

Antidiabetic GLP-1 Receptor Agonists Have Neuroprotective Properties in Experimental Animal Models of Alzheimer’s Disease
 , , , and
, , , and
Abstract
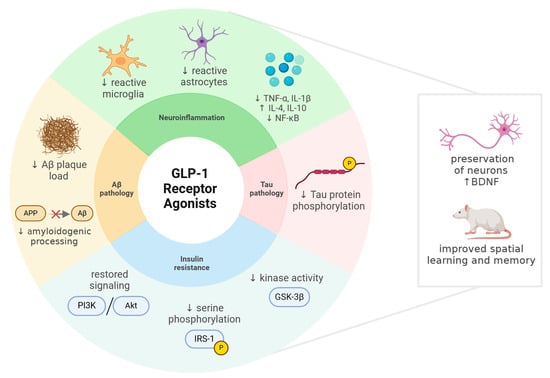
1. Introduction
2. Alzheimer’s Disease and Type 2 Diabetes Mellitus (T2DM)
3. Insulin Resistance in Alzheimer’s Disease
4. Glucagon-like Peptide-1 and Its Analogues
4.1. Sources and Physiological Effects of GLP-1
4.2. Functional Features and Therapeutic Benefits of GLP-1RAs
4.3. Incretin-Based Multi-Agonists
5. GLP-1R Activation in the Context of Neuroprotection
5.1. GLP-1 and GLP-1RAs Exert Specific Neuroprotective Effects
5.2. GLP-1RAs Down-Regulate Chronic Oxidative Stress of the CNS
6. GLP-1RAs in Experimental Models of AD
6.1. Exenatide

{kind=link}
{kind=link}
| Study | Animal Model | Route of Administration and Dosage | Results | ||
|---|---|---|---|---|---|
| Behavior | Biochemistry | Histology | |||
| [168] | 3xTg-AD mice ± ip. STZ | micro-osmotic pump at 3.5 pM/kg/min rate (sc.), 16 weeks | n.d. | ↓ APP, soluble Aβ oligomer in the hippocampus no diff. in total Tau | ↓ Aβ plaque load in the hippocampus |
| [164] | APP/PS1 mice | 25 nmol/kg ip., daily for 3 weeks | MWM: ↓ escape latency and distance moved, ↑ time in target quadrant; | ↓ Ser363-, Ser 312-p-IRS-1, and p-JNK in the hippocampus ↓ soluble Aβ | ↓ Aβ plaque load in the cerebral cortex |
| [175] | C57BL/6J mice, intrahippocampal LPS, and ip. STZ | 10 µg/kg ip., daily for 28 days | MWM: ↓ escape latency and ↑ time in target quadrant; | ↓ COX1, COX2, CD45, NF-κB in the hippocampus | ↓ Iba-1+ microglia and GFAP+ astrocyte in the hippocampus; ↑ Tyr-hydroxylase (in locus coeruleus) and serotonin immunoreactivity (in raphe nucleus) |
| [182] | Wistar rats, icv. STZ | 10 μg/kg sc., twice a day, 14 days | MWM: ↓ escape latency and distance moved, ↑ time in target quadrant | ↓ Ser396-, Thr181-p-Tau ↑ Ser9-p-GSK-3β, ↓ Tyr216-p- GSK-3β and total GSK-3β | ↑ density of CA1 normal neurons |
| [167] | PS1-KI and 3xTg-AD mice | 500 µg/kg ip., daily, 5 days a week, 9 months | MWM: no statistically significant difference (both strains) | no modification of COX (both strains), ↑ forward LDH activity (PS1-KI) in brain homogenate | no reduction in hippocampal Aβ load and hyperphosphorylated-Tau (3xTg-AD) |
| [174] | CD1 mice, icv. Aβ1-42 | 0.2 mg/kg sc., five doses given at 3-h intervals; | n.d. | ↑ the plasma membrane GluR1 and ADAM10 in the neocortex | n.d. |
| [176] | Sprague Dawley rats, icv. STZ | 20 µg/kg ip., daily for 14 days | PAL: ↑ latency | ↓ TNF-α, ↑ ChAT | ↑ neuron count in CA1 and CA3 regions |
| [94] | C57BL/6 mice, intrahippocampal Aβ31-35 | single dose of 0.5 nmol intranasal or 0.05 nmol intrahippocampal drug, 30 min prior to Aβ31-35 | MWM: ↓ escape latency and ↑ time in target quadrant; ↑ locomotor activity | n.d. | n.d. |
| [179] | Sprague Dawley rats, icv. Aβ1-42 | single dose of 0.02 nmol, 0.2 nmol, 2 nmol drug, 15 min before the Aβ1-42 | MWM: ↓ escape latency and distance moved, ↑ time in target quadrant (0.2 and 2 nmol) | ↑ Bcl2 mRNA, ↓ Bax mRNA and caspase-3 in the hippocampus (0.2 and 2 nmol) | n.d. |
| [162] | APP/PS1 mice | 25 nmol/kg sc., twice a day, 4 weeks | MWM: ↓ escape latency, ↑ time in target quadrant and number of target crossings | ↓ GnT-III, GlcNAc and ↑ Ser473-p-Akt, Ser9-p-GSK-3β, β-catenin in the hippocampus | ↓ GnT-III expression in cortex and hippocampus |
| [163] | 5xFAD mice | 100 µg/kg sc., twice a day, 16 weeks | MWM: ↓ escape latency, ↑ number of target crossings | ↑ PSD95 and SYN and normalized mitochondrial dynamics in the hippocampus | ↓ Aβ plaque number, ↑ specific surface area of mitochondria in the CA1 region |
| [166] | 3xTg-AD mice ± 6 months-long high fat diet (HFD) | 500 µg/kg ip. for five days per week, 3 months; | No difference in MWM and ORT | ↑ BDNF level and improved it’s signaling (HFD and non-HFD) ↑ Ser1101-p-IRS-1 (HFD), ↓ NF-κB and PPARα/γ (HFD) | No difference in Aβ and p-Tau load in the CA1 region |
| [183] | Tg2576 mice | 0.075 μg intranasal, 6 days a week, 8 months ± 0.43 × 10−3 IU insulin | MWM: ↓ escape latency (monotherapy) | no difference in hippocampal Aβ1-42 ↓ IRS1, INSR, AKT1, AKT3, CTNNB1, GSK-3β, IGF-1R mRNA in the hippocampus (combination only) | n.d. |
| [177] | Wistