
Small-Molecule Inhibitors of Amyloid Beta: Insights from Molecular Dynamics—Part A: Endogenous Compounds and Repurposed Drugs


Abstract
:1. Introduction
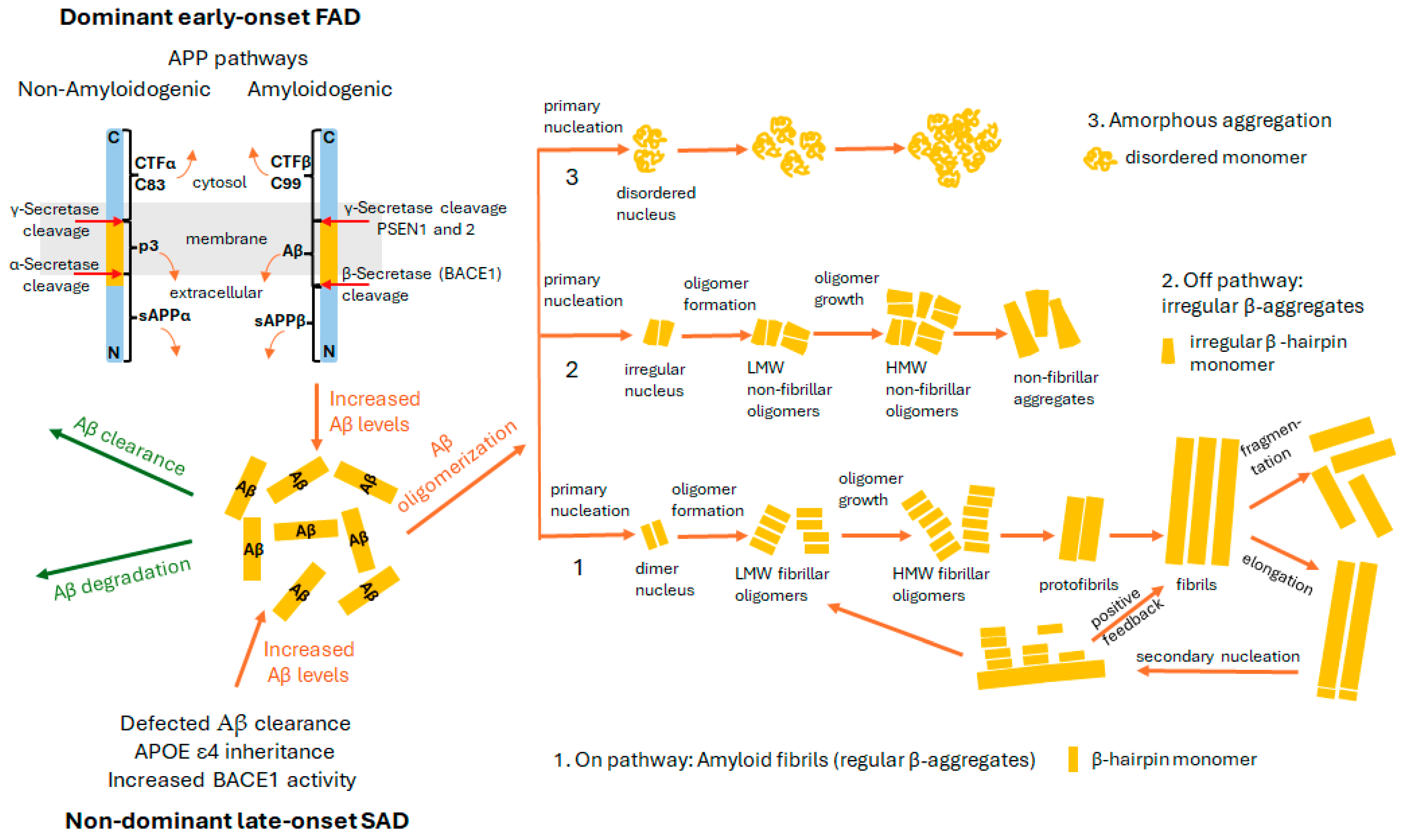
2. Amyloid Hypothesis
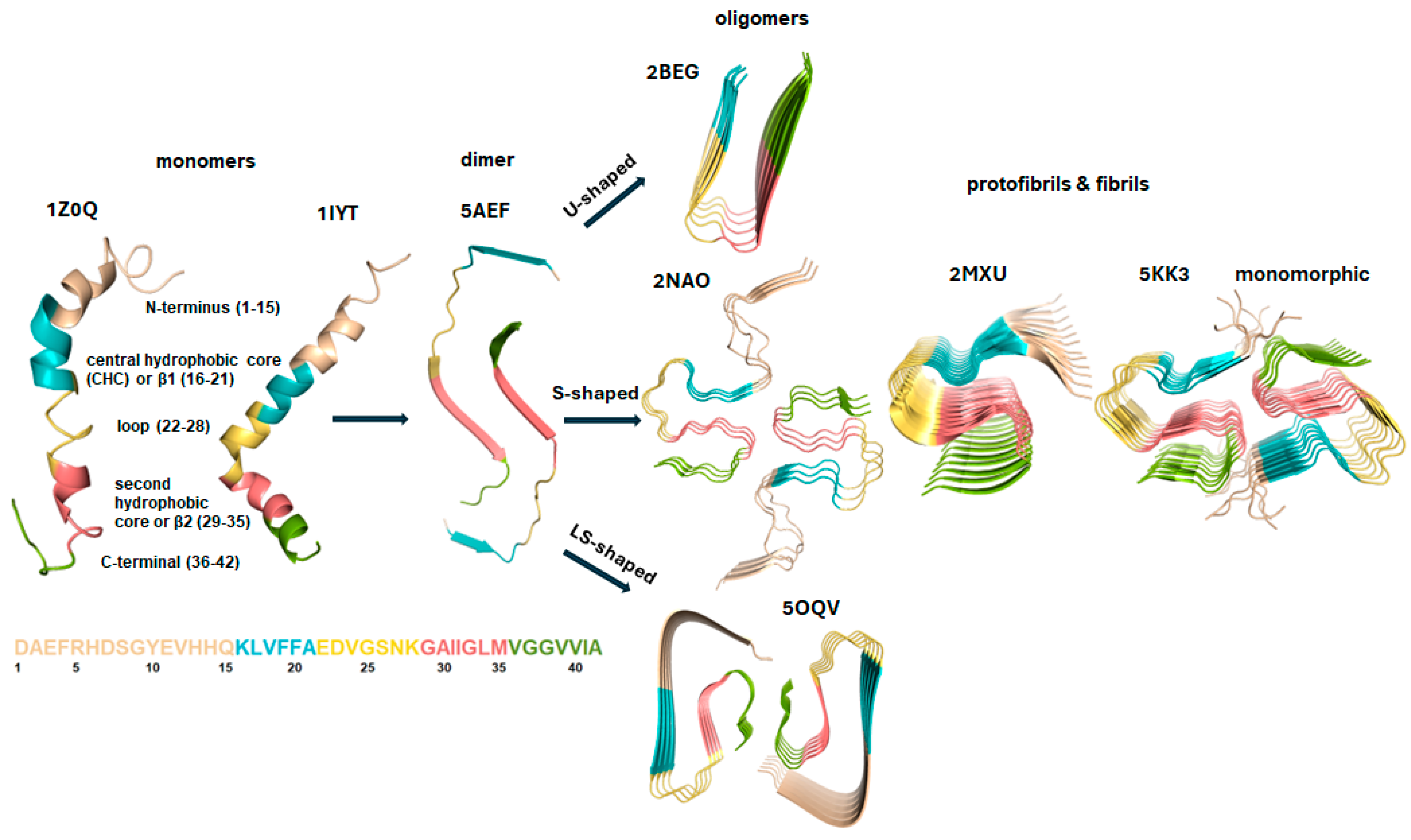
3. Structural Characteristics and Forms of Monomers, Dimers, and Oligomers
4. Interactions Leading to and Stabilizing Aβ Aggregation
5. Molecular Dynamics Simulations: Challenges and Prospects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Examined FFs | Criteria for Comparison | Studied System/s | Sampling Method/ Water Model | Ref. |
|---|---|---|---|---|
| AMBER94, AMBER96, AMBER99, AMBER99-ILDN, AMBER03, AMBER12SB, AMBER14SB, GROMOS43a1, GROMOS43a2GROMOS45a3, GROMOS53a5, GROMOS53a6, GROMOS54a7, CHARM22*, CHARRMM36, CHARRMM36m, and OPLS-AA | global reaction coordinates, secondary structure content, fibril population and formation | 100 dimeric Aβ16–22 | cMD/TIP3P for all FFs, except for GROMOS FFs―SPC, CHARM22*―mTIP3P, and OPLS―TIP4P | [117] |
| ff99SB, ff14SB, FF14SB_IDPs, CHARMM36, CHARMM36m | chemical shifts, secondary structure content, nonbonded energy component, Enonbonded | monomeric Aβ42 | cMD and REMD/TIP3P | [118] |
| ff99IDPs, ff14IDPs, ff14IDPSFF, ff03w, CHARMM36m, and CHARMM22* | chemical shits, J-couplings, global reaction coordinates, secondary structure content | RS-peptide, HEWL19, HIV-rev, Aβ40, Aβ42, phosphodiesterase-γ, CspTm, and ubiquitin | cMD/for ff99IDPs, ff14IDPs, ff14IDPSFF―TIP3P, for ff03w―TIP4P-2005; and for CHARMM FFs―CHARMM-modified TIP3P | [113] |
| Gromos54a7, OPLS-AA, AMBER03ws, CHARMM22*, and AMBER99SB*ILDN | oligomer formation kinetics, in terms of dissociation constant, KD and ∆G, and collision acceptance probability | monomeric forms and six monomers of each of the three peptides: Aβ16–22, one non-amyloidogenic mutant (F19V, F20V), and the Aβ16–22 (F19L) mutant, which exhibits rapid fibrillogenesis | SPC for Gromos54a7, TIP4P/2005 for AMBER03ws, TIP4P for OPLS-AA, TIP4P-Ew for CHARMM22* and AMBER99SB*ILDN | [119] |
| OPLS, AMBER99SB, AMBER99SB*ILDN, AMBER99SBILDN-NMR and CHARMM22* | local NMR observables, including chemical shifts, J-couplings, and residual dipolar couplings (RDCs) | monomeric Aβ1–42 | REMD/TIP4P-Ew for AMBER99SB, AMBER99SB*ILDN, AM-BER99SBILDN-NMR and CHARMM22*, TIP3P for OPLS | [116] |
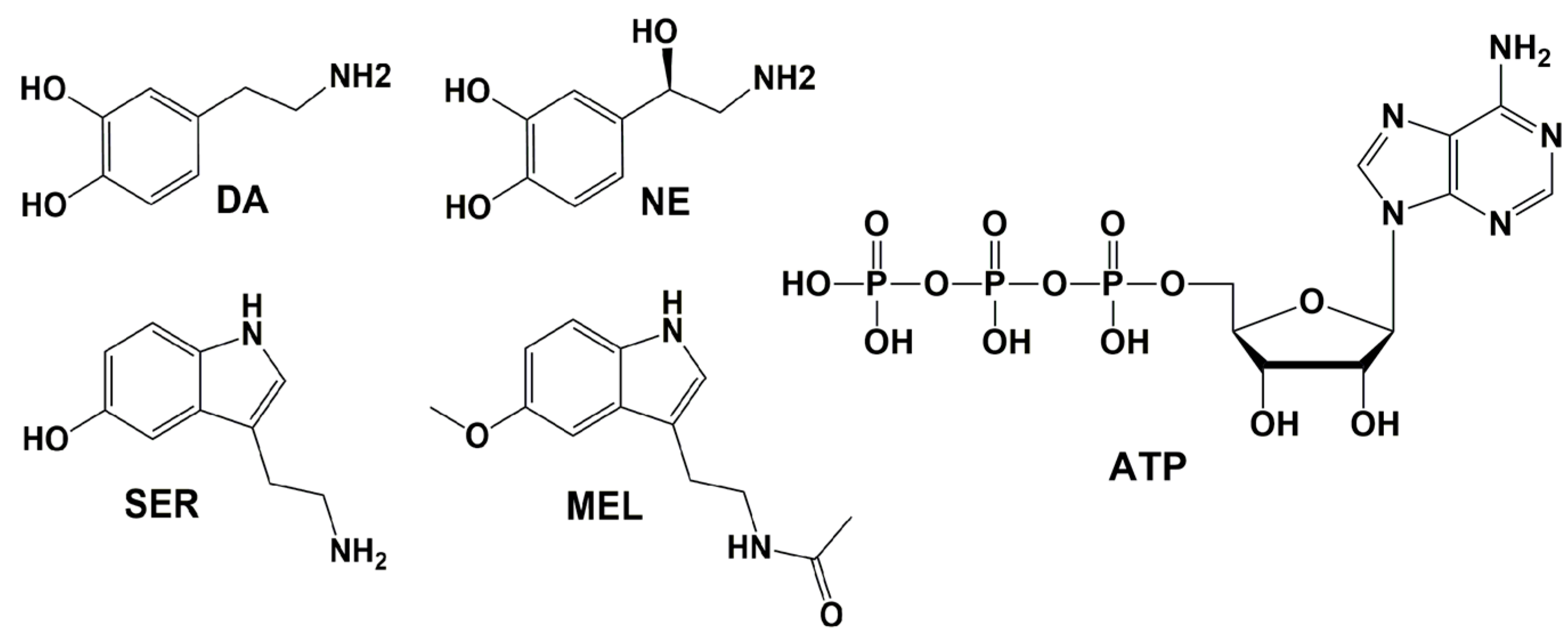
6. Endogenous Compounds Inhibiting Aβ
6.1. Dopamine (DA) and Norepinephrine (NE)
| FF/Water Model | Duration per System, ns | Aβ Length/PDB ID/Type (Monomer/Dimer/ (Proto-)Fibril) | Inhibitor * | Main Findings | Ref. |
|---|---|---|---|---|---|
| REMD/GROMOS 57a7/SPC | 50 per replica | Aβ1–40/2LMN/decamer, protofibril | DA | preferably binds to β-2 and N-terminal; significantly affects the oligomer’s double-layer structure | [201] |
| AMBER99SBILDN/TIP3P | 5 × 500 per system | Aβ1–42/5OQV/pentamer | (1) 5 DA+* (molecular ratio 1:1) (2) 10 DA+ (2:1) (3) 50 DA+ (10:1) (4) 40 DA+ + 10 DA0 (10:1) | (1) binds to H6-E11 (turn-1 region); weak disruptive effect; (2) binds to H6-E11, the F4-L34-V36 hydrophobic core, the turn-2 region (F20, E22 and D23), and the C-terminal residues I41 and A42; stronger disruptive effect (3) binds mainly to the outer surface, decreasing the flexibility of the Aβ protofibril and stabilizing it (4) DA0 molecules bind to the inner surface of the protofibril, primarily to the F4-L34-V36 hydrophobic core; π-π stacking with DA+ increases their inner surface binding, resulting in a disruptive effect of the DA0 and DA+ mixture | [202] |
| REMD/AMBER99SBILDN/TIP3P | 950 per replica (48 replicas) | (1) Aβ1–42/1IYT/two monomers placed in three orientations: parallel, antiparallel and perpendicular | 10 DA+ (molar ratio 5:1) | inhibits the dimerization | [202] |
| REMD/AMBER99SBILDN/TIP3P | 300 per replica (54 replicas) | Aβ1–42/1Z0Q/dimer | 20 NE (molar ratio 10:1) | suppresses and reduces the interpeptide β-sheet content; five dominant BS were identified; main contact residues: hydrophobic interactions with L17, I31, and I41; π-π stacking with Y4, F10, and F20; H-bonds with D1, E3, D7, E11, E22, and D23; cation-π interactions with R5 | [203] |
| AMBER99SBILDN/TIP3P | 2 × 1000 (2 × 1 µs) | Aβ1–42/5OQV/pentamer | 100 NE | β-sheet content decreased, while coil content increased; the number of fibril H-bonds decreased; H-bonds formed with D1, A2, D23, and A42; destabilizes the preformed fibril | [203] |
| AMBER99SBILDN/TIP3P | 3 × 500 (per system) | Aβ1–42/5OQV/tetramer | (1) 20 NE+ (molar ration 5:1) (2) 20 NE0 (molar ration 5:1) | (1) destabilizes the protofibril; primarily disrupts the D1-H14 region; decreases β-sheet content; increases the kink angle around Y10; disrupts H6-E11 H-bonds and K28-A42 salt bridges; forms H-bonds with E3, D7, E11, Q15, E22, and D23; engages in π-π stacking with H6, H13, and F20; (2) disrupts the protofibril; decreases β-sheet content in dispersed regions, including A2–H6, A21–G22, S26, and M35–V36; primarily forms hydrophobic interactions and π-π stacking with F4, R5, H6, Y10, H13, Q15, F20 and L34. | [204] |
| AMBER99SBILDN/TIP3P | 3 × 500 | Aβ1–42/5OQV/tetramer | 40 SER (molar ratio 10:1) | disrupts fibrils by decreasing β-sheet content at the N-terminal (D1-Y10); main contact residues include F4, H6, Y10, H13, Q15, and L34; dominant interactions involve π-π stacking with F4, H6, Y10, and H13; disrupts fibril-stabilizing contacts A2-V36 and F4-L34 at core-1; as well as salt bridges between L34-A42. | [205] |
| AMBER99SBILDN/TIP3P | 3 × 500 | Aβ1–42/5OQV/tetramer | 40 MEL (molar ratio 10:1) | disrupts fibrils by decreasing β-sheet content at the N-terminal (D1–Y10) and C-terminal (D23–A42); two BSs were identified: (1) at the N-terminal (F4, H6, Y10, H13, H14, Q15, L17, and F19) and (2) at the C-terminal (N27, I31, I32, L34, and V36); dominant interactions include π-π stacking with F4, H6, Y10, H13, H14, and F19, as well as hydrophobic interactions with N27, I31, I32, L34 and V36; disrupts fibril-stabilizing contacts A2-V36 and F4-L34 at core-1, L17-I31 at core-2, and I32-M35 at core-3, along with salt bridges between L34 andA42 | [205] |
| AMBER14SB/TIP3P | (1) 500 (2) 500 (3) 3 × 500 | Aβ16–22/random pentamer | (1) 20 ATP (molar ratio 4:1) (2) 25 ATP (molar ratio 5:1) (3) 30 ATP (molar ratio 6:1) | with increasing ATP concentration, (i) β-sheet content decreased, while turn, bend, and coil contents increased, preventing oligomerization; (ii) the ATP-F π-π stacking disrupted the F-F interpeptide interactions; (iii) peptide-peptide H-bonds decreased, while ATP-peptide H-bonds increased; | [206] |
| AMBER99SB-ILDN/TIP3P AMBER-FB15/TIP3P-FB | 2 × 500 (for each FF) | Aβ16–22/random pentamer | 30 ATP (molar ratio 6:1) | β-sheet content decreases sharply; interpeptide F-F interactions are reduced; peptide-peptide H-bonds decrease abruptly, preventing β-sheets formation; | [206] |
| AMBER14SB/TIP3P AMBER99SB-ILDN/TIP3P AMBER-FB15/TIP3P-FB | 500 (for each FF) | Aβ16–22/dimer | 16 ATP (molar ratio 8:1) | unfavored dimerization | [206] |
| AMBER14SB/TIP3P AMBER99SB-ILDN/TIP3P AMBER-FB15/TIP3P-FB | 500 | Aβ16–22/prefibrillar pentamer | 150 ATP (molecular ration 30:1) | destabilizes β-sheet content and promotes disaggregation; completely disaggregates fibrils in the AMBER14SB and AMBER-FB15 FFs; a significant decreases inter-peptide H-bonds | [206] |
6.2. Serotonin (SER) and Melatonin (MEL)
6.3. Adenosine Triphosphate (ATP)
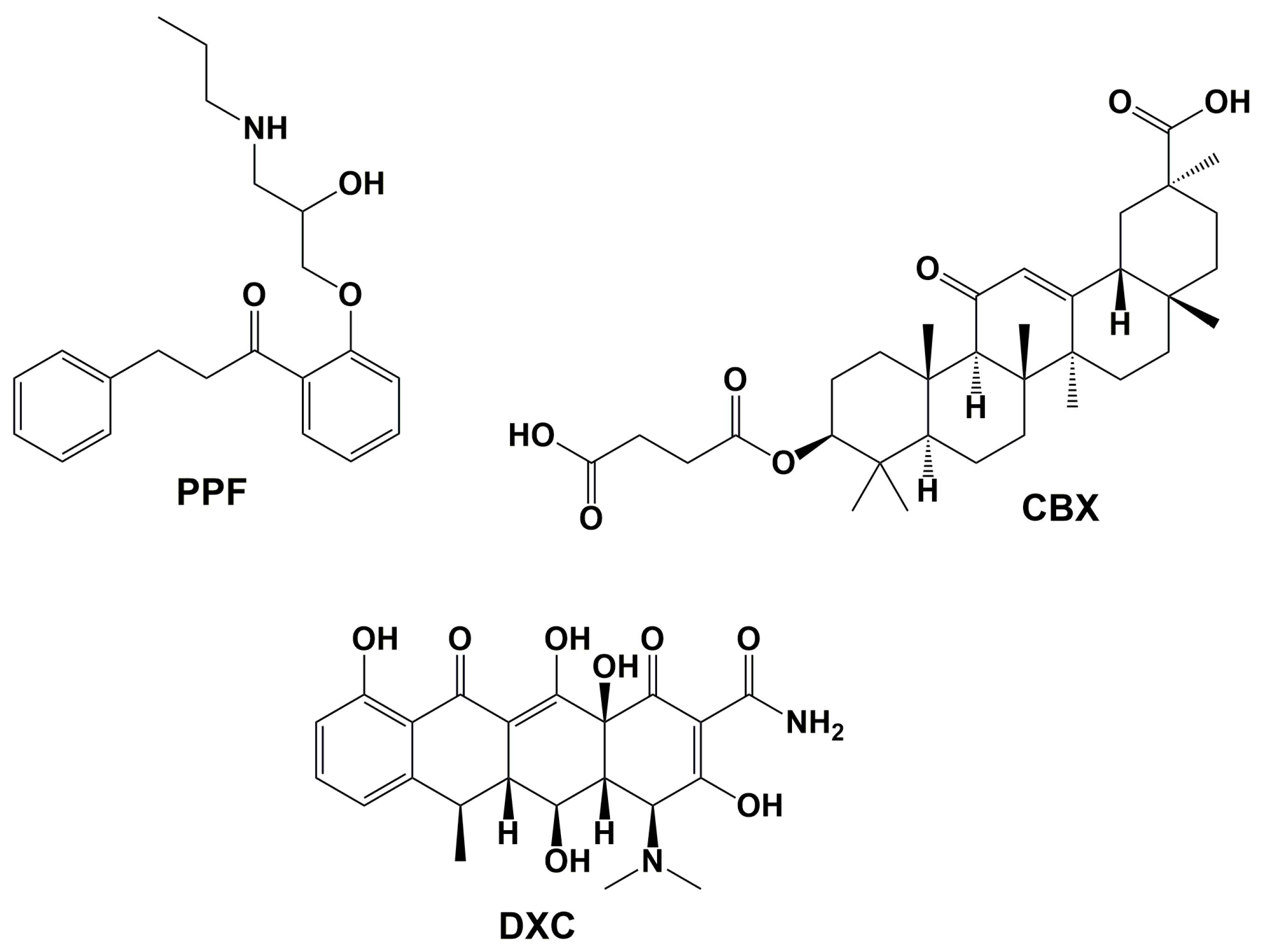
