
Targeting Plasmodium falciparum Schizont Egress Antigen-1 in Infected Red Blood Cells: Docking-Based Fingerprinting, Density Functional Theory, Molecular Dynamics Simulations, and Binding Free Energy Analysis


Abstract
:1. Introduction
2. Results
2.1. Protein Structure Preparation and Validation
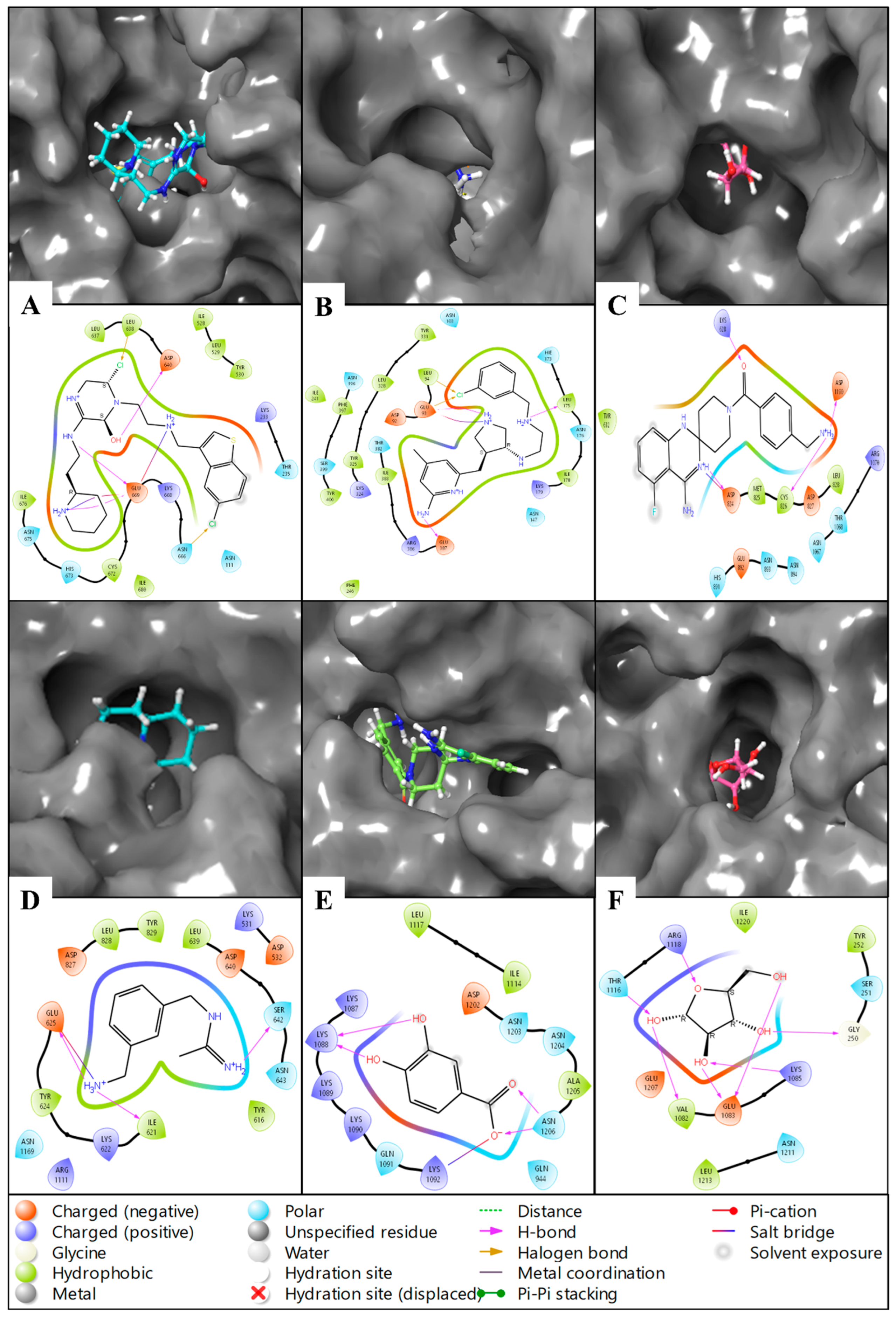
2.2. Molecular Docking Studies
2.3. Molecular Interaction Fingerprints
2.4. Pharmacokinetic Studies
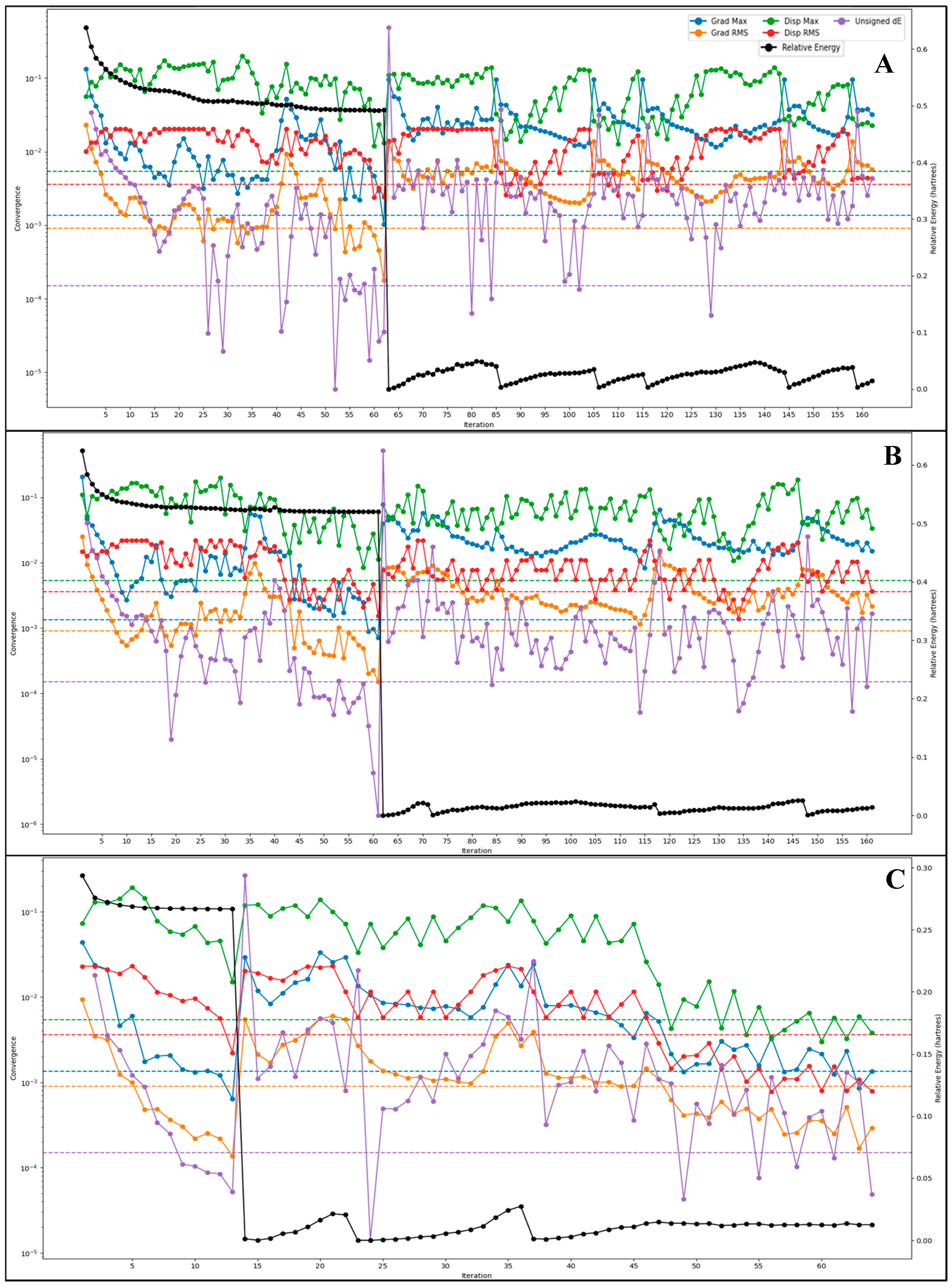
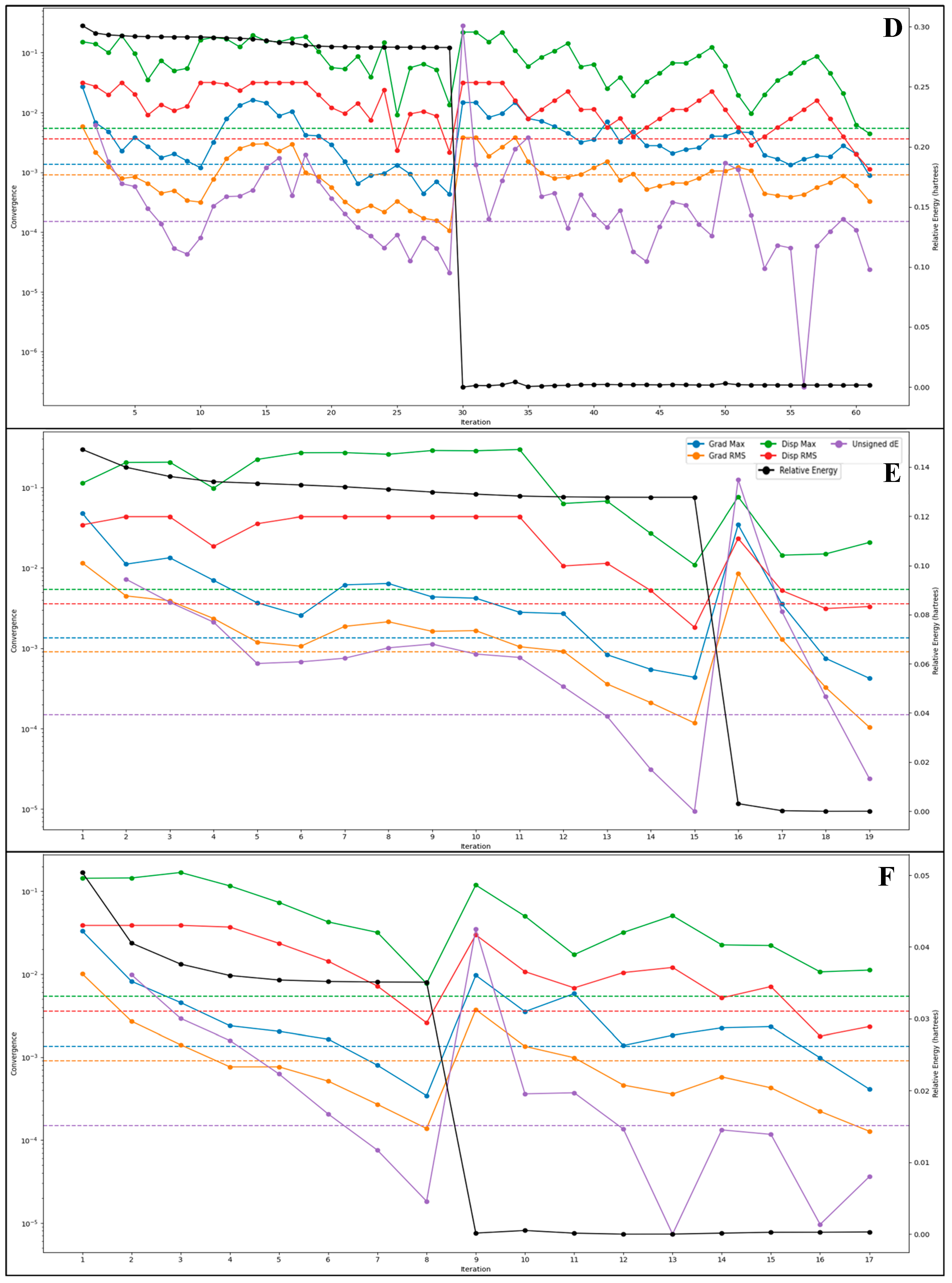
2.5. DFT-Based Compound Optimisation Studies
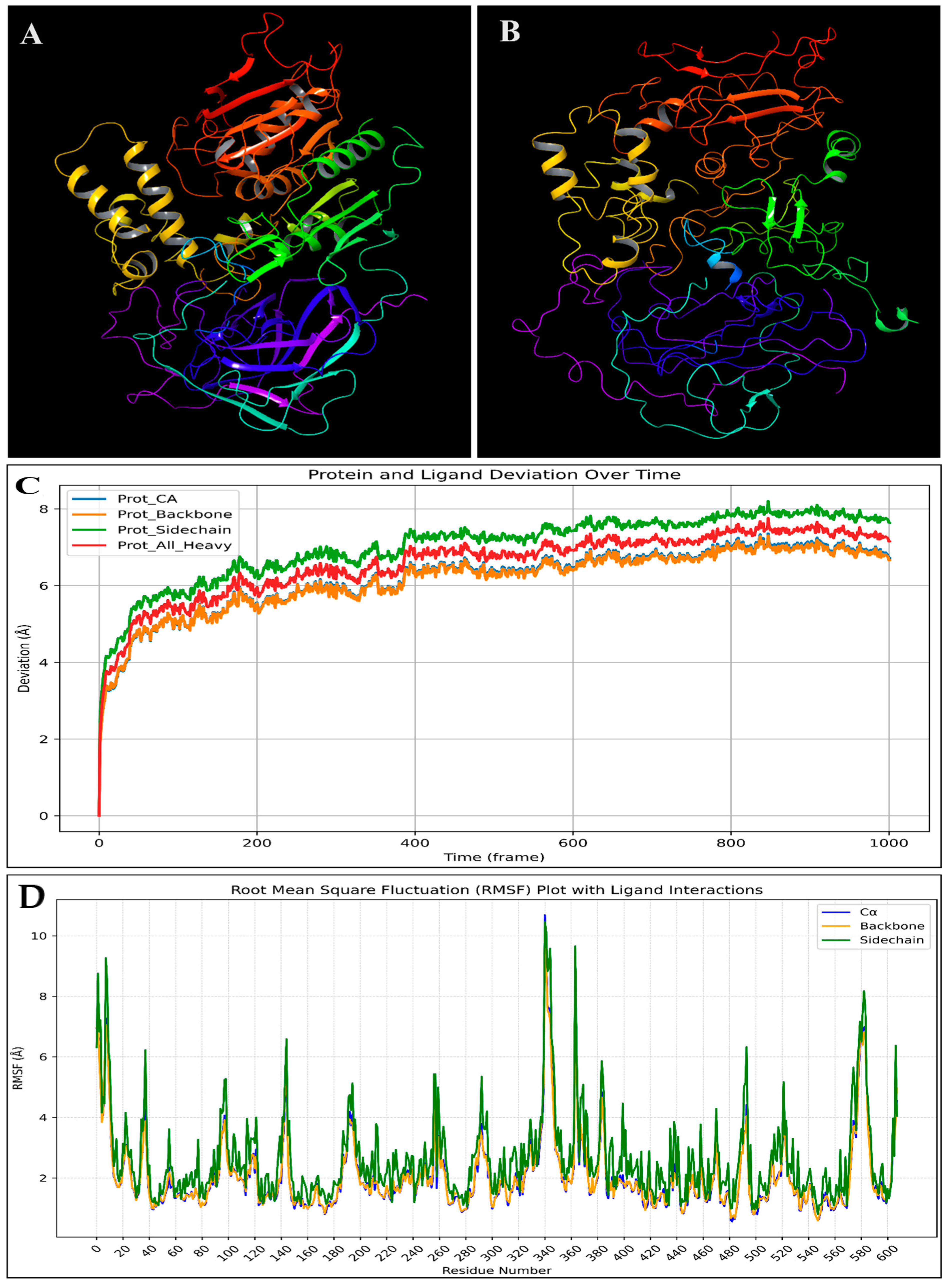
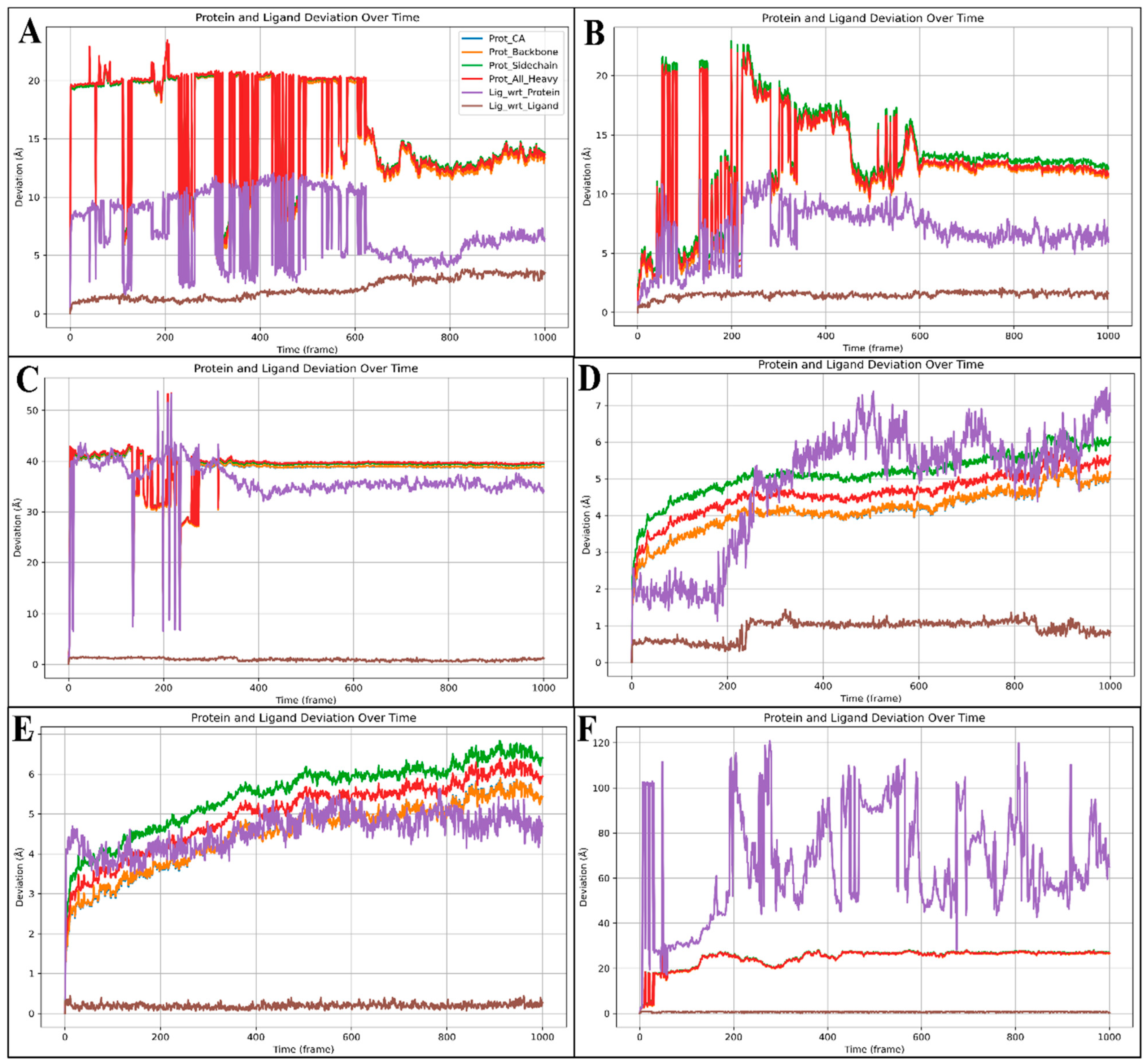
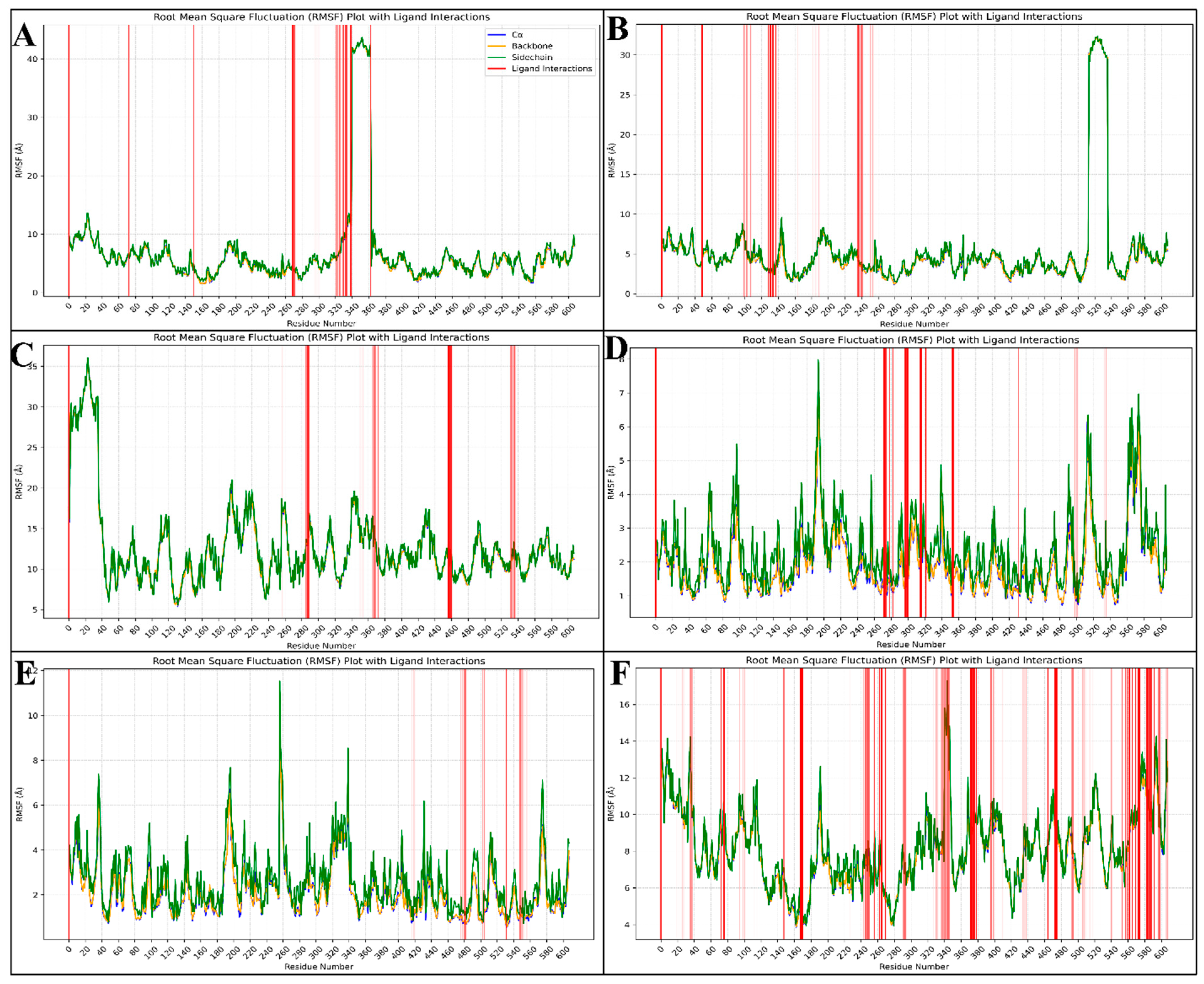
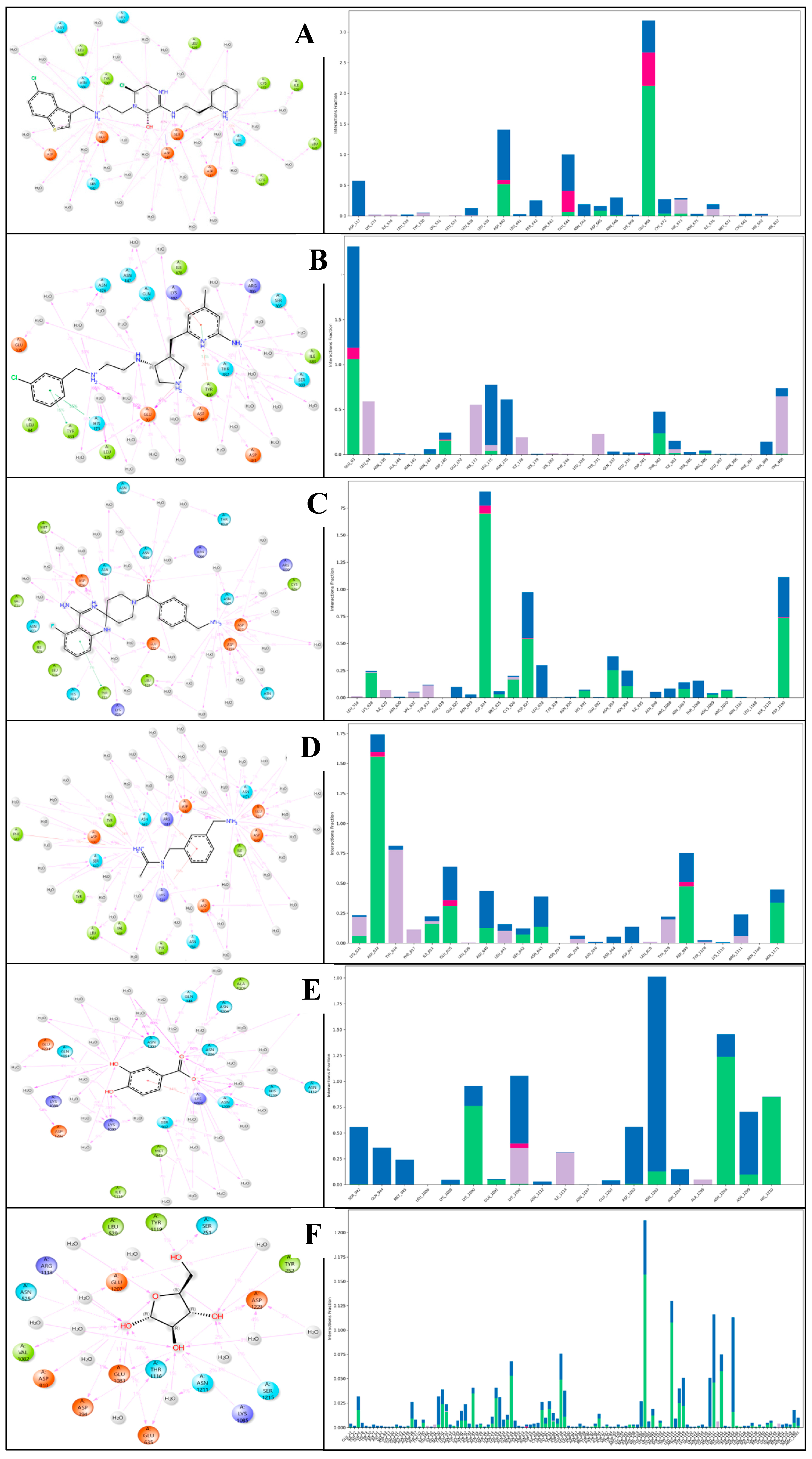
2.6. Molecular Dynamics Simulation
2.6.1. Root-Mean-Square Deviation (RMSD)
2.6.2. Root-Mean-Square Fluctuations (RMSF)
2.6.3. Simulation Interaction Diagram (SID) Studies
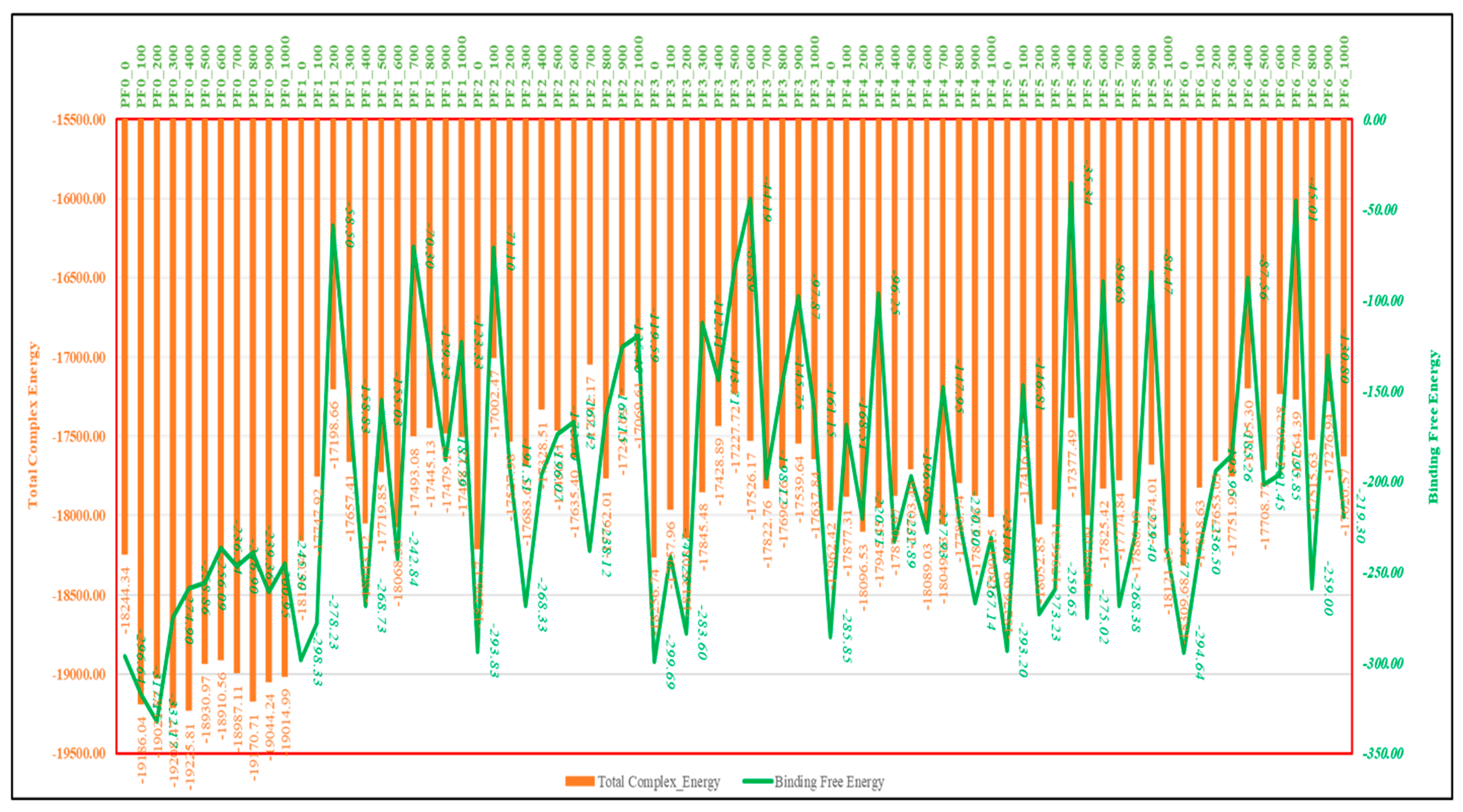
2.7. MM\GBSA Studies
3. Discussion
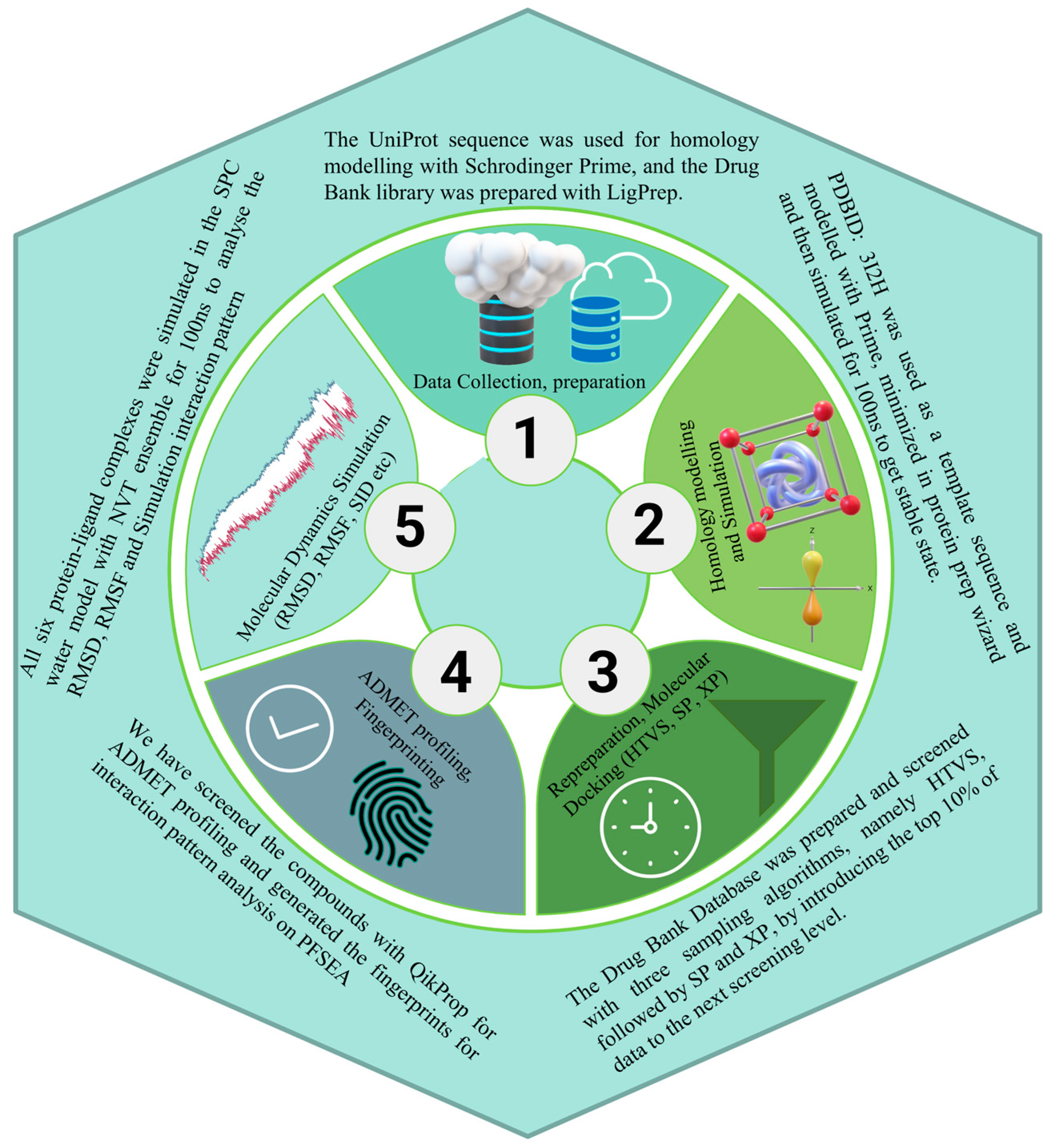
4. Methods
4.1. PfSEA-1 Sequence Downloading, Modelling, and Preparation
4.2. Molecular Dynamics Simulation and Validation
4.3. Ligand Library Preparation
4.4. Molecular Docking with Simulation-Minimised Structure
4.5. Interaction Fingerprinting and Pharmacokinetics of Identified Candidates
4.6. Optimisation Studies with Density Functional Theory
4.7. Molecular Dynamics Simulation and MM\GBSA Computations
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zareen, S.; Rehman, H.U.; Gul, N.; Zareen, H.; Hisham, M.; Ullah, I.; Saeed, K. Malaria is still a life threatening disease review. J. Entomol. Zool. Stud. 2016, 105, 105–112. [Google Scholar]
- Balmith, M.; Basson, C.; Brand, S.J. The Malaria Burden: A South African Perspective. J. Trop. Med. 2024, 2024, 6619010. [Google Scholar] [CrossRef] [PubMed]
- Varo, R.; Chaccour, C.; Bassat, Q. Update on malaria. Med. Clínica Engl. Ed. 2020, 155, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Kolawole, E.O.; Ayeni, E.T.; Abolade, S.A.; Ugwu, S.E.; Awoyinka, T.B.; Ofeh, A.S.; Okolo, B.O. Malaria endemicity in Sub-Saharan Africa: Past and present issues in public health. Microbes Infect. Dis. 2023, 4, 242–251. [Google Scholar] [CrossRef]
