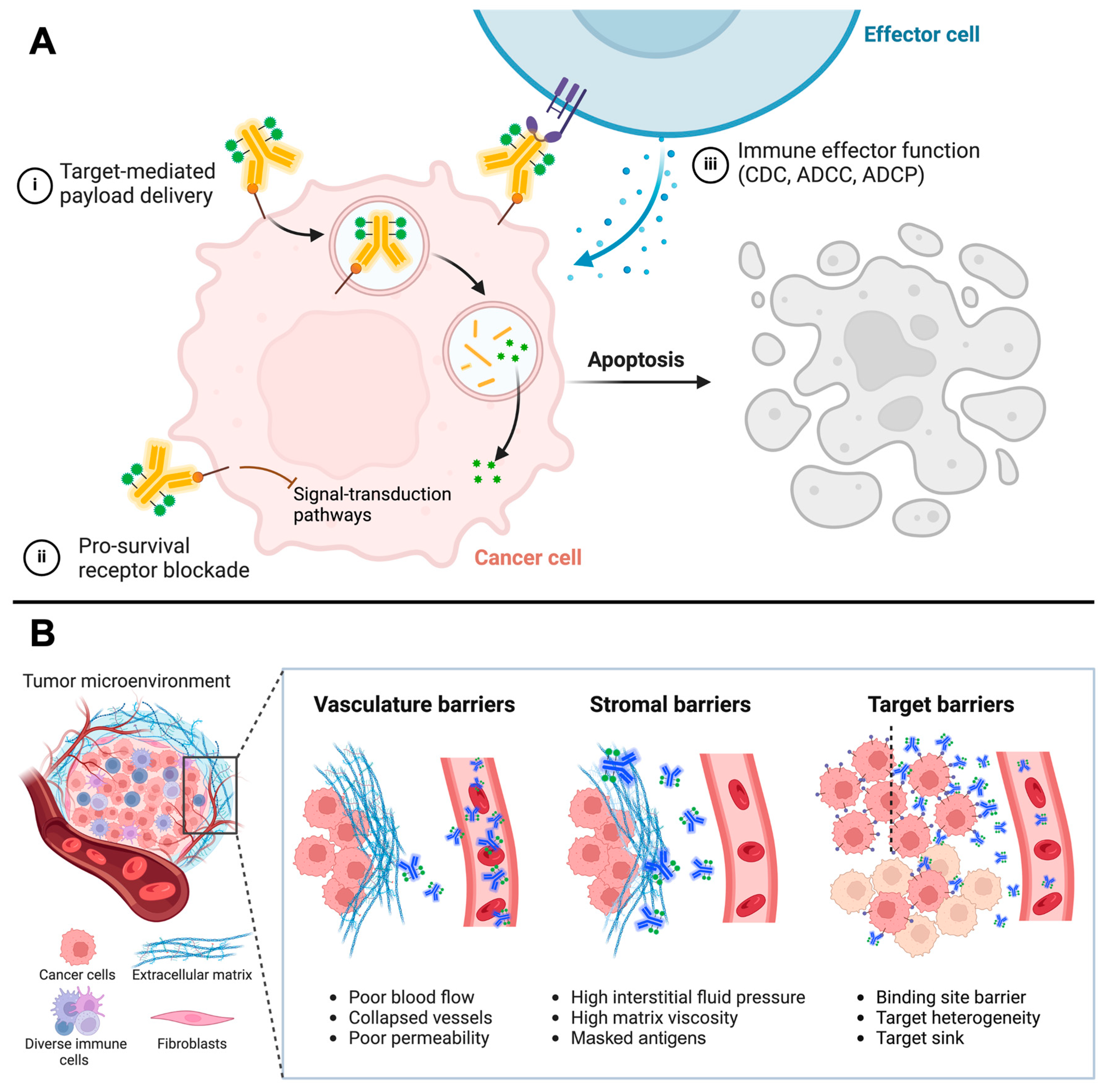
2. Antibodies’ Role in ADCs
3. Characteristics of Promising ADC Targets in Solid Tumors
4. Influence of Target Expression Levels on Efficacy
5. Tumor Types and Genetic Factors
6. Improving Conjugation and Linker Chemistry to Manage Platform Toxicity
7. Next-Generation Carriers to Manage Tumor Penetration and Systemic Toxicity
8. Summary and Concluding Remarks
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colombo, R.; Rich, J.R. The Therapeutic Window of Antibody Drug Conjugates: A Dogma in Need of Revision. Cancer Cell 2022, 40, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; Anami, Y.; Ha, S.Y.Y.; Yamazaki, C.M. Exploring the next Generation of Antibody–Drug Conjugates. Nat. Rev. Clin. Oncol. 2024, 21, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and Challenges for the next Generation of Antibody–Drug Conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Larson, S.M.; Carrasquillo, J.A.; Cheung, N.-K.V.; Press, O.W. Radioimmunotherapy of Human Tumours. Nat. Rev. Cancer 2015, 15, 347. [Google Scholar] [CrossRef]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s Magic Bullet Concept: 100 Years of Progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Ashman, N.; Bargh, J.D.; Spring, D.R. Non-Internalising Antibody–Drug Conjugates. Chem. Soc. Rev. 2022, 51, 9182–9202. [Google Scholar] [CrossRef]
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; de Sauvage, F.J.; Eaton, D.; Elkins, K.; Elliott, J.M.; et al. Antibody-Drug Conjugates for the Treatment of Non-Hodgkin’s Lymphoma: Target and Linker-Drug Selection. Cancer Res. 2009, 69, 2358–2364. [Google Scholar] [CrossRef]
- Dijoseph, J.F.; Dougher, M.M.; Armellino, D.C.; Kalyandrug, L.; Kunz, A.; Boghaert, E.R.; Hamann, P.R.; Damle, N.K. CD20-Specific Antibody-Targeted Chemotherapy of Non-Hodgkin’s B-Cell Lymphoma Using Calicheamicin-Conjugated Rituximab. Cancer Immunol. Immunother. 2007, 56, 1107–1117. [Google Scholar] [CrossRef]
- Govindan, S.V.; Cardillo, T.M.; Moon, S.-J.; Hansen, H.J.; Goldenberg, D.M. CEACAM5-Targeted Therapy of Human Colonic and Pancreatic Cancer Xenografts with Potent Labetuzumab-SN-38 Immunoconjugates. Clin. Cancer Res. 2009, 15, 6052–6061. [Google Scholar] [CrossRef]
- Moon, S.-J.; Govindan, S.V.; Cardillo, T.M.; D’Souza, C.A.; Hansen, H.J.; Goldenberg, D.M. Antibody Conjugates of 7-Ethyl-10-Hydroxycamptothecin (SN-38) for Targeted Cancer Chemotherapy. J. Med. Chem. 2008, 51, 6916–6926. [Google Scholar] [CrossRef]
- Junttila, T.T.; Li, G.; Parsons, K.; Phillips, G.L.; Sliwkowski, M.X. Trastuzumab-DM1 (T-DM1) Retains All the Mechanisms of Action of Trastuzumab and Efficiently Inhibits Growth of Lapatinib Insensitive Breast Cancer. Breast Cancer Res. Treat. 2011, 128, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J.; Lazar, G.A. Next Generation Antibody Drugs: Pursuit of the “High-Hanging Fruit”. Nat. Rev. Clin. Oncol. 2017, 17, 197–223. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Dumontet, C.; Reichert, J.M.; Senter, P.D.; Lambert, J.M.; Beck, A. Antibody–Drug Conjugates Come of Age in Oncology. Nat. Rev. Drug Discov. 2023, 22, 641–661. [Google Scholar] [CrossRef]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab Ozogamicin, a Potent and Selective Anti-CD33 Antibody-Calicheamicin Conjugate for Treatment of Acute Myeloid Leukemia. Bioconjug. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef]