7. Repurposed Drugs Inhibiting Aβ
7.1. Propafenone (PPF)
7.2. Carbenoxolone (CBX)
7.3. Doxycycline (DXC)
| FF/Water Model | Duration per System, ns | Aβ Length/PDB ID/Type (Monomer/Dimer/ (Proto-)Fibril) | Inhibitor | Main Findings | Ref. |
|---|---|---|---|---|---|
| GROMOS96 43a1/SPC | 4 × 25 | Aβ9–40/2LMN/dodecamer Aβ1–40/2M4J/nonamer | PPF | Mainly forms contacts with hydrophobic residues; β-content decreases; | [222] |
| GROMOS96/SPC | 10 | (1) Aβ1–42/1IYT/monomer (2)Aβ17–42/2BEG/pentamer, protofibril | CBX | (1) reduces α-helix and β-sheet secondary structure; increases unstructured contend; forms contacts with F4, R5, H6, Y10, V12, H14, Q15, K16 V18, F19, and A30; forms H-bonds with R5, Q15, and F4; (2) reduces β-sheet secondary structure; increases unstructured content; forms contacts with L17, V18, F19, F20, A21, D23, K38, L34, V36, V40, and I32; forms H-bonds with F19 and D23; disrupts the salt bridge between D23 and K38. | [228] |
| aMD/ff14SB/TIP3P | 3 × 1000 (1 µs) | (1) Aβ11–42/2MXU/pentamer (2) Aβ1–42/5OQV/pentamer | 5 DXC (molar ratio 1:1) | (1) destabilizes the hydrophobic core (N15-A30); three main BSs: (i) near the M35 side chain; (ii) between I32 and L34; and (iii) between L17 and F19; (2) two BSs were identified: (i) near E1, V39, and I41; and (ii) at K16, V18, and F20. | [244] |
8. Mechanisms of Aβ Inhibition in Drug Discovery
9. Challenges and Perspectives in MD Simulations
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations and Acronyms
| aa | amino acids |
| AD | Alzheimer’s disease |
| AICD | APP intracellular domain |
| aMD | accelerated molecular dynamics |
| APP | amyloid-β (A4) precursor protein |
| ATP | adenosine triphosphate |
| BACE1 | β—secretase 1, β- site amyloid precursor protein (APP) cleavaging enzyme 1 |
| BBB | blood–brain barrier |
| BS | binding site |
| CBX | Carbenoxolone |
| CG | coarse-grained |
| CGM | coarse-grained model |
| cMD | classical/conventional molecular dynamics |
| CTF | C-terminal fragment of APP |
| DA | dopamine |
| FAD | familial Alzheimer’s disease |
| FF | force field |
| IDP | intrinsically disordered proteins |
| LMW | low molecular weight |
| LMWO | low molecular weight oligomers |
| MD | molecular dynamics |
| MEL | melatonin |
| NE | norepinephrine |
| NMR | nuclear magnetic resonance |
| PPF | Propafenone |
| RE | replica exchange |
| REM | replica exchange method |
| REMD | replica exchange molecular dynamics |
| REXAMD | replica exchange accelerated molecular dynamics |
| ROS | reactive oxygen species |
| SAD | sporadic Alzheimer’s disease |
| SER | serotonin |
| SPC | simple point charge water model |
| STDR | simulated tempering distributed replica |
| TIP3P | transferable intermolecular potential 3-point water model |
| TMD | transmembrane domain |
References
- GBD 2019 Collaborators Global Mortality from Dementia: Application of a New Method and Results from the Global Burden of Disease Study 2019. A&D Transl. Res. Clin. Interv. 2021, 7, e12200. [CrossRef]
- World Health Organization. Dementia. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 20 March 2023).
- Ghiso, J.; Frangione, B. Amyloidosis and Alzheimer’s Disease. Adv. Drug Deliv. Rev. 2002, 54, 1539–1551. [Google Scholar] [CrossRef]
- Hamley, I.W. The Amyloid Beta Peptide: A Chemist’s Perspective. Role in Alzheimer’s and Fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef] [PubMed]
- Cvetković-Dožić, D.; Skender-Gazibara, M.; Dožić, S. Neuropathological Hallmarks of Alzheimer’s Disease. Arch. Oncol. 2001, 9, 195–199. [Google Scholar]
- Zhang, W.; Pei, J.; Lai, L. Computational Multitarget Drug Design. J. Chem. Inf. Model. 2017, 57, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Bodea, L.-G.; Martinez-Marmol, R.; Meunier, F.A.; Götz, J. Amyloid-β and Tau Complexity—Towards Improved Biomarkers and Targeted Therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T. On Neurodegenerative Diseases, Models, and Treatment Strategies: Lessons Learned and Lessons Forgotten a Generation Following the Cholinergic Hypothesis. Exp. Neurol. 2000, 163, 495–529. [Google Scholar] [CrossRef]
- Terry, A.V.; Buccafusco, J.J. The Cholinergic Hypothesis of Age and Alzheimer’s Disease-Related Cognitive Deficits: Recent Challenges and Their Implications for Novel Drug Development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Winek, K.; Soreq, H.; Meisel, A. Regulators of Cholinergic Signaling in Disorders of the Central Nervous System. J. Neurochem. 2021, 158, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s Disease Hypothesis and Related Therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The Complexity of Tau in Alzheimer’s Disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Kabir, M.T.; Rahman, M.S.; Behl, T.; Jeandet, P.; Ashraf, G.M.; Najda, A.; Bin-Jumah, M.N.; El-Seedi, H.R.; Abdel-Daim, M.M. Revisiting the Amyloid Cascade Hypothesis: From Anti-Aβ Therapeutics to Auspicious New Ways for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 5858. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau Pathology Is an Initiating Factor in Sporadic Alzheimer’s Disease. Alzheimer’s & Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Ando, K.; Laporte, V.; Dedecker, R.; Suain, V.; Authelet, M.; Héraud, C.; Pierrot, N.; Yilmaz, Z.; Octave, J.-N.; et al. Lack of Tau Proteins Rescues Neuronal Cell Death and Decreases Amyloidogenic Processing of APP in APP/PS1 Mice. Am. J. Pathol. 2012, 181, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.-M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s Disease–Linked Presenilin 1 Variants Elevate Aβ1–42/1–40 Ratio In Vitro and In Vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.T.; Uddin, M.S.; Setu, J.R.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Exploring the Role of PSEN Mutations in the Pathogenesis of Alzheimer’s Disease. Neurotox. Res. 2020, 38, 833–849. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-Avidity Binding to Beta-Amyloid and Increased Frequency of Type 4 Allele in Late-Onset Familial Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; St. George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of Apolipoprotein E Allele Ε4 with Late-onset Familial and Sporadic Alzheimer’s Disease. Neurology 1993, 43, 1467. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. Thirty Years of Alzheimer’s Disease Genetics: The Implications of Systematic Meta-Analyses. Nat. Rev. Neurosci. 2008, 9, 768–778. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS β-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Yang, L.-B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.-L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated β-Secretase Expression and Enzymatic Activity Detected in Sporadic Alzheimer Disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef]
- Dawkins, E.; Small, D.H. Insights into the Physiological Function of the β-amyloid Precursor Protein: Beyond Alzheimer’s Disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef]
- Halim, A.; Brinkmalm, G.; Rüetschi, U.; Westman-Brinkmalm, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Larson, G.; Nilsson, J. Site-Specific Characterization of Threonine, Serine, and Tyrosine Glycosylations of Amyloid Precursor Protein/Amyloid β-Peptides in Human Cerebrospinal Fluid. Proc. Natl. Acad. Sci. USA 2011, 108, 11848–11853. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Thompson, R.; Zhang, H.; Xu, H. APP Processing in Alzheimer’s Disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef]