- Kurtis, J.D.; Raj, D.K.; Michelow, I.C.; Park, S.; Nixon, C.E.; McDonald, E.A.; Nixon, C.P.; Pond-Tor, S.; Jha, A.; Taliano, R.J. Maternally-derived antibodies to schizont egress antigen-1 and protection of infants from severe malaria. Clin. Infect. Dis. 2019, 68, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Arya, H.; Sahu, W.; Reddy, K.S.; Nimesh, S.; Alotaibi, B.S.; Hakami, M.A.; Almasoudi, H.H.; Hessien, K.B.G.; Hasan, M.R. Green synthesized silver nanoparticles of Terminalia bellirica leaves extract: Synthesis, characterization, in-silico studies, and antimalarial activity. Artif. Cells Nanomed. Biotechnol. 2024, 52, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.O.; Oso, O.V.; Obeagu, E.I.; Adeyemo, A.T. Malaria vaccine: Prospects and challenges. Madonna Univ. J. Med. Health Sci. ISSN 2814-3035 2022, 2, 22–40. [Google Scholar]
- Rogerson, S.J.; Beeson, J.G.; Laman, M.; Poespoprodjo, J.R.; William, T.; Simpson, J.A.; Price, R.N. Identifying and combating the impacts of COVID-19 on malaria. BMC Med. 2020, 18, 239. [Google Scholar] [CrossRef] [PubMed]
- Voß, Y.; Klaus, S.; Guizetti, J.; Ganter, M. Plasmodium schizogony, a chronology of the parasite’s cell cycle in the blood stage. PLoS Pathog. 2023, 19, e1011157. [Google Scholar] [CrossRef]
- Chandley, P.; Ranjan, R.; Kumar, S.; Rohatgi, S. Host-parasite interactions during Plasmodium infection: Implications for immunotherapies. Front. Immunol. 2023, 13, 1091961. [Google Scholar] [CrossRef] [PubMed]
- Etefia, E.; Inyang-Etoh, P. Malaria vaccine development: Challenges and prospects. Med. Pharm. J. 2023, 2, 28–42. [Google Scholar] [CrossRef]
- Raj, D.K.; Nixon, C.P.; Nixon, C.E.; Dvorin, J.D.; DiPetrillo, C.G.; Pond-Tor, S.; Wu, H.-W.; Jolly, G.; Pischel, L.; Lu, A. Antibodies to PfSEA-1 block parasite egress from RBCs and protect against malaria infection. Science 2014, 344, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Laurens, M.B.; Plowe, C.V. Malaria Vaccines. In Infectious Diseases; Springer: Berlin/Heidelberg, Germany, 2023; pp. 123–154. [Google Scholar]