- DiJoseph, J.F.; Armellino, D.C.; Boghaert, E.R.; Khandke, K.; Dougher, M.M.; Sridharan, L.; Kunz, A.; Hamann, P.R.; Gorovits, B.; Udata, C.; et al. Antibody-Targeted Chemotherapy with CMC-544: A CD22-Targeted Immunoconjugate of Calicheamicin for the Treatment of B-Lymphoid Malignancies. Blood 2004, 103, 1807–1814. [Google Scholar] [CrossRef]
- Herbener, P.; Schönfeld, K.; König, M.; Germer, M.; Przyborski, J.M.; Bernöster, K.; Schüttrumpf, J. Functional Relevance of in Vivo Half Antibody Exchange of an IgG4 Therapeutic Antibody-Drug Conjugate. PLoS ONE 2018, 13, e0195823. [Google Scholar] [CrossRef]
- Zhang, A.; Fang, J.; Chou, R.Y.-T.; Bondarenko, P.V.; Zhang, Z. Conformational Difference in Human IgG2 Disulfide Isoforms Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Biochemistry 2015, 54, 1956–1962. [Google Scholar] [CrossRef]
- Carter, P.; Smith, L.; Ryan, M. Identification and Validation of Cell Surface Antigens for Antibody Targeting in Oncology. Endocr. Relat. Cancer 2004, 11, 659–687. [Google Scholar] [CrossRef]
- Bodyak, N.D.; Mosher, R.; Yurkovetskiy, A.V.; Yin, M.; Bu, C.; Conlon, P.R.; Demady, D.R.; DeVit, M.J.; Gumerov, D.R.; Gurijala, V.R.; et al. The Dolaflexin-Based Antibody–Drug Conjugate XMT-1536 Targets the Solid Tumor Lineage Antigen SLC34A2/NaPi2b. Mol. Cancer Ther. 2021, 20, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.Y.; Do, P.; Goswami, S.; Nunes, J.; Chiang, C.-L.; Elgamal, S.; Ventura, A.M.; Cheney, C.; Zapolnik, K.; Williams, E.; et al. The ROR1 Antibody-Drug Conjugate HuXBR1-402-G5-PNU Effectively Targets ROR1+ Leukemia. Blood Adv. 2021, 5, 3152–3162. [Google Scholar] [CrossRef] [PubMed]
- Press, M.F.; Slamon, D.J.; Flom, K.J.; Park, J.; Zhou, J.-Y.; Bernstein, L. Evaluation of HER-2/Neu Gene Amplification and Overexpression: Comparison of Frequently Used Assay Methods in a Molecularly Characterized Cohort of Breast Cancer Specimens. J. Clin. Oncol. 2002, 20, 3095–3105. [Google Scholar] [CrossRef]
- Okajima, D.; Yasuda, S.; Maejima, T.; Karibe, T.; Sakurai, K.; Aida, T.; Toki, T.; Yamaguchi, J.; Kitamura, M.; Kamei, R.; et al. Datopotamab Deruxtecan, a Novel TROP2-Directed Antibody-Drug Conjugate, Demonstrates Potent Antitumor Activity by Efficient Drug Delivery to Tumor Cells. Mol. Cancer Ther. 2021, 20, 2329–2340. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; O’Donnell, P.H.; Balar, A.V.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma after Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. 2019, 37, 2592–2600. [Google Scholar] [CrossRef]
- Mersana Therapeutics Announces Topline Data from UPLIFT Clinical Trial in Patients with Platinum-Resistant Ovarian Cancer and Strategic Reprioritization. Available online: https://ir.mersana.com/news-releases/news-release-details/mersana-therapeutics-announces-topline-data-uplift-clinical (accessed on 22 January 2025).
- Dempsey, N.; Rosenthal, A.; Dabas, N.; Kropotova, Y.; Lippman, M.; Bishopric, N.H. Trastuzumab-Induced Cardiotoxicity: A Review of Clinical Risk Factors, Pharmacologic Prevention, and Cardiotoxicity of Other HER2-Directed Therapies. Breast Cancer Res. Treat. 2021, 188, 21–36. [Google Scholar] [CrossRef]