- Zerr, I. (Ed.) Understanding Alzheimer’s Disease; InTech: London, UK, 2013; ISBN 978-953-51-1009-5. [Google Scholar]
- Chen, G.; Xu, T.; Yan, Y.; Zhou, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Gandy, S. The Role of Cerebral Amyloid β Accumulation in Common Forms of Alzheimer Disease. J. Clin. Investig. 2005, 115, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid Precursor Protein Processing and Bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Urban, A.S.; Pavlov, K.V.; Kamynina, A.V.; Okhrimenko, I.S.; Arseniev, A.S.; Bocharov, E.V. Structural Studies Providing Insights into Production and Conformational Behavior of Amyloid-β Peptide Associated with Alzheimer’s Disease Development. Molecules 2021, 26, 2897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; McLaughlin, R.; Goodyer, C.; LeBlanc, A. Selective Cytotoxicity of Intracellular Amyloid β Peptide1–42 through P53 and Bax in Cultured Primary Human Neurons. J. Cell Biol. 2002, 156, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.C.; Kulig, W.; Poojari, C.; Rog, T.; Strodel, B. Physiologically-Relevant Levels of Sphingomyelin, but Not GM1, Induces a β-Sheet-Rich Structure in the Amyloid-β(1-42) Monomer. Biochim. Biophys. Acta (BBA)-Biomembr. 2018, 1860, 1709–1720. [Google Scholar] [CrossRef]
- Gargari, S.A.; Barzegar, A. Simulations on the Dual Effects of Flavonoids as Suppressors of Aβ42 Fibrillogenesis and Destabilizers of Mature Fibrils. Sci. Rep. 2020, 10, 16636. [Google Scholar] [CrossRef]
- Du, W.-J.; Guo, J.-J.; Gao, M.-T.; Hu, S.-Q.; Dong, X.-Y.; Han, Y.-F.; Liu, F.-F.; Jiang, S.; Sun, Y. Brazilin Inhibits Amyloid β-Protein Fibrillogenesis, Remodels Amyloid Fibrils and Reduces Amyloid Cytotoxicity. Sci. Rep. 2015, 5, 7992. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Aucoin, D.; Ahmed, M.; Ziliox, M.; Van Nostrand, W.E.; Smith, S.O. Capping of Aβ42 Oligomers by Small Molecule Inhibitors. Biochemistry 2014, 53, 7893–7903. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, R. The Toxicity and Polymorphism of β-Amyloid Oligomers. Int. J. Mol. Sci. 2020, 21, 4477. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of Amyloid-β42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [PubMed]
- Linse, S. Monomer-Dependent Secondary Nucleation in Amyloid Formation. Biophys. Rev. 2017, 9, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Scheidt, T.; Łapińska, U.; Kumita, J.R.; Whiten, D.R.; Klenerman, D.; Wilson, M.R.; Cohen, S.I.A.; Linse, S.; Vendruscolo, M.; Dobson, C.M.; et al. Secondary Nucleation and Elongation Occur at Different Sites on Alzheimer’s Amyloid-β Aggregates. Sci. Adv. 2019, 5, eaau3112. [Google Scholar] [CrossRef]
- Ferrone, F.A.; Hofrichter, J.; Eaton, W.A. Kinetics of Sickle Hemoglobin Polymerization. J. Mol. Biol. 1985, 183, 611–631. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.A.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Nucleated Polymerization with Secondary Pathways. II. Determination of Self-Consistent Solutions to Growth Processes Described by Non-Linear Master Equations. J. Chem. Phys. 2011, 135, 065106. [Google Scholar] [CrossRef] [PubMed]
- Tabaton, M.; Nunzi, M.G.; Xue, R.; Usiak, M.; Autiliogambetti, L.; Gambetti, P. Soluble Amyloid β-Protein Is a Marker of Alzheimer Amyloid in Brain But Not in Cerebrospinal Fluid. Biochem. Biophys. Res. Commun. 1994, 200, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Frackowiak, J.; Zoltowska, A.; Wisniewski, H.M. Non-Fibrillar β-Amyloid Protein Is Associated with Smooth Muscle Cells of Vessel Walls in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 1994, 53, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β Protein Dimers Isolated Directly from Alzheimer’s Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Chae, S.; Kim, J.H.; Barald, K.F.; Park, J.Y.; Lee, S.-H. Neurotoxic Amyloid Beta Oligomeric Assemblies Recreated in Microfluidic Platform with Interstitial Level of Slow Flow. Sci. Rep. 2013, 3, 1921. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Decourt, B.; Boumelhem, F.; Pope, E.D.; Shi, J.; Mari, Z.; Sabbagh, M.N. Critical Appraisal of Amyloid Lowering Agents in AD. Curr. Neurol. Neurosci. Rep. 2021, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A Critical Appraisal of Amyloid-β-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and Kinetic Basis for the Selectivity of Aducanumab for Aggregated Forms of Amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef]
- Wu, W.; Ji, Y.; Wang, Z.; Wu, X.; Li, J.; Gu, F.; Chen, Z.; Wang, Z. The FDA-Approved Anti-Amyloid-β Monoclonal Antibodies for the Treatment of Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Eur. J. Med. Res. 2023, 28, 544. [Google Scholar] [CrossRef]
- Pagano, K.; Tomaselli, S.; Molinari, H.; Ragona, L. Natural Compounds as Inhibitors of Aβ Peptide Aggregation: Chemical Requirements and Molecular Mechanisms. Front. Neurosci. 2020, 14, 619667. [Google Scholar] [CrossRef]
- Boopathi, S.; Poma, A.B.; Garduño-Juárez, R. An Overview of Several Inhibitors for Alzheimer’s Disease: Characterization and Failure. Int. J. Mol. Sci. 2021, 22, 10798. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liao, W.; Huang, D.; Ou, M.; Chen, T.; Wang, X.; Zhao, R.; Zhang, L.; Mei, L.; Liu, J.; et al. Current Strategies of Detecting Aβ Species and Inhibiting Aβ Aggregation: Status and Prospects. Coord. Chem. Rev. 2023, 495, 215375. [Google Scholar] [CrossRef]
- Zhao, Y.; Rao, P.P.N. Small Molecules N-Phenylbenzofuran-2-Carboxamide and N -Phenylbenzo[b]Thiophene-2-Carboxamide Promote Beta-Amyloid (Aβ42) Aggregation and Mitigate Neurotoxicity. ACS Chem. Neurosci. 2023, 14, 4185–4198. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Herbst, M.; Wiglenda, T.; Friedrich, R.P.; Boeddrich, A.; Schiele, F.; Kleckers, D.; Lopez Del Amo, J.M.; Grüning, B.A.; Wang, Q.; et al. Small-Molecule Conversion of Toxic Oligomers to Nontoxic β-Sheet–Rich Amyloid Fibrils. Nat. Chem. Biol. 2012, 8, 93–101. [Google Scholar] [CrossRef]
- Luo, J.; Yu, C.-H.; Yu, H.; Borstnar, R.; Kamerlin, S.C.L.; Gräslund, A.; Abrahams, J.P.; Wärmländer, S.K.T.S. Cellular Polyamines Promote Amyloid-Beta (Aβ) Peptide Fibrillation and Modulate the Aggregation Pathways. ACS Chem. Neurosci. 2013, 4, 454–462. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, N.E.C.; Do, T.D.; Tro, M.; LaPointe, N.E.; Feinstein, S.C.; Shea, J.-E.; Bowers, M.T. Opposing Effects of Cucurbit[7]Uril and 1,2,3,4,6-Penta-O-Galloyl-β-d-Glucopyranose on Amyloid β25–35 Assembly. ACS Chem. Neurosci. 2016, 7, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Sonzini, S.; Stanyon, H.F.; Scherman, O.A. Decreasing Amyloid Toxicity through an Increased Rate of Aggregation. Phys. Chem. Chem. Phys. 2017, 19, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Qian, C.; Yang, T.; Wang, Y.; Luo, J.; Zhang, C.; Wang, X.; Wang, X.; Guo, Z. Small Molecule-Mediated Co-Assembly of Amyloid-β Oligomers Reduces Neurotoxicity through Promoting Non-Fibrillar Aggregation. Chem. Sci. 2020, 11, 7158–7169. [Google Scholar] [CrossRef]
- Mohamed, T.; Gujral, S.S.; Rao, P.P.N. Tau Derived Hexapeptide AcPHF6 Promotes Beta-Amyloid (Aβ) Fibrillogenesis. ACS Chem. Neurosci. 2018, 9, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Bryngelson, J.D.; Onuchic, J.N.; Socci, N.D.; Wolynes, P.G. Funnels, Pathways, and the Energy Landscape of Protein Folding: A Synthesis. Proteins 1995, 21, 167–195. [Google Scholar] [CrossRef]