- Ogwang, R. Development and Characterization of Human Monoclonal Antibodies with Functional Activity Against Plasmodium Falciparum Merozoites. Ph.D. Thesis, The Open University, Milton Keynes, UK, 2023. [Google Scholar]
- Maestro, S. Maestro; Schrödinger, LLC.: New York, NY, USA, 2020; Volume 2020. [Google Scholar]
- Chandrasekaran, B.; Abed, S.N.; Al-Attraqchi, O.; Kuche, K.; Tekade, R.K. Computer-aided prediction of pharmacokinetic (ADMET) properties. In Dosage Form Design Parameters; Elsevier: Amsterdam, The Netherlands, 2018; pp. 731–755. [Google Scholar]
- QikProp, S. Schrödinger Release 2017; Maestro LLC.: New York, NY, USA, 2017. [Google Scholar]
- Poluri, K.M.; Gulati, K.; Tripathi, D.K.; Nagar, N. Protein-Protein Interactions in Host–Pathogen Interactions. In Protein-Protein Interactions: Pathophysiological and Therapeutic Aspects: Volume II; Springer: Berlin/Heidelberg, Germany, 2023; pp. 207–264. [Google Scholar]
- Vyas, V.; Ukawala, R.; Ghate, M.; Chintha, C. Homology modeling a fast tool for drug discovery: Current perspectives. Indian J. Pharm. Sci. 2012, 74, 1. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; McGinnis, S.; Madden, T.L. BLAST: Improvements for better sequence analysis. Nucleic Acids Res. 2006, 34 (Suppl. S2), W6–W9. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, D.; Nance, M.R.; Gao, D.; Ko, M.-C.; Macdonald, J.; Tamburi, P.; Yoon, D.; Landry, D.M.; Woods, J.H.; Zhan, C.-G. Structural analysis of thermostabilizing mutations of cocaine esterase. Protein Eng. Des. Sel. 2010, 23, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2016, 45, D271–D281. [Google Scholar] [PubMed]
- Sahu, A.; Ahmad, S.; Imtiyaz, K.; Kizhakkeppurath Kumaran, A.; Islam, M.; Raza, K.; Easwaran, M.; Kurukkan Kunnath, A.; Rizvi, M.A.; Verma, S. In-silico and in-vitro study reveals Ziprasidone as a potential aromatase inhibitor against breast carcinoma. Sci. Rep. 2023, 13, 16545. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Ahmad, S.; Yadav, M.K.; Raza, K.; Kamal, M.A.; Akhtar, S. Structure-based virtual screening, molecular docking, molecular dynamics simulation, and metabolic reactivity studies of quinazoline derivatives for their anti-EGFR activity against tumor angiogenesis. Curr. Med. Chem. 2024, 31, 595–619. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Karwasra, R.; Ahmad, S.; Bano, N.; Qazi, S.; Raza, K.; Singh, S.; Varma, S. Macrophage-targeted punicalagin nanoengineering to alleviate methotrexate-induced neutropenia: A molecular docking, DFT, and MD simulation analysis. Molecules 2022, 27, 6034. [Google Scholar] [CrossRef]
- Kaul, T.; Eswaran, M.; Ahmad, S.; Thangaraj, A.; Jain, R.; Kaul, R.; Raman, N.M.; Bharti, J. Probing the effect of a plus 1bp frameshift mutation in protein-DNA interface of domestication gene, NAMB1, in wheat. J. Biomol. Struct. Dyn. 2019, 38, 3633–3647. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Ahmad, S.; Bano, N.; Qazi, S.; Yadav, M.K.; Ahmad, N.; Raza, K. Multitargeted molecular dynamic understanding of butoxypheser against SARS-CoV-2: An in silico study. Nat. Prod. Commun. 2022, 17, 1934578X221115499. [Google Scholar] [CrossRef]
- Ahmad, S.; Dahiya, V.; Vibhuti, A.; Pandey, R.P.; Tripathi, M.K.; Yadav, M.K. Therapeutic protein-based vaccines. In Protein-Based Therapeutics; Springer Nature: Singapore, 2023; pp. 355–384. [Google Scholar]
- Ahmad, S.; Kishan, A.; Chitkara, P.; Asiri, S.A.; Eswaran, M.; Mehta, S.; Alam, M. Natural Product-Based Drug Designing for Treatment of Human Parasitic Diseases. In Natural Product Based Drug Discovery Against Human Parasites: Opportunities and Challenges; Springer: Berlin/Heidelberg, Germany, 2023; pp. 37–59. [Google Scholar]
- McDonald, I. NpT-ensemble Monte Carlo calculations for binary liquid mixtures. Mol. Phys. 1972, 23, 41–58. [Google Scholar] [CrossRef]
- Release, S. Schrödinger Suite 2017-1 Protein Preparation Wizard; Epik, Schrödinger, LLC.: New York, NY, USA, 2017. [Google Scholar]
- Almasoudi, H.H.; Hakami, M.A.; Alhazmi, A.Y.; Makkawi, M.; Alasmari, S.; Alghamdi, Y.S.; Mashraqi, M.M. Unveiling the multitargeted repurposing potential of taxifolin (dihydroquercetin) in cervical cancer: An extensive MM\GBSA-based screening, and MD simulation study. Med. Oncol. 2023, 40, 218. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34 (Suppl. S1), D668–D672. [Google Scholar] [CrossRef]