- Yu, M.; Zhou, L.; Cao, M.; Ji, C.; Zheng, Y. Post-Marketing Drug Safety Surveillance of Enfortumab Vedotin: An Observational Pharmacovigilance Study Based on a Real-World Database. Front. Immunol. 2024, 15, 1397692. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef]
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; González-Martín, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 609–619. [Google Scholar] [CrossRef]
- King, G.T.; Eaton, K.D.; Beagle, B.R.; Zopf, C.J.; Wong, G.Y.; Krupka, H.I.; Hua, S.Y.; Messersmith, W.A.; El-Khoueiry, A.B. A Phase 1, Dose-Escalation Study of PF-06664178, an Anti-Trop-2/Aur0101 Antibody-Drug Conjugate in Patients with Advanced or Metastatic Solid Tumors. Investig. New Drugs 2018, 36, 836–847. [Google Scholar] [CrossRef]
- Fang, W.; Cheng, Y.; Chen, Z.; Wang, W.; Yin, Y.; Li, Y.; Xu, H.; Li, X.; Wainberg, Z.A.; Yu, G.; et al. SKB264 (TROP2-ADC) for the Treatment of Patients with Advanced NSCLC: Efficacy and Safety Data from a Phase 2 Study. J. Clin. Oncol. 2023, 41, 9114. [Google Scholar] [CrossRef]
- O’Donoghue, J.A.; Smith-Jones, P.M.; Humm, J.L.; Ruan, S.; Pryma, D.A.; Jungbluth, A.A.; Divgi, C.R.; Carrasquillo, J.A.; Pandit-Taskar, N.; Fong, Y.; et al. 124I-HuA33 Antibody Uptake Is Driven by A33 Antigen Concentration in Tissues from Colorectal Cancer Patients Imaged by Immuno-PET. J. Nucl. Med. 2011, 52, 1878–1885. [Google Scholar] [CrossRef] [PubMed]
- Polson, A.G.; Ho, W.Y.; Ramakrishnan, V. Investigational Antibody-Drug Conjugates for Hematological Malignancies. Expert Opin. Investig. Drugs 2011, 20, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Kung Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN-CD33A: A Novel CD33-Targeting Antibody-Drug Conjugate Using a Pyrrolobenzodiazepine Dimer Is Active in Models of Drug-Resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Damelin, M.; Zhong, W.; Myers, J.; Sapra, P. Evolving Strategies for Target Selection for Antibody-Drug Conjugates. Pharm. Res. 2015, 32, 3494–3507. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and Safety of Trastuzumab as a Single Agent in First-Line Treatment of HER2-Overexpressing Metastatic Breast Cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef]
- Krop, I.E.; LoRusso, P.; Miller, K.D.; Modi, S.; Yardley, D.; Rodriguez, G.; Guardino, E.; Lu, M.; Zheng, M.; Girish, S.; et al. A Phase II Study of Trastuzumab Emtansine in Patients with Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer Who Were Previously Treated with Trastuzumab, Lapatinib, an Anthracycline, a Taxane, and Capecitabine. J. Clin. Oncol. 2012, 30, 3234–3241. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Bang, Y.-J.; Iwasa, S.; Sugimoto, N.; Ryu, M.-H.; Sakai, D.; Chung, H.C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. 1422MO Trastuzumab Deruxtecan (T-DXd; DS-8201) in Patients with HER2-Low, Advanced Gastric or Gastroesophageal Junction (GEJ) Adenocarcinoma: Results of the Exploratory Cohorts in the Phase II, Multicenter, Open-Label DESTINY-Gastric01 Study. Ann. Oncol. 2020, 31, S899–S900. [Google Scholar] [CrossRef]