- Onuchic, J.N.; Wolynes, P.G. Theory of Protein Folding. Curr. Opin. Struct. Biol. 2004, 14, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; Chan, H.S. From Levinthal to Pathways to Funnels. Nat. Struct. Mol. Biol. 1997, 4, 10–19. [Google Scholar] [CrossRef]
- Reynolds, N.P.; Adamcik, J.; Berryman, J.T.; Handschin, S.; Zanjani, A.A.H.; Li, W.; Liu, K.; Zhang, A.; Mezzenga, R. Competition between Crystal and Fibril Formation in Molecular Mutations of Amyloidogenic Peptides. Nat. Commun. 2017, 8, 1338. [Google Scholar] [CrossRef]
- Adamcik, J.; Mezzenga, R. Amyloid Polymorphism in the Protein Folding and Aggregation Energy Landscape. Angew. Chem. Int. Ed. 2018, 57, 8370–8382. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.J.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fändrich, M.; et al. Half a Century of Amyloids: Past, Present and Future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef] [PubMed]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D Structure of Alzheimer’s Amyloid-β(1–42) Fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Rohou, A.; Lasker, K.; Yadav, J.K.; Schiene-Fischer, C.; Fändrich, M.; Grigorieff, N. Peptide Dimer Structure in an Aβ(1–42) Fibril Visualized with Cryo-EM. Proc. Natl. Acad. Sci. USA 2015, 112, 11858–11863. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1–42) Fibril Structure Illuminates Self-Recognition and Replication of Amyloid in Alzheimer’s Disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril Structure of Amyloid-β(1–42) by Cryo–Electron Microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-Resolution Structure of a Disease-Relevant Aβ(1–42) Amyloid Fibril. Proc. Natl. Acad. Sci. USA 2016, 113. [Google Scholar] [CrossRef] [PubMed]
- Eanes, E.D.; Glenner, G.G. X-RAY DIFFRACTION STUDIES ON AMYLOID FILAMENTS. J. Histochem. Cytochem. 1968, 16, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, D.A.; Abraham, C.; Selkoe, D.J. X-Ray Diffraction from Intraneuronal Paired Helical Filaments and Extraneuronal Amyloid Fibers in Alzheimer Disease Indicates Cross-Beta Conformation. Proc. Natl. Acad. Sci. USA 1986, 83, 503–507. [Google Scholar] [CrossRef]
- Sunde, M.; Blake, C. The Structure of Amyloid Fibrils by Electron Microscopy and X-Ray Diffraction. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1997; Volume 50, pp. 123–159. ISBN 978-0-12-034250-1. [Google Scholar]
- Serpell, L.C. Alzheimer’s Amyloid Fibrils: Structure and Assembly. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2000, 1502, 16–30. [Google Scholar] [CrossRef]
- Makin, O.S.; Serpell, L.C. Structures for Amyloid Fibrils. FEBS J. 2005, 272, 5950–5961. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural Conversion of Neurotoxic Amyloid-β1–42 Oligomers to Fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Härd, T. Protein Engineering to Stabilize Soluble Amyloid β-protein Aggregates for Structural and Functional Studies. FEBS J. 2011, 278, 3884–3892. [Google Scholar] [CrossRef]
- Kowalewski, T.; Holtzman, D.M. In Situ Atomic Force Microscopy Study of Alzheimer’s β-Amyloid Peptide on Different Substrates: New Insights into Mechanism of β-Sheet Formation. Proc. Natl. Acad. Sci. USA 1999, 96, 3688–3693. [Google Scholar] [CrossRef]
- Adler, J.; Scheidt, H.A.; Krüger, M.; Thomas, L.; Huster, D. Local Interactions Influence the Fibrillation Kinetics, Structure and Dynamics of Aβ(1–40) but Leave the General Fibril Structure Unchanged. Phys. Chem. Chem. Phys. 2014, 16, 7461–7471. [Google Scholar] [CrossRef]
- Korn, A.; McLennan, S.; Adler, J.; Krueger, M.; Surendran, D.; Maiti, S.; Huster, D. Amyloid β (1–40) Toxicity Depends on the Molecular Contact between Phenylalanine 19 and Leucine 34. ACS Chem. Neurosci. 2018, 9, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Bonhommeau, S.; Talaga, D.; Hunel, J.; Cullin, C.; Lecomte, S. Tip-Enhanced Raman Spectroscopy to Distinguish Toxic Oligomers from Aβ 1–42 Fibrils at the Nanometer Scale. Angew. Chem. Int. Ed. 2017, 56, 1771–1774. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Yau, W.-M.; Luo, Y.; Mattson, M.P.; Tycko, R. Antiparallel β-Sheet Architecture in Iowa-Mutant β-Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2012, 109, 4443–4448. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Tran, J.; Jiang, L.; Guo, Z. A New Structural Model of Alzheimer’s Aβ42 Fibrils Based on Electron Paramagnetic Resonance Data and Rosetta Modeling. J. Struct. Biol. 2016, 194, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Paparcone, R.; Pires, M.A.; Buehler, M.J. Mutations Alter the Geometry and Mechanical Properties of Alzheimer’s Aβ(1−40) Amyloid Fibrils. Biochemistry 2010, 49, 8967–8977. [Google Scholar] [CrossRef]
- Petkova, A.T.; Yau, W.-M.; Tycko, R. Experimental Constraints on Quaternary Structure in Alzheimer’s β-Amyloid Fibrils. Biochemistry 2006, 45, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.-M.; Tycko, R. Molecular Structural Basis for Polymorphism in Alzheimer’s β-Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [PubMed]
- Friedemann, M.; Helk, E.; Tiiman, A.; Zovo, K.; Palumaa, P.; Tõugu, V. Effect of Methionine-35 Oxidation on the Aggregation of Amyloid-β Peptide. Biochem. Biophys. Rep. 2015, 3, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Gonnelli, L.; Luchinat, C.; Mao, J.; Nesi, A. A New Structural Model of Aβ40 Fibrils. J. Am. Chem. Soc. 2011, 133, 16013–16022. [Google Scholar] [CrossRef]
- Paravastu, A.K.; Petkova, A.T.; Tycko, R. Polymorphic Fibril Formation by Residues 10–40 of the Alzheimer’s β-Amyloid Peptide. Biophys. J. 2006, 90, 4618–4629. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A Structural Model for Alzheimer’s β-Amyloid Fibrils Based on Experimental Constraints from Solid State NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M. Molecular Dynamics Simulations of Biomolecules. Acc. Chem. Res. 2002, 35, 321–323. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular Dynamics Simulations of Biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and Testing of the GROMOS Force-Field Versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Luo, R.; Chen, H.-F. The IDP-Specific Force Field ff14IDPSFF Improves the Conformer Sampling of Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2017, 57, 1166–1178. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Wang, W.; Ye, W.; Ji, D.; Luo, R.; Chen, H. ff14IDPs Force Field Improving the Conformation Sampling of Intrinsically Disordered Proteins. Chem. Biol. Drug Des. 2017, 89, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, H.; Yang, S.; Luo, R.; Chen, H.-F. Well-Balanced Force Field Ff 03 CMAP for Folded and Disordered Proteins. J. Chem. Theory Comput. 2019, 15, 6769–6780. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Liu, H.; Song, D.; Zhang, Y.; Yang, S.; Luo, R.; Chen, H.-F. Extensive Tests and Evaluation of the CHARMM36IDPSFF Force Field for Intrinsically Disordered Proteins and Folded Proteins. Phys. Chem. Chem. Phys. 2019, 21, 21918–21931. [Google Scholar] [CrossRef] [PubMed]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a Molecular Dynamics Force Field for Both Folded and Disordered Protein States. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Ji, D.; Wang, W.; Luo, R.; Chen, H.-F. Test and Evaluation of ff99IDPs Force Field for Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2015, 55, 1021–1029. [Google Scholar] [CrossRef]