- Yadav, M.K.; Ahmad, S.; Raza, K.; Kumar, S.; Eswaran, M.; Pasha, K.M.M. Predictive modeling and therapeutic repurposing of natural compounds against the receptor-binding domain of SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 41, 1527–1539. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Pasha Km, M.; Raza, K.; Rafeeq, M.M.; Habib, A.H.; Eswaran, M.; Yadav, M.K. Reporting dinaciclib and theodrenaline as a multitargeted inhibitor against SARS-CoV-2: An in-silico study. J. Biomol. Struct. Dyn. 2023, 41, 4013–4023. [Google Scholar] [CrossRef]
- Release, S. Receptor Grid Generation; Schrödinger, LLC.: New York, NY, USA, 2019. [Google Scholar]
- Release, S. Glide; Schrödinger, LLC.: New York, NY, USA, 2020. [Google Scholar]
- Tripathi, M.K.; Ahmad, S.; Tyagi, R.; Dahiya, V.; Yadav, M.K. Fundamentals of molecular modeling in drug design. In Computer Aided Drug Design (CADD): From Ligand-Based Methods to Structure-Based Approaches; Elsevier: Amsterdam, The Netherlands, 2022; pp. 125–155. [Google Scholar]
- Ahmad, S.; Singh, A.P.; Bano, N.; Raza, K.; Singh, J.; Medigeshi, G.R.; Pandey, R.; Gautam, H.K. Integrative analysis discovers Imidurea as dual multitargeted inhibitor of CD69, CD40, SHP2, lysozyme, GATA3, cCBL, and S-cysteinase from SARS-CoV-2 and M. tuberculosis. Int. J. Biol. Macromol. 2024, 270, 132332. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Almasoudi, H.H.; Nahari, M.H.; Alhazmi, A.Y.M.; Almasabi, S.H.A.; Al-Mansour, F.S.H.; Hakami, M.A. Delineating Pixantrone Maleate’s adroit activity against cervical cancer proteins through multitargeted docking-based MM\GBSA, QM-DFT and MD simulation. PLoS ONE 2023, 18, e0295714. [Google Scholar] [CrossRef] [PubMed]
- Almasoudi, H.H.; Mashraqi, M.M.; Alshamrani, S.A.; Alharthi, A.A.; Alsalmi, O.; Nahari, M.H.; Al-Mansour, F.S.H.; Alhazmi, A.Y.M. Structure-Based In Silico Approaches Reveal IRESSA as a Multitargeted Breast Cancer Regulatory, Signalling, and Receptor Protein Inhibitor. Pharmaceuticals 2024, 17, 208. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy Component | Before MDS (kcal/mol) | After MDS (kcal/mol) |
|---|---|---|
| Total Energy of the System | 1.12 × 104 | −5.57 × 104 |