- Bardia, A.; Barrios, C.; Dent, R.; Hu, X.; O’Shaughnessy, J.; Yonemori, K.; Darilay, A.; Boston, S.; Liu, Y.; Patel, G.; et al. Abstract OT-03-09: Trastuzumab Deruxtecan (T-DXd; DS-8201) vs. Investigator’s Choice of Chemotherapy in Patients with Hormone Receptor-Positive (HR+), HER2 Low Metastatic Breast Cancer Whose Disease Has Progressed on Endocrine Therapy in the Metastatic Setting: A Randomized, Global Phase 3 Trial (DESTINY-Breast06). Cancer Res. 2021, 81, OT-03-09. [Google Scholar]
- Siena, S.; Di Bartolomeo, M.; Raghav, K.; Masuishi, T.; Loupakis, F.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Fakih, M.; Elez, E.; et al. Trastuzumab Deruxtecan (DS-8201) in Patients with HER2-Expressing Metastatic Colorectal Cancer (DESTINY-CRC01): A Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2021, 22, 779–789. [Google Scholar] [CrossRef]
- Nagayama, A.; Vidula, N.; Ellisen, L.; Bardia, A. Novel Antibody-Drug Conjugates for Triple Negative Breast Cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920915980. [Google Scholar] [CrossRef]
- Bardia, A.; Tolaney, S.M.; Punie, K.; Loirat, D.; Oliveira, M.; Kalinsky, K.; Zelnak, A.; Aftimos, P.; Dalenc, F.; Sardesai, S.; et al. Biomarker Analyses in the Phase III ASCENT Study of Sacituzumab Govitecan versus Chemotherapy in Patients with Metastatic Triple-Negative Breast Cancer. Ann. Oncol. 2021, 32, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Klümper, N.; Tran, N.K.; Zschäbitz, S.; Hahn, O.; Büttner, T.; Roghmann, F.; Bolenz, C.; Zengerling, F.; Schwab, C.; Nagy, D.; et al. NECTIN4 Amplification Is Frequent in Solid Tumors and Predicts Enfortumab Vedotin Response in Metastatic Urothelial Cancer. J. Clin. Oncol. 2024, 42, 2446–2455. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Durán, I.; Lee, J.-L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C.; et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N. Engl. J. Med. 2021, 384, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Zschäbitz, S.; Biernath, N.; Hilser, T.; Höllein, A.; Zengerling, F.; Cascucelli, J.; Paffenholz, P.; Seidl, D.; Lutz, C.; Schlack, K.; et al. Enfortumab Vedotin in Metastatic Urothelial Carcinoma: Survival and Safety in a European Multicenter Real-World Patient Cohort. Eur. Urol. Open Sci. 2023, 53, 31–37. [Google Scholar] [CrossRef]
- Koshkin, V.S.; Henderson, N.; James, M.; Natesan, D.; Freeman, D.; Nizam, A.; Su, C.T.; Khaki, A.R.; Osterman, C.K.; Glover, M.J.; et al. Efficacy of Enfortumab Vedotin in Advanced Urothelial Cancer: Analysis from the Urothelial Cancer Network to Investigate Therapeutic Experiences (UNITE) Study. Cancer 2022, 128, 1194–1205. [Google Scholar] [CrossRef]
- Chu, C.E.; Sjöström, M.; Egusa, E.A.; Gibb, E.A.; Badura, M.L.; Zhu, J.; Koshkin, V.S.; Stohr, B.A.; Meng, M.V.; Pruthi, R.S.; et al. Heterogeneity in NECTIN4 Expression across Molecular Subtypes of Urothelial Cancer Mediates Sensitivity to Enfortumab Vedotin. Clin. Cancer Res. 2021, 27, 5123–5130. [Google Scholar] [CrossRef]
- Klümper, N.; Ralser, D.J.; Ellinger, J.; Roghmann, F.; Albrecht, J.; Below, E.; Alajati, A.; Sikic, D.; Breyer, J.; Bolenz, C.; et al. Membranous NECTIN-4 Expression Frequently Decreases during Metastatic Spread of Urothelial Carcinoma and Is Associated with Enfortumab Vedotin Resistance. Clin. Cancer Res. 2023, 29, 1496–1505. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Bang, Y.-J. HER2-Targeted Therapies—A Role beyond Breast Cancer. Nat. Rev. Clin. Oncol. 2019, 17, 33–48. [Google Scholar] [CrossRef]
- Camidge, D.R.; Bar, J.; Horinouchi, H.; Goldman, J.W.; Moiseenko, F.V.; Filippova, E.; Cicin, I.; Bradbury, P.A.; Daaboul, N.; Tomasini, P.; et al. Telisotuzumab Vedotin (Teliso-V) Monotherapy in Patients (Pts) with Previously Treated c-Met–Overexpressing (OE) Advanced Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2022, 40, 9016. [Google Scholar] [CrossRef]
- Coates, J.T.; Sun, S.; Leshchiner, I.; Thimmiah, N.; Martin, E.E.; McLoughlin, D.; Danysh, B.P.; Slowik, K.; Jacobs, R.A.; Rhrissorrakrai, K.; et al. Parallel Genomic Alterations of Antigen and Payload Targets Mediate Polyclonal Acquired Clinical Resistance to Sacituzumab Govitecan in Triple-Negative Breast Cancer. Cancer Discov. 2021, 11, 2436–2445. [Google Scholar] [CrossRef]
- Hudziak, R.M.; Schlessinger, J.; Ullrich, A. Increased Expression of the Putative Growth Factor Receptor P185HER2 Causes Transformation and Tumorigenesis of NIH 3T3 Cells. Proc. Natl. Acad. Sci. USA 1987, 84, 7159–7163. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, P.P.; Pierce, J.H.; Kraus, M.H.; Segatto, O.; King, C.R.; Aaronson, S.A. ErbB-2 Is a Potent Oncogene When Overexpressed in NIH/3T3 Cells. Science 1987, 237, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the Potential of Antibody-Drug Conjugates for Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Baxter, L.T. Mechanisms of Heterogeneous Distribution of Monoclonal Antibodies and Other Macromolecules in Tumors: Significance of Elevated Interstitial Pressure. Cancer Res. 1988, 48, 7022–7032. [Google Scholar]
- Baxter, L.T.; Jain, R.K. Transport of Fluid and Macromolecules in Tumors. IV. A Microscopic Model of the Perivascular Distribution. Microvasc. Res. 1991, 41, 252–272. [Google Scholar] [CrossRef]
- Juweid, M.; Neumann, R.; Paik, C.; Perez-Bacete, M.J.; Sato, J.; van Osdol, W.; Weinstein, J.N. Micropharmacology of Monoclonal Antibodies in Solid Tumors: Direct Experimental Evidence for a Binding Site Barrier. Cancer Res. 1992, 52, 5144–5153. [Google Scholar]
- Graff, C.P.; Wittrup, K.D. Theoretical Analysis of Antibody Targeting of Tumor Spheroids: Importance of Dosage for Penetration, and Affinity for Retention. Cancer Res. 2003, 63, 1288–1296. [Google Scholar]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.-H.; Sliwkowski, M.X.; et al. Phase I Study of Trastuzumab-DM1, an HER2 Antibody-Drug Conjugate, given Every 3 Weeks to Patients with HER2-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [CrossRef]
- Ocean, A.J.; Starodub, A.N.; Bardia, A.; Vahdat, L.T.; Isakoff, S.J.; Guarino, M.; Messersmith, W.A.; Picozzi, V.J.; Mayer, I.A.; Wegener, W.A.; et al. Sacituzumab Govitecan (IMMU-132), an Anti-Trop-2-SN-38 Antibody-Drug Conjugate for the Treatment of Diverse Epithelial Cancers: Safety and Pharmacokinetics. Cancer 2017, 123, 3843–3854. [Google Scholar] [CrossRef]