- Best, R.B.; Mittal, J. Protein Simulations with an Optimized Water Model: Cooperative Helix Formation and Temperature-Induced Unfolded State Collapse. J. Phys. Chem. B 2010, 114, 14916–14923. [Google Scholar] [CrossRef]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. How Robust Are Protein Folding Simulations with Respect to Force Field Parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef] [PubMed]
- Samantray, S.; Yin, F.; Kav, B.; Strodel, B. Different Force Fields Give Rise to Different Amyloid Aggregation Pathways in Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 6462–6475. [Google Scholar] [CrossRef]
- Rahman, M.U.; Rehman, A.U.; Liu, H.; Chen, H.-F. Comparison and Evaluation of Force Fields for Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2020, 60, 4912–4923. [Google Scholar] [CrossRef] [PubMed]
- Shabane, P.S.; Izadi, S.; Onufriev, A.V. General Purpose Water Model Can Improve Atomistic Simulations of Intrinsically Disordered Proteins. J. Chem. Theory Comput. 2019, 15, 2620–2634. [Google Scholar] [CrossRef] [PubMed]
- Amber24. Available online: https://ambermd.org/AmberModels_proteins.php (accessed on 19 February 2025).
- Carballo-Pacheco, M.; Strodel, B. Comparison of Force Fields for Alzheimer’s A: A Case Study for Intrinsically Disordered Proteins. Protein Sci. 2017, 26, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Man, V.H.; He, X.; Derreumaux, P.; Ji, B.; Xie, X.-Q.; Nguyen, P.H.; Wang, J. Effects of All-Atom Molecular Mechanics Force Fields on Amyloid Peptide Assembly: The Case of Aβ16–22 Dimer. J. Chem. Theory Comput. 2019, 15, 1440–1452. [Google Scholar] [CrossRef] [PubMed]
- Krupa, P.; Quoc Huy, P.D.; Li, M.S. Properties of Monomeric Aβ42 Probed by Different Sampling Methods and Force Fields: Role of Energy Components. J. Chem. Phys. 2019, 151, 055101. [Google Scholar] [CrossRef]
- Carballo-Pacheco, M.; Ismail, A.E.; Strodel, B. On the Applicability of Force Fields To Study the Aggregation of Amyloidogenic Peptides Using Molecular Dynamics Simulations. J. Chem. Theory Comput. 2018, 14, 6063–6075. [Google Scholar] [CrossRef] [PubMed]
- Minary, P.; Tuckerman, M.E.; Martyna, G.J. Long Time Molecular Dynamics for Enhanced Conformational Sampling in Biomolecular Systems. Phys. Rev. Lett. 2004, 93, 150201. [Google Scholar] [CrossRef]
- Lei, H.; Duan, Y. Improved Sampling Methods for Molecular Simulation. Curr. Opin. Struct. Biol. 2007, 17, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Darve, E.; Rodríguez-Gómez, D.; Pohorille, A. Adaptive Biasing Force Method for Scalar and Vector Free Energy Calculations. J. Chem. Phys. 2008, 128, 144120. [Google Scholar] [CrossRef]
- Perez, D.; Uberuaga, B.P.; Shim, Y.; Amar, J.G.; Voter, A.F. Chapter 4 Accelerated Molecular Dynamics Methods: Introduction and Recent Developments. In Annual Reports in Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2009; Volume 5, pp. 79–98. ISBN 978-0-444-53359-3. [Google Scholar]
- Zuckerman, D.M. Equilibrium Sampling in Biomolecular Simulations. Annu. Rev. Biophys. 2011, 40, 41–62. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Mohammad, T.; Hasan, G.M.; Hassan, M.I. Advancements in Docking and Molecular Dynamics Simulations Towards Ligand-Receptor Interactions and Structure-Function Relationships. CTMC 2018, 18, 1755–1768. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Deuflhard, P.; Hermans, J.; Leimkuhler, B.; Mark, A.E.; Reich, S.; Skeel, R.D. (Eds.) Computational Molecular Dynamics: Challenges, Methods, Ideas: Proceedings of the 2nd International Symposium on Algorithms for Macromolecular Modelling, Berlin, May 21–24, 1997; Lecture Notes in Computational Science and Engineering; Springer: Berlin/Heidelberg, Germany, 1999; Volume 4, ISBN 978-3-540-63242-9. [Google Scholar]
- Tuckerman, M.E.; Martyna, G.J. Understanding Modern Molecular Dynamics: Techniques and Applications. J. Phys. Chem. B 2000, 104, 159–178. [Google Scholar] [CrossRef]
- Becker, O.M.; MacKerell, A.D., Jr.; Roux, B.; Watanabe, M. Chapter 3: Dynamics Methods. In Computational Biochemistry and Biophysics; Becker, O.M., Watanabe, M., Eds.; Marcel Dekker, Inc.: Basel, Switzerland, 2001; ISBN 0-8247-0455-X. [Google Scholar]
- Rauscher, S.; Neale, C.; Pomès, R. Simulated Tempering Distributed Replica Sampling, Virtual Replica Exchange, and Other Generalized-Ensemble Methods for Conformational Sampling. J. Chem. Theory Comput. 2009, 5, 2640–2662. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, J.; Hospital, A.; Goñi, R.; Orozco, M. Molecular Dynamics Simulations: Advances and Applications. AABC 2015, 37–47. [Google Scholar] [CrossRef]
- Huang, X.; Bowman, G.R.; Pande, V.S. Convergence of Folding Free Energy Landscapes via Application of Enhanced Sampling Methods in a Distributed Computing Environment. J. Chem. Phys. 2008, 128, 205106. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, U.H.E. Parallel Tempering Algorithm for Conformational Studies of Biological Molecules. Chem. Phys. Lett. 1997, 281, 140–150. [Google Scholar] [CrossRef]
- Hansmann, U.H.E.; Okamoto, Y. New Monte Carlo Algorithms for Protein Folding. Curr. Opin. Struct. Biol. 1999, 9, 177–183. [Google Scholar] [CrossRef]
- Nagasima, T.; Sugita, Y.; Mitsutake, A.; Okamoto, Y. Generalized-Ensemble Simulations of Spin Systems and Protein Systems. Comput. Phys. Commun. 2002, 146, 69–76. [Google Scholar] [CrossRef]
- Lyubartsev, A.P.; Martsinovski, A.A.; Shevkunov, S.V.; Vorontsov-Velyaminov, P.N. New Approach to Monte Carlo Calculation of the Free Energy: Method of Expanded Ensembles. J. Chem. Phys. 1992, 96, 1776–1783. [Google Scholar] [CrossRef]
- Marinari, E.; Parisi, G. Simulated Tempering: A New Monte Carlo Scheme. Europhys. Lett. 1992, 19, 451–458. [Google Scholar] [CrossRef]
- Swendsen, R.H.; Wang, J.-S. Replica Monte Carlo Simulation of Spin-Glasses. Phys. Rev. Lett. 1986, 57, 2607–2609. [Google Scholar] [CrossRef]
- Geyer, C.J. Markov Chain Monte Carlo Maximum Likelihood. In Proceedings of the 23rd Symposium on the Interface, Washington, DC, USA, 21–24 April 1991; pp. 156–163. [Google Scholar]
- Geyer, C.J.; Thompson, E.A. Annealing Markov Chain Monte Carlo with Applications to Ancestral Inference. J. Am. Stat. Assoc. 1995, 90, 909–920. [Google Scholar] [CrossRef]
- Tesi, M.C.; Janse Van Rensburg, E.J.; Orlandini, E.; Whittington, S.G. Monte Carlo Study of the Interacting Self-Avoiding Walk Model in Three Dimensions. J. Stat. Phys. 1996, 82, 155–181. [Google Scholar] [CrossRef]
- Hukushima, K.; Nemoto, K. Exchange Monte Carlo Method and Application to Spin Glass Simulations. J. Phys. Soc. Jpn. 1996, 65, 1604–1608. [Google Scholar] [CrossRef]
- Marinari, E.; Parisi, G.; Ruiz-Lorenzo, J.J. Numerical Simulations of Spin Glass Systems. In Series on Directions in Condensed Matter Physics; World Scientific: Singapore, 1997; Volume 12, pp. 59–98. ISBN 978-981-02-3183-5. [Google Scholar]
- Sugita, Y.; Okamoto, Y. Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Opps, S.B.; Schofield, J. Extended State-Space Monte Carlo Methods. Phys. Rev. E 2001, 63, 056701. [Google Scholar] [CrossRef]
- Kofke, D.A. On the Acceptance Probability of Replica-Exchange Monte Carlo Trials. J. Chem. Phys. 2002, 117, 6911–6914. [Google Scholar] [CrossRef]
- Rhee, Y.M.; Pande, V.S. Multiplexed-Replica Exchange Molecular Dynamics Method for Protein Folding Simulation. Biophys. J. 2003, 84, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Paschek, D.; García, A.E. Reversible Temperature and Pressure Denaturation of a Protein Fragment: A Replica Exchange Molecular Dynamics Simulation Study. Phys. Rev. Lett. 2004, 93, 238105. [Google Scholar] [CrossRef] [PubMed]