| Total Potential Energy | 1.12 × 104 | −5.57 × 104 |
| Total Kinetic Energy | 0 | 0 |
| Temperature of the System | 0 K | 0 K |
| Bond Stretch Energy | 5.37 × 102 | 5.63 × 102 |
| Angle Bending Energy | 2.96 × 103 | 1.55 × 103 |
| Torsion Angle Energy | 2.12 × 103 | 8.85 × 102 |
| Restraining Energy for Torsions | 0 | 0 |
| 1,4 Lennard-Jones Energy | 3.68 × 103 | 2.86 × 103 |
| 1,4 Electrostatic Energy | 8.69 × 102 | 8.46 × 102 |
| Lennard-Jones Energy | −2.17 × 103 | −9.96 × 103 |
| Electrostatic Energy | −2.92 × 103 | −5.34 × 104 |
| H-bond Energy | 0 | 0 |
| S No | Drug Bank | Drug Name | Docking Score | MMGBSA dG Bind Coulomb | MMGBSA dG Bind Covalent | MMGBSA dG Bind Hbond |
|---|---|---|---|---|---|---|
| 1 | DB07521 | Chlobenethyzenol | −8.107 | −445.18 | 4.98 | −1.95 |
| 2 | DB08019 | Ametchomine | −7.005 | −319.81 | 4.06 | −3.53 |
| 3 | DB08750 | Amiflupipquamine | −6.94 | −385.43 | 1.88 | −2.69 |
| 4 | DB02044 | Ambenzyne | −6.505 | −375.08 | 0.98 | −3.04 |
| 5 | DB03946 | Dihycid | −6.225 | 78.17 | 6.11 | −3.87 |
| 6 | DB03142 | Alparabinos | −4.481 | −40.44 | 8.54 | −4.65 |
| S No | Drug Bank | Drug Name | Prime Hbond | Prime vdW | Ligand Efficiency | Ligand Efficiency sa |
| 1 | DB07521 | Chlobenethyzenol | −328.93 | 4.67284 × 1020 | −0.262 | −0.822 |
| 2 | DB08019 | Ametchomine | −330.51 | 4.67284 × 1020 | −0.269 | −0.798 |
| 3 | DB08750 | Amiflupipquamine | −329.67 | 4.67284 × 1020 | −0.257 | −0.771 |
| 4 | DB02044 | Ambenzyne | −330.02 | 4.67284 × 1020 | −0.5 | −1.177 |
| 5 | DB03946 | Dihycid | −330.85 | 4.67284 × 1020 | −0.566 | −1.258 |
| 6 | DB03142 | Alparabinos | −331.63 | 4.67284 × 1020 | −0.448 | −0.965 |
| Descriptors | Standard | Alparabinos | Dihycid | Ambenzyne | Amiflupipquamine | Ametchomine | Chlobenethyzenol |
|---|---|---|---|---|---|---|---|
| #stars | 0–5 | 0 | 0 | 0 | 0 | 0 | 5 |
| #amine | 0–1 | 3 | 3 | 1 | 1 | 0 | 0 |
| #amidine | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| #acid | 0–1 | 0 | 0 | 0 | 0 | 1 | 0 |
| #amide | 0–1 | 0 | 0 | 0 | 0 | 0 | 0 |
| #rotor | 0–15 | 10 | 9 | 4 | 5 | 3 | 5 |
| #rtvFG | 0–2 | 1 | 0 | 0 | 0 | 0 | 1 |
| CNS | −2 (inact), +2 (act) | 1 | 0 | −1 | 0 | −2 | −2 |