- Doi, T.; Shitara, K.; Naito, Y.; Shimomura, A.; Fujiwara, Y.; Yonemori, K.; Shimizu, C.; Shimoi, T.; Kuboki, Y.; Matsubara, N.; et al. Safety, Pharmacokinetics, and Antitumour Activity of Trastuzumab Deruxtecan (DS-8201), a HER2-Targeting Antibody-Drug Conjugate, in Patients with Advanced Breast and Gastric or Gastro-Oesophageal Tumours: A Phase 1 Dose-Escalation Study. Lancet Oncol. 2017, 18, 1512–1522. [Google Scholar] [CrossRef]
- Tarantino, P.; Ricciuti, B.; Pradhan, S.M.; Tolaney, S.M. Optimizing the Safety of Antibody–Drug Conjugates for Patients with Solid Tumours. Nat. Rev. Clin. Oncol. 2023, 20, 558–576. [Google Scholar] [CrossRef] [PubMed]
- Szijj, P.A.; Bahou, C.; Chudasama, V. Minireview: Addressing the Retro-Michael Instability of Maleimide Bioconjugates. Drug Discov. Today Technol. 2018, 30, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Peters, T., Jr. All About Albumin. Biochemistry, Genetics, and Medical Applications; Numerous Figures and Tables; Academic Press, Inc.: San Diego, CA, USA, 1995; p. XX. 432p. [Google Scholar]
- Zorzi, A.; Linciano, S.; Angelini, A. Non-Covalent Albumin-Binding Ligands for Extending the Circulating Half-Life of Small Biotherapeutics. Medchemcomm 2019, 10, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, J.; Bern, M.; Sand, K.M.K.; Grevys, A.; Dalhus, B.; Sandlie, I.; Andersen, J.T. Human and Mouse Albumin Bind Their Respective Neonatal Fc Receptors Differently. Sci. Rep. 2018, 8, 14648. [Google Scholar] [CrossRef]
- Lhospice, F.; Brégeon, D.; Belmant, C.; Dennler, P.; Chiotellis, A.; Fischer, E.; Gauthier, L.; Boëdec, A.; Rispaud, H.; Savard-Chambard, S.; et al. Site-Specific Conjugation of Monomethyl Auristatin E to Anti-CD30 Antibodies Improves Their Pharmacokinetics and Therapeutic Index in Rodent Models. Mol. Pharm. 2015, 12, 1863–1871. [Google Scholar] [CrossRef]
- Pillow, T.H.; Tien, J.; Parsons-Reponte, K.L.; Bhakta, S.; Li, H.; Staben, L.R.; Li, G.; Chuh, J.; Fourie-O’Donohue, A.; Darwish, M.; et al. Site-Specific Trastuzumab Maytansinoid Antibody–Drug Conjugates with Improved Therapeutic Activity through Linker and Antibody Engineering. J. Med. Chem. 2014, 57, 7890–7899. [Google Scholar] [CrossRef]
- Anami, Y.; Otani, Y.; Xiong, W.; Ha, S.Y.Y.; Yamaguchi, A.; Rivera-Caraballo, K.A.; Zhang, N.; An, Z.; Kaur, B.; Tsuchikama, K. Homogeneity of Antibody-Drug Conjugates Critically Impacts the Therapeutic Efficacy in Brain Tumors. Cell Rep. 2022, 39, 110839. [Google Scholar] [CrossRef]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-Specific Conjugation of a Cytotoxic Drug to an Antibody Improves the Therapeutic Index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Yeh, R.; O’Donoghue, J.A.; Jayaprakasam, V.S.; Mauguen, A.; Min, R.; Park, S.; Brockway, J.P.; Bromberg, J.F.; Zhi, W.I.; Robson, M.E.; et al. First-in-Human Evaluation of Site-Specifically Labeled 89Zr-Pertuzumab in Patients with HER2-Positive Breast Cancer. J. Nucl. Med. 2024, 65, 386–393. [Google Scholar] [CrossRef]
- Fang, J.; Guo, L.; Zhang, Y.; Guo, Q.; Wang, M.; Wang, X. The Target Atlas for Antibody-Drug Conjugates across Solid Cancers. Cancer Gene Ther. 2024, 31, 273–284. [Google Scholar] [CrossRef]