- Rathore, N.; Chopra, M.; De Pablo, J.J. Optimal Allocation of Replicas in Parallel Tempering Simulations. J. Chem. Phys. 2005, 122, 024111. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica Exchange with Solute Tempering: A Method for Sampling Biological Systems in Explicit Water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef]
- Li, H.; Li, G.; Berg, B.A.; Yang, W. Finite Reservoir Replica Exchange to Enhance Canonical Sampling in Rugged Energy Surfaces. J. Chem. Phys. 2006, 125, 144902. [Google Scholar] [CrossRef]
- Trebst, S.; Troyer, M.; Hansmann, U.H.E. Optimized Parallel Tempering Simulations of Proteins. J. Chem. Phys. 2006, 124, 174903. [Google Scholar] [CrossRef] [PubMed]
- Brenner, P.; Sweet, C.R.; VonHandorf, D.; Izaguirre, J.A. Accelerating the Replica Exchange Method through an Efficient All-Pairs Exchange. J. Chem. Phys. 2007, 126, 074103. [Google Scholar] [CrossRef] [PubMed]
- Okur, A.; Roe, D.R.; Cui, G.; Hornak, V.; Simmerling, C. Improving Convergence of Replica-Exchange Simulations through Coupling to a High-Temperature Structure Reservoir. J. Chem. Theory Comput. 2007, 3, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Rick, S.W. Replica Exchange with Dynamical Scaling. J. Chem. Phys. 2007, 126, 054102. [Google Scholar] [CrossRef]
- Ruscio, J.Z.; Fawzi, N.L.; Head-Gordon, T. How Hot? Systematic Convergence of the Replica Exchange Method Using Multiple Reservoirs. J. Comput. Chem. 2010, 31, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Rauscher, S.; Pomès, R. Simulated Tempering Distributed Replica Sampling: A Practical Guide to Enhanced Conformational Sampling. J. Phys.Conf. Ser. 2010, 256, 012011. [Google Scholar] [CrossRef]
- Chen, J.; Im, W.; Brooks, C.L. Application of Torsion Angle Molecular Dynamics for Efficient Sampling of Protein Conformations. J. Comput. Chem. 2005, 26, 1565–1578. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.; Lavery, R.; Mousseau, N.; Zakrzewska, K.; Derreumaux, P. ARTIST: An Activated Method in Internal Coordinate Space for Sampling Protein Energy Landscapes. Proteins 2006, 63, 967–975. [Google Scholar] [CrossRef]
- Trylska, J.; Tozzini, V.; McCammon, J.A. Exploring Global Motions and Correlations in the Ribosome. Biophys. J. 2005, 89, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.-J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Brooks, C.L.; Khandogin, J. Recent Advances in Implicit Solvent-Based Methods for Biomolecular Simulations. Curr. Opin. Struct. Biol. 2008, 18, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Grubmüller, H. Predicting Slow Structural Transitions in Macromolecular Systems: Conformational Flooding. Phys. Rev. E 1995, 52, 2893–2906. [Google Scholar] [CrossRef] [PubMed]
- Lange, O.F.; Schäfer, L.V.; Grubmüller, H. Flooding in GROMACS: Accelerated Barrier Crossings in Molecular Dynamics. J. Comput. Chem. 2006, 27, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Huber, T.; Torda, A.E.; Van Gunsteren, W.F. Local Elevation: A Method for Improving the Searching Properties of Molecular Dynamics Simulation. J. Comput.-Aided Mol. Des. 1994, 8, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping Free-Energy Minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef]
- Bussi, G.; Laio, A.; Parrinello, M. Equilibrium Free Energies from Nonequilibrium Metadynamics. Phys. Rev. Lett. 2006, 96, 090601. [Google Scholar] [CrossRef] [PubMed]
- Leone, V.; Marinelli, F.; Carloni, P.; Parrinello, M. Targeting Biomolecular Flexibility with Metadynamics. Curr. Opin. Struct. Biol. 2010, 20, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Darve, E.; Pohorille, A. Calculating Free Energies Using Average Force. J. Chem. Phys. 2001, 115, 9169–9183. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Beutler, T.C.; Van Gunsteren, W.F. The Computation of a Potential of Mean Force: Choice of the Biasing Potential in the Umbrella Sampling Technique. J. Chem. Phys. 1994, 100, 1492–1497. [Google Scholar] [CrossRef]
- Voter, A.F. A Method for Accelerating the Molecular Dynamics Simulation of Infrequent Events. J. Chem. Phys. 1997, 106, 4665–4677. [Google Scholar] [CrossRef]
- Voter, A.F. Hyperdynamics: Accelerated Molecular Dynamics of Infrequent Events. Phys. Rev. Lett. 1997, 78, 3908–3911. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated Molecular Dynamics: A Promising and Efficient Simulation Method for Biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Hamelberg, D. A Statistical Analysis of the Precision of Reweighting-Based Simulations. J. Chem. Phys. 2008, 129, 034103. [Google Scholar] [CrossRef] [PubMed]
- Fajer, M.; Hamelberg, D.; McCammon, J.A. Replica-Exchange Accelerated Molecular Dynamics (REXAMD) Applied to Thermodynamic Integration. J. Chem. Theory Comput. 2008, 4, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- Wereszczynski, J.; McCammon, J.A. Using Selectively Applied Accelerated Molecular Dynamics to Enhance Free Energy Calculations. J. Chem. Theory Comput. 2010, 6, 3285–3292. [Google Scholar] [CrossRef] [PubMed]
- Sinko, W.; De Oliveira, C.A.F.; Pierce, L.C.T.; McCammon, J.A. Protecting High Energy Barriers: A New Equation to Regulate Boost Energy in Accelerated Molecular Dynamics Simulations. J. Chem. Theory Comput. 2012, 8, 17–23. [Google Scholar] [CrossRef]
- Wang, Y.; Harrison, C.B.; Schulten, K.; McCammon, J.A. Implementation of Accelerated Molecular Dynamics in NAMD. Comput. Sci. Disc. 2011, 4, 015002. [Google Scholar] [CrossRef] [PubMed]
- Pawnikar, S.; Bhattarai, A.; Wang, J.; Miao, Y. Binding Analysis Using Accelerated Molecular Dynamics Simulations and Future Perspectives. AABC 2022, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced Sampling Techniques in Molecular Dynamics Simulations of Biological Systems. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Harpole, T.J.; Delemotte, L. Conformational Landscapes of Membrane Proteins Delineated by Enhanced Sampling Molecular Dynamics Simulations. Biochim. Biophys. Acta (BBA) Biomembr. 2018, 1860, 909–926. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, G.S.; Stegailov, V.V. Efficiency of Classical Molecular Dynamics Algorithms on Supercomputers. Math. Models Comput. Simul. 2016, 8, 734–743. [Google Scholar] [CrossRef]
- Jung, J.; Kobayashi, C.; Kasahara, K.; Tan, C.; Kuroda, A.; Minami, K.; Ishiduki, S.; Nishiki, T.; Inoue, H.; Ishikawa, Y.; et al. New Parallel Computing Algorithm of Molecular Dynamics for Extremely Huge Scale Biological Systems. J. Comput. Chem. 2021, 42, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Lu, D.; Yan, Y.; Hu, S.; Liu, R.; Tan, G.; Sun, N.; Jiang, W.; Liu, L.; Chen, Y.; et al. Extending the Limit of Molecular Dynamics with Ab Initio Accuracy to 10 Billion Atoms. In Proceedings of the 27th ACM SIGPLAN Symposium on Principles and Practice of Parallel Programming, Seoul, Republic of Korea, 2–6 April 2022; ACM: New York, NY, USA, 2022; pp. 205–218. [Google Scholar]
- Shaw, D.E.; Adams, P.J.; Azaria, A.; Bank, J.A.; Batson, B.; Bell, A.; Bergdorf, M.; Bhatt, J.; Butts, J.A.; Correia, T.; et al. Anton 3: Twenty Microseconds of Molecular Dynamics Simulation before Lunch. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis. St. Louis, MO, USA, 14–19 November 2021; ACM: New York, NY, USA, 2021; pp. 1–11. [Google Scholar]
- Top500 The List. Available online: https://top500.org/lists/top500/2023/06/ (accessed on 20 June 2023).