| mol MW | 130.0–725.0 | 484.486 | 373.928 | 367.425 | 177.249 | 154.122 | 150.131 |
| dipole | 1.0–12.5 | 3.471 | 3.578 | 5.54 | 4.115 | 5.953 | 1.757 |
| SASA | 300.0–1000.0 | 833.794 | 598.359 | 641.741 | 448.687 | 331.243 | 312.149 |
| FOSA | 0.0–750.0 | 406.592 | 283.638 | 175.215 | 157.785 | 0 | 127.898 |
| FISA | 7.0–330.0 | 88.112 | 114.794 | 163.364 | 145.123 | 206.617 | 184.251 |
| PISA | 0.0–450.0 | 167.583 | 151.102 | 268.468 | 145.779 | 124.626 | 0 |
| WPSA | 0.0–175.0 | 171.507 | 48.825 | 34.694 | 0 | 0 | 0 |
| volume-1 | 500.0–2000.0 | 1480.343 | 1152.562 | 1138.44 | 710.665 | 505.322 | 484.044 |
| donorHB | 0.0–6.0 | 4 | 5 | 5 | 4 | 3 | 4 |
| accptHB | 2.0–20.0 | 8.2 | 6 | 6 | 2.5 | 3.5 | 8.5 |
| dip^2/V | 0.0–0.13 | 0.008138 | 0.011108 | 0.0269578 | 0.0238265 | 0.0701263 | 0.0063776 |
| ACxDN^.5/SA | 0.0–0.05 | 0.0196691 | 0.022422 | 0.0209063 | 0.0111436 | 0.0183013 | 0.0544611 |
| glob | 0.75–0.95 | 0.7533806 | 0.8884775 | 0.8216352 | 0.8583498 | 0.9262546 | 0.9551232 |
| QPpolrz | 13.0–70.0 | 48.269 | 35.636 | 39.402 | 20.468 | 13.333 | 10.007 |
| QPlogPC16 | 4.0–18.0 | 16.213 | 12.378 | 12.739 | 7.576 | 6.013 | 5.298 |
| QPlogPoct | 8.0–35.0 | 26.522 | 22.398 | 24.217 | 13.742 | 11.442 | 13.225 |
| QPlogPw | 4.0–45.0 | 15.255 | 14.233 | 16.564 | 9.996 | 9.949 | 14.851 |
| QPlogPo/w | −2.0–6.5 | 3.292 | 1.375 | 2.017 | 0.269 | 0.031 | −1.745 |
| QPlogS | −6.5–0.5 | −3.545 | 0.263 | −3.599 | −0.885 | −0.798 | −0.705 |
| CIQPlogS | −6.5–0.5 | −3.161 | −1.696 | −3.845 | −0.857 | −1.429 | −0.314 |
| QPlogHERG | concern below −5 | −8.5 | −6.236 | −6.425 | −5.245 | −1.512 | −2.258 |
| QPPCaco | <25 poor, >500 great | 22.441 | 12.532 | 69.763 | 103.895 | 27.552 | 177.276 |
| QPlogBB | −3.0–1.2 | 0.144 | −0.032 | −0.807 | −0.653 | −1.223 | −1.084 |
| QPPMDCK | <25 poor, >500 great | 96.199 | 10.905 | 47.685 | 47.346 | 12.965 | 76.248 |
| QPlogKp | −8.0–−1.0 | −5.718 | −6.364 | −5.263 | −7.326 | −4.6 | −4.435 |
| IP(eV) | 7.9–10.5 | 8.49 | 8.367 | 8.515 | 8.408 | 9.292 | 10.868 |
| EA(eV) | −0.9–1.7 | 0.737 | 0.131 | 0.649 | 0.265 | 0.609 | −2.286 |
| #metab | 1–8 | 5 | 6 | 3 | 4 | 2 | 3 |
| QPlogKhsa | −1.5–1.5 | 0.411 | 0.025 | 0.131 | −0.456 | −0.9 | −0.839 |