- Flynn, P.; Suryaprakash, S.; Grossman, D.; Panier, V.; Wu, J. The Antibody-Drug Conjugate Landscape. Nat. Rev. Drug Discov. 2024, 23, 577–578. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Wang, Z.; Wang, Y. Bispecific Antibody Drug Conjugates: Making 1+1>2. Acta Pharm. Sin. B 2024, 14, 1965–1986. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Guo, K.; Peng, J.; Sun, J.; Xu, T. JSKN003, A Novel Biparatopic ANTI-HER2 Antibody-Drug Conjugate, Exhibits Potent Antitumor Efficacy. Antib. Ther. 2023, 6, tbad014.009. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, J.; Shen, L.; Liu, J.; Huang, J.; Zhuang, Z.; Yin, Y.; Wang, X.; Wang, X.; Wu, J. Evaluation of the Safety, Pharmacokinetics, and Efficacy of JSKN003 in Patients with Advanced Solid Tumors: A Phase I/II Clinical Study. J. Clin. Oncol. 2024, 42, 3031. [Google Scholar] [CrossRef]
- Wan, W.; Zhao, S.; Zhuo, S.; Zhang, Y.; Chen, L.; Li, G.; Renshaw, B.; Khalili, J.S.; Xiao, S.; Zhu, Y. Abstract 2642: BL-B01D1, a Novel EGFR×HER3-Targeting ADC, Demonstrates Robust Anti-Tumor Efficacy in Preclinical Evaluation. Cancer Res. 2023, 83, 2642. [Google Scholar] [CrossRef]
- Kumagai, S.; Koyama, S.; Nishikawa, H. Antitumour Immunity Regulated by Aberrant ERBB Family Signalling. Nat. Rev. Cancer 2021, 21, 181–197. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, Y.; Zhao, Y.; Fang, W.; Zhao, H.; Huang, Y.; Yang, Y.; Chen, L.; Hou, X.; Zou, W.; et al. BL-B01D1, a First-in-Class EGFRxHER3 Bispecific Antibody-Drug Conjugate (ADC), in Patients with Locally Advanced or Metastatic Solid Tumor: Results from a First-in-Human Phase 1 Study. J. Clin. Oncol. 2023, 41, 3001. [Google Scholar] [CrossRef]
- Ye, D.; Bian, X.; Yang, T.; Jiang, S.; Cao, M.; Hua, X.; Xiao, S.; Wang, H.; Zhu, H.; Zhu, Y. 1959O BL-B01D1, an EGFR x HER3 Bispecific Antibody-Drug Conjugate (ADC), in Patients with Locally Advanced or Metastatic Urothelial Carcinoma (UC). Ann. Oncol. 2024, 35, S1133. [Google Scholar] [CrossRef]
- Yang, X.; Nishimiya, D.; Löchte, S.; Jude, K.M.; Borowska, M.; Savvides, C.S.; Dougan, M.; Su, L.; Zhao, X.; Piehler, J.; et al. Facile Repurposing of Peptide-MHC-Restricted Antibodies for Cancer Immunotherapy. Nat. Biotechnol. 2023, 41, 932–943. [Google Scholar] [CrossRef]
- Autio, K.A.; Boni, V.; Humphrey, R.W.; Naing, A. Probody Therapeutics: An Emerging Class of Therapies Designed to Enhance on-Target Effects with Reduced off-Tumor Toxicity for Use in Immuno-Oncology. Clin. Cancer Res. 2020, 26, 984–989. [Google Scholar] [CrossRef]
- Jhaveri, K.; Han, H.; Dotan, E.; Oh, D.-Y.; Ferrario, C.; Tolcher, A.; Lee, K.-W.; Liao, C.-Y.; Kang, Y.-K.; Kim, Y.H.; et al. 460MO Preliminary Results from a Phase I Study Using the Bispecific, Human Epidermal Growth Factor 2 (HER2)-Targeting Antibody-Drug Conjugate (ADC) Zanidatamab Zovodotin (ZW49) in Solid Cancers. Ann. Oncol. 2022, 33, S749–S750. [Google Scholar] [CrossRef]
- Pegram, M.D.; Hamilton, E.P.; Tan, A.R.; Storniolo, A.M.; Balic, K.; Rosenbaum, A.I.; Liang, M.; He, P.; Marshall, S.; Scheuber, A.; et al. First-in-Human, Phase 1 Dose-Escalation Study of Biparatopic Anti-HER2 Antibody-Drug Conjugate MEDI4276 in Patients with HER2-Positive Advanced Breast or Gastric Cancer. Mol. Cancer Ther. 2021, 20, 1442–1453. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Choi, H.; Jin, Z.; Liu, L.; Lu, J.; Stokell, D.J.; Murphy, A.T.; Dunn, K.W.; Martinez, M.M.; Feng, Y. Reducing Target Binding Affinity Improves the Therapeutic Index of Anti-MET Antibody-Drug Conjugate in Tumor Bearing Animals. PLoS ONE 2024, 19, e0293703. [Google Scholar] [CrossRef]