- Lumi. Available online: https://www.lumi-supercomputer.eu/efficient-molecular-dynamics-simulations-on-lumi/ (accessed on 5 February 2025).
- Shirts, M.; Pande, V.S. Screen Savers of the World Unite! Science 2000, 290, 1903–1904. [Google Scholar] [CrossRef] [PubMed]
- Folding@home. Available online: http://foldingathome.org (accessed on 6 February 2025).
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 Simulations Go Exascale to Predict Dramatic Spike Opening and Cryptic Pockets across the Proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]
- Lemkul, J.A.; Bevan, D.R. The Role of Molecular Simulations in the Development of Inhibitors of Amyloid β-Peptide Aggregation for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2012, 3, 845–856. [Google Scholar] [CrossRef]
- Nasica-Labouze, J.; Nguyen, P.H.; Sterpone, F.; Berthoumieu, O.; Buchete, N.-V.; Coté, S.; De Simone, A.; Doig, A.J.; Faller, P.; Garcia, A.; et al. Amyloid β Protein and Alzheimer’s Disease: When Computer Simulations Complement Experimental Studies. Chem. Rev. 2015, 115, 3518–3563. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.H.; Morales, L.G.F.; Basurto, J.C.; Hernández, M.C.R. Molecular Docking and Molecular Dynamics Simulation to Evaluate Compounds That Avoid the Amyloid Beta 1-42 Aggregation. In Computational Modeling of Drugs Against Alzheimer’s Disease; Roy, K., Ed.; Neuromethods; Springer: New York, NY, USA, 2018; Volume 132, pp. 229–248. ISBN 978-1-4939-7403-0. [Google Scholar]
- Low, K.J.Y.; Venkatraman, A.; Mehta, J.S.; Pervushin, K. Molecular Mechanisms of Amyloid Disaggregation. J. Adv. Res. 2022, 36, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.A. Dopamine, Learning and Motivation. Nat. Rev. Neurosci. 2004, 5, 483–494. [Google Scholar] [CrossRef]
- Berridge, C.W.; Waterhouse, B.D. The Locus Coeruleus–Noradrenergic System: Modulation of Behavioral State and State-Dependent Cognitive Processes. Brain Res. Rev. 2003, 42, 33–84. [Google Scholar] [CrossRef]
- Murchison, C.F.; Zhang, X.-Y.; Zhang, W.-P.; Ouyang, M.; Lee, A.; Thomas, S.A. A Distinct Role for Norepinephrine in Memory Retrieval. Cell 2004, 117, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Anti-Parkinsonian Agents Have Anti-Amyloidogenic Activity for Alzheimer’s β-Amyloid Fibrils in Vitro. Neurochem. Int. 2006, 48, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Huong, V.T.; Shimanouchi, T.; Shimauchi, N.; Yagi, H.; Umakoshi, H.; Goto, Y.; Kuboi, R. Catechol Derivatives Inhibit the Fibril Formation of Amyloid-β Peptides. J. Biosci. Bioeng. 2010, 109, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Tavanti, F.; Pedone, A.; Menziani, M.C. Computational Insight into the Effect of Natural Compounds on the Destabilization of Preformed Amyloid-β(1–40) Fibrils. Molecules 2018, 23, 1320. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Zhan, C.; Lao, Z.; Li, F.; Dong, X.; Wei, G. A Comprehensive Insight into the Mechanisms of Dopamine in Disrupting Aβ Protofibrils and Inhibiting Aβ Aggregation. ACS Chem. Neurosci. 2021, 12, 4007–4019. [Google Scholar] [CrossRef]
- Zou, Y.; Qian, Z.; Chen, Y.; Qian, H.; Wei, G.; Zhang, Q. Norepinephrine Inhibits Alzheimer’s Amyloid-β Peptide Aggregation and Destabilizes Amyloid-β Protofibrils: A Molecular Dynamics Simulation Study. ACS Chem. Neurosci. 2019, 10, 1585–1594. [Google Scholar] [CrossRef]
- Gao, D.; Wan, J.; Zou, Y.; Gong, Y.; Dong, X.; Xu, Z.; Tang, J.; Wei, G.; Zhang, Q. The Destructive Mechanism of Aβ1–42 Protofibrils by Norepinephrine Revealed via Molecular Dynamics Simulations. Phys. Chem. Chem. Phys. 2022, 24, 19827–19836. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Zhan, C.; Zou, Y.; Qian, Z.; Wei, G.; Zhang, Q. Serotonin and Melatonin Show Different Modes of Action on Aβ42 Protofibril Destabilization. ACS Chem. Neurosci. 2021, 12, 799–809. [Google Scholar] [CrossRef]
- Pal, S.; Paul, S. ATP Controls the Aggregation of Aβ16–22 Peptides. J. Phys. Chem. B 2020, 124, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.J.; Noristani, H.N.; Verkhratsky, A. The Serotonergic System in Ageing and Alzheimer’s Disease. Prog. Neurobiol. 2012, 99, 15–41. [Google Scholar] [CrossRef]
- Porter, R.J.; Lunn, B.S.; O’Brien, J.T. Effects of Acute Tryptophan Depletion on Cognitive Function in Alzheimer’s Disease and in the Healthy Elderly. Psychol. Med. 2003, 33, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Wester, P.; Backstrom, I.; Gottfries, J.; Oreland, L.; Stenstrom, A.; Winblad, B. The Regional Distribution of Dopamine and Serotonin Uptake and Transmitter Concentrations in the Human Brain. Neurochem. Int. 1987, 10, 445–450. [Google Scholar] [CrossRef]
- Hornedo-Ortega, R.; Da Costa, G.; Cerezo, A.B.; Troncoso, A.M.; Richard, T.; Garcia-Parrilla, M.C. In Vitro Effects of Serotonin, Melatonin, and Other Related Indole Compounds on Amyloid-β Kinetics and Neuroprotection. Mol. Nutr. Food Res. 2018, 62, 1700383. [Google Scholar] [CrossRef] [PubMed]
- Karasek, M. Melatonin, Human Aging, and Age-Related Diseases. Exp. Gerontol. 2004, 39, 1723–1729. [Google Scholar] [CrossRef]
- Sack, R.L.; Lewy, A.J.; Erb, D.L.; Vollmer, W.M.; Singer, C.M. Human Melatonin Production Decreases with Age. J. Pineal Res. 1986, 3, 379–388. [Google Scholar] [CrossRef]
- Skene, D. Melatonin Rhythmicity: Effect of Age and Alzheimer’s Disease. Exp. Gerontol. 2003, 38, 199–206. [Google Scholar] [CrossRef]
- Skene, D.J.; Vivien-Roels, B.; Sparks, D.L.; Hunsaker, J.C.; Pévet, P.; Ravid, D.; Swaab, D.F. Daily Variation in the Concentration of Melatonin and 5-Methoxytryptophol in the Human Pineal Gland: Effect of Age and Alzheimer’s Disease. Brain Res. 1990, 528, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Huang, Q.-X.; Yang, S.-S.; Chu, J.; Wang, J.-Z.; Tian, Q. Melatonin in Alzheimer’s Disease. Int. J. Mol. Sci. 2013, 14, 14575–14593. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, P.; Ren, L.; Hu, C.; Bi, J. Protective Effect of Melatonin on Soluble Aβ1–42-Induced Memory Impairment, Astrogliosis, and Synaptic Dysfunction via the Musashi1/Notch1/Hes1 Signaling Pathway in the Rat Hippocampus. Alzheimer’s Res. Ther. 2016, 8, 40. [Google Scholar] [CrossRef]
- Poeggeler, B.; Miravalle, L.; Zagorski, M.G.; Wisniewski, T.; Chyan, Y.-J.; Zhang, Y.; Shao, H.; Bryant-Thomas, T.; Vidal, R.; Frangione, B.; et al. Melatonin Reverses the Profibrillogenic Activity of Apolipoprotein E4 on the Alzheimer Amyloid Aβ Peptide. Biochemistry 2001, 40, 14995–15001. [Google Scholar] [CrossRef] [PubMed]
- Pappolla, M.; Bozner, P.; Soto, C.; Shao, H.; Robakis, N.K.; Zagorski, M.; Frangione, B.; Ghiso, J. Inhibition of Alzheimer β-Fibrillogenesis by Melatonin. J. Biol. Chem. 1998, 273, 7185–7188. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a Biological Hydrotrope. Science 2017, 356, 753–756. [Google Scholar] [CrossRef]
- Kuramochi, M.; Nakamura, M.; Takahashi, H.; Komoriya, T.; Takita, T.; Pham, N.T.K.; Yasukawa, K.; Yoshimune, K. Adenosine Triphosphate Induces Amorphous Aggregation of Amyloid β by Increasing Aβ Dynamics. Sci. Rep. 2024, 14, 8134. [Google Scholar] [CrossRef] [PubMed]
- Coskuner, O.; Murray, I.V.J. Adenosine Triphosphate (ATP) Reduces Amyloid-β Protein Misfolding in Vitro. JAD 2014, 41, 561–574. [Google Scholar] [CrossRef]