| HumanOralAbs | N/A | 2 | 2 | 3 | 2 | 2 | 2 |
| % HumanOralAbs | >80% is high, <25% is poor | 70.402 | 54.65 | 71.754 | 64.615 | 52.903 | 56.972 |
| SAfluorine | 0.0–100.0 | 0 | 0 | 34.694 | 0 | 0 | 0 |
| SAamideO | 0.0–35.0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PSA | 7.0–200.0 | 74.425 | 78.123 | 99.369 | 73.945 | 93.526 | 97.786 |
| #NandO | 2–15 | 6 | 5 | 6 | 3 | 4 | 5 |
| RuleOfFive | maximum is 4 | 0 | 0 | 0 | 0 | 0 | 0 |
| RuleOfThree | maximum is 3 | 1 | 1 | 0 | 0 | 0 | 0 |
| #ringatoms | N/A | 21 | 17 | 21 | 6 | 6 | 5 |
| #in34 | N/A | 0 | 0 | 0 | 0 | 0 | 0 |
| #in56 | N/A | 21 | 17 | 21 | 6 | 6 | 5 |
| #noncon | N/A | 8 | 4 | 5 | 0 | 0 | 4 |
| #nonHatm | N/A | 31 | 26 | 27 | 13 | 11 | 10 |
| Jm | N/A | 0.000264 | 0.296407 | 0.000505 | 0.001091 | 0.616045 | 1.086211 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almasoudi, H.H.; Nahari, M.H. Targeting Plasmodium falciparum Schizont Egress Antigen-1 in Infected Red Blood Cells: Docking-Based Fingerprinting, Density Functional Theory, Molecular Dynamics Simulations, and Binding Free Energy Analysis. Pharmaceuticals 2025, 18, 237. https://doi.org/10.3390/ph18020237
Almasoudi HH, Nahari MH. Targeting Plasmodium falciparum Schizont Egress Antigen-1 in Infected Red Blood Cells: Docking-Based Fingerprinting, Density Functional Theory, Molecular Dynamics Simulations, and Binding Free Energy Analysis. Pharmaceuticals. 2025; 18(2):237. https://doi.org/10.3390/ph18020237
Chicago/Turabian StyleAlmasoudi, Hassan H., and Mohammed H. Nahari. 2025. "Targeting Plasmodium falciparum Schizont Egress Antigen-1 in Infected Red Blood Cells: Docking-Based Fingerprinting, Density Functional Theory, Molecular Dynamics Simulations, and Binding Free Energy Analysis" Pharmaceuticals 18, no. 2: 237. https://doi.org/10.3390/ph18020237
APA StyleAlmasoudi, H. H., & Nahari, M. H. (2025). Targeting Plasmodium falciparum Schizont Egress Antigen-1 in Infected Red Blood Cells: Docking-Based Fingerprinting, Density Functional Theory, Molecular Dynamics Simulations, and Binding Free Energy Analysis. Pharmaceuticals, 18(2), 237. https://doi.org/10.3390/ph18020237