- Cilliers, C.; Menezes, B.; Nessler, I.; Linderman, J.; Thurber, G.M. Improved Tumor Penetration and Single-Cell Targeting of Antibody-Drug Conjugates Increases Anticancer Efficacy and Host Survival. Cancer Res. 2018, 78, 758–768. [Google Scholar] [CrossRef]
- Singh, A.P.; Guo, L.; Verma, A.; Wong, G.G.-L.; Thurber, G.M.; Shah, D.K. Antibody Coadministration as a Strategy to Overcome Binding-Site Barrier for ADCs: A Quantitative Investigation. AAPS J. 2020, 22, 28. [Google Scholar] [CrossRef]
- Medina Pérez, V.M.; Baselga, M.; Schuhmacher, A.J. Single-Domain Antibodies as Antibody-Drug Conjugates: From Promise to Practice-A Systematic Review. Cancers 2024, 16, 2681. [Google Scholar] [CrossRef]
- Khera, E.; Cilliers, C.; Bhatnagar, S.; Thurber, G.M. Computational Transport Analysis of Antibody-Drug Conjugate Bystander Effects and Payload Tumoral Distribution: Implications for Therapy. Mol. Syst. Des. Eng. 2018, 3, 73–88. [Google Scholar] [CrossRef]
- Tsumura, R.; Manabe, S.; Takashima, H.; Koga, Y.; Yasunaga, M.; Matsumura, Y. Influence of the dissociation rate constant on the intra-tumor distribution of antibody-drug conjugate against tissue factor. J. Control. Release 2018, 284, 49–56. [Google Scholar] [CrossRef]
- Henry, K.A.; MacKenzie, C.R. Antigen Recognition by Single-Domain Antibodies: Structural Latitudes and Constraints. MAbs 2018, 10, 815–826. [Google Scholar] [CrossRef]
- Chatalic, K.L.S.; Veldhoven-Zweistra, J.; Bolkestein, M.; Hoeben, S.; Koning, G.A.; Boerman, O.C.; de Jong, M.; van Weerden, W.M. A Novel 111In-Labeled Anti-Prostate-Specific Membrane Antigen Nanobody for Targeted SPECT/CT Imaging of Prostate Cancer. J. Nucl. Med. 2015, 56, 1094–1099. [Google Scholar] [CrossRef]
- Zinn, S.; Vazquez-Lombardi, R.; Zimmermann, C.; Sapra, P.; Jermutus, L.; Christ, D. Advances in Antibody-Based Therapy in Oncology. Nat. Cancer 2023, 4, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Razzaghdoust, A.; Rahmatizadeh, S.; Mofid, B.; Muhammadnejad, S.; Parvin, M.; Torbati, P.M.; Basiri, A. Data-Driven Discovery of Molecular Targets for Antibody-Drug Conjugates in Cancer Treatment. Biomed Res. Int. 2021, 2021, 2670573. [Google Scholar] [CrossRef] [PubMed]
- Rubahamya, B.; Dong, S.; Thurber, G.M. Clinical Translation of Antibody Drug Conjugate Dosing in Solid Tumors from Preclinical Mouse Data. Sci. Adv. 2024, 10, eadk1894. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaguchi, A.; Manning, H.C. Diverse Roles of Antibodies in Antibody–Drug Conjugates. Pharmaceuticals 2025, 18, 180. https://doi.org/10.3390/ph18020180
Yamaguchi A, Manning HC. Diverse Roles of Antibodies in Antibody–Drug Conjugates. Pharmaceuticals. 2025; 18(2):180. https://doi.org/10.3390/ph18020180
Chicago/Turabian StyleYamaguchi, Aiko, and H. Charles Manning. 2025. "Diverse Roles of Antibodies in Antibody–Drug Conjugates" Pharmaceuticals 18, no. 2: 180. https://doi.org/10.3390/ph18020180
APA StyleYamaguchi, A., & Manning, H. C. (2025). Diverse Roles of Antibodies in Antibody–Drug Conjugates. Pharmaceuticals, 18(2), 180. https://doi.org/10.3390/ph18020180