- Ngo, S.T.; Fang, S.-T.; Huang, S.-H.; Chou, C.-L.; Huy, P.D.Q.; Li, M.S.; Chen, Y.-C. Anti-Arrhythmic Medication Propafenone a Potential Drug for Alzheimer’s Disease Inhibiting Aggregation of Aβ: In Silico and in Vitro Studies. J. Chem. Inf. Model. 2016, 56, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, G.R.; Swiergiel, A.H. Properties of Gap Junction Blockers and Their Behavioural, Cognitive and Electrophysiological Effects: Animal and Human Studies. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2009, 33, 181–198. [Google Scholar] [CrossRef]
- Rozental, R.; Srinivas, M.; Spray, D.C. How to Close a Gap Junction Channel: Efficacies and Potencies of Uncoupling Agents. In Connexin Methods and Protocols; Humana Press: Totowa, NJ, USA, 2000; Volume 154, pp. 447–476. ISBN 978-1-59259-043-8. [Google Scholar]
- Hellmich, H.L.; Rojo, D.R.; Micci, M.-A.; Sell, S.L.; Boone, D.R.; Crookshanks, J.M.; DeWitt, D.S.; Masel, B.E.; Prough, D.S. Pathway Analysis Reveals Common Pro-Survival Mechanisms of Metyrapone and Carbenoxolone after Traumatic Brain Injury. PLoS ONE 2013, 8, e53230. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.; Nehru, B. Inhibition of Neuroinflammation and Mitochondrial Dysfunctions by Carbenoxolone in the Rotenone Model of Parkinson’s Disease. Mol. Neurobiol. 2015, 51, 209–219. [Google Scholar] [CrossRef]
- Sandeep, T.C.; Yau, J.L.W.; MacLullich, A.M.J.; Noble, J.; Deary, I.J.; Walker, B.R.; Seckl, J.R. 11β-Hydroxysteroid Dehydrogenase Inhibition Improves Cognitive Function in Healthy Elderly Men and Type 2 Diabetics. Proc. Natl. Acad. Sci. USA 2004, 101, 6734–6739. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Nehru, B.; Saini, A. Inhibition of Alzheimer’s Amyloid-Beta Aggregation in-Vitro by Carbenoxolone: Insight into Mechanism of Action. Neurochem. Int. 2017, 108, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Bellotti, V.; Gianni, A.M.; Merlini, G. New Drug Therapy of Amyloidoses: Resorption of AL-Type Deposits with 4′-Iodo-4′-Deoxydoxorubicin. Blood 1995, 86, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Ascari, E.; Amboldi, N.; Bellotti, V.; Arbustini, E.; Perfetti, V.; Ferrari, M.; Zorzoli, I.; Marinone, M.G.; Garini, P. Interaction of the Anthracycline 4′-Iodo-4′-Deoxydoxorubicin with Amyloid Fibrils: Inhibition of Amyloidogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 2959–2963. [Google Scholar] [CrossRef]
- Palha, J.A.; Ballinari, D.; Amboldi, N.; Cardoso, I.; Fernandes, R.; Bellotti, V.; Merlini, G.; Saraiva, M.J. 4′-Iodo-4′-Deoxydoxorubicin Disrupts the Fibrillar Structure of Transthyretin Amyloid. Am. J. Pathol. 2000, 156, 1919–1925. [Google Scholar] [CrossRef]
- Tagliavini, F.; McArthur, R.A.; Canciani, B.; Giaccone, G.; Porro, M.; Bugiani, M.; Lievens, P.M.-J.; Bugiani, O.; Peri, E.; Dall’Ara, P.; et al. Effectiveness of Anthracycline Against Experimental Prion Disease in Syrian Hamsters. Science 1997, 276, 1119–1121. [Google Scholar] [CrossRef] [PubMed]
- Diomede, L.; Cassata, G.; Fiordaliso, F.; Salio, M.; Ami, D.; Natalello, A.; Doglia, S.M.; De Luigi, A.; Salmona, M. Tetracycline and Its Analogues Protect Caenorhabditis Elegans from β Amyloid-Induced Toxicity by Targeting Oligomers. Neurobiol. Dis. 2010, 40, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Santamaria, G.; La Vitola, P.; Brandi, E.; Grandi, F.; Viscomi, A.R.; Beeg, M.; Gobbi, M.; Salmona, M.; Ottonello, S.; et al. Doxycycline Counteracts Neuroinflammation Restoring Memory in Alzheimer’s Disease Mouse Models. Neurobiol. Aging 2018, 70, 128–139. [Google Scholar] [CrossRef]
- Forloni, G.; Colombo, L.; Girola, L.; Tagliavini, F.; Salmona, M. Anti-amyloidogenic Activity of Tetracyclines: Studies in Vitro. FEBS Lett. 2001, 487, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, I.; Merlini, G.; Saraiva, M.J. 4 ′-iodo-4′-Deoxydoxorubicin and Tetracyclines Disrupt Transthyretin Amyloid Fibrils in Vitro Producing Noncytotoxic Species: Screening for TTR Fibril Disrupters. FASEB J. 2003, 17, 803–809. [Google Scholar] [CrossRef]
- Cardoso, I.; Saraiva, M.J. Doxycycline Disrupts Transthyretin Amyloid: Evidence from Studies in a FAP Transgenic Mice Model. FASEB J. 2006, 20, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Obici, L.; Cortese, A.; Lozza, A.; Lucchetti, J.; Gobbi, M.; Palladini, G.; Perlini, S.; Saraiva, M.J.; Merlini, G. Doxycycline plus Tauroursodeoxycholic Acid for Transthyretin Amyloidosis: A Phase II Study. Amyloid 2012, 19, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Stoilova, T.; Colombo, L.; Forloni, G.; Tagliavini, F.; Salmona, M. A New Face for Old Antibiotics: Tetracyclines in Treatment of Amyloidoses. J. Med. Chem. 2013, 56, 5987–6006. [Google Scholar] [CrossRef]
- Airoldi, C.; Colombo, L.; Manzoni, C.; Sironi, E.; Natalello, A.; Doglia, S.M.; Forloni, G.; Tagliavini, F.; Del Favero, E.; Cantù, L.; et al. Tetracycline Prevents Aβ Oligomer Toxicity through an Atypical Supramolecular Interaction. Org. Biomol. Chem. 2011, 9, 463–472. [Google Scholar] [CrossRef]
- Montagna, G.; Cazzulani, B.; Obici, L.; Uggetti, C.; Giorgetti, S.; Porcari, R.; Ruggiero, R.; Mangione, P.P.; Brambilla, M.; Lucchetti, J.; et al. Benefit of Doxycycline Treatment on Articular Disability Caused by Dialysis Related Amyloidosis. Amyloid 2013, 20, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, S.; Raimondi, S.; Pagano, K.; Relini, A.; Bucciantini, M.; Corazza, A.; Fogolari, F.; Codutti, L.; Salmona, M.; Mangione, P.; et al. Effect of Tetracyclines on the Dynamics of Formation and Destructuration of β2-Microglobulin Amyloid Fibrils. J. Biol. Chem. 2011, 286, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.E.; Ren, R.; Toraldo, G.; SooHoo, P.; Guan, J.; O’Hara, C.; Jasuja, R.; Trinkaus-Randall, V.; Liao, R.; Connors, L.H.; et al. Doxycycline Reduces Fibril Formation in a Transgenic Mouse Model of AL Amyloidosis. Blood 2011, 118, 6610–6617. [Google Scholar] [CrossRef]
- Gautieri, A.; Beeg, M.; Gobbi, M.; Rigoldi, F.; Colombo, L.; Salmona, M. The Anti-Amyloidogenic Action of Doxycycline: A Molecular Dynamics Study on the Interaction with Aβ42. Int. J. Mol. Sci. 2019, 20, 4641. [Google Scholar] [CrossRef]
- Kamal, M.; Knox, J.; Horne, R.I.; Tiwari, O.S.; Burns, A.R.; Han, D.; Levy, D.; Laor Bar-Yosef, D.; Gazit, E.; Vendruscolo, M.; et al. A Rapid in Vivo Pipeline to Identify Small Molecule Inhibitors of Amyloid Aggregation. Nat. Commun. 2024, 15, 8311. [Google Scholar] [CrossRef] [PubMed]
- Vasilev, B.; Atanasova, M. A (Comprehensive) Review of the Application of Quantitative Structure–Activity Relationship (QSAR) in the Prediction of New Compounds with Anti-Breast Cancer Activity. Appl. Sci. 2025, 15, 1206. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atanasova, M. Small-Molecule Inhibitors of Amyloid Beta: Insights from Molecular Dynamics—Part A: Endogenous Compounds and Repurposed Drugs. Pharmaceuticals 2025, 18, 306. https://doi.org/10.3390/ph18030306
Atanasova M. Small-Molecule Inhibitors of Amyloid Beta: Insights from Molecular Dynamics—Part A: Endogenous Compounds and Repurposed Drugs. Pharmaceuticals. 2025; 18(3):306. https://doi.org/10.3390/ph18030306
Chicago/Turabian StyleAtanasova, Mariyana. 2025. "Small-Molecule Inhibitors of Amyloid Beta: Insights from Molecular Dynamics—Part A: Endogenous Compounds and Repurposed Drugs" Pharmaceuticals 18, no. 3: 306. https://doi.org/10.3390/ph18030306
APA StyleAtanasova, M. (2025). Small-Molecule Inhibitors of Amyloid Beta: Insights from Molecular Dynamics—Part A: Endogenous Compounds and Repurposed Drugs. Pharmaceuticals, 18(3), 306. https://doi.org/10.3390/ph18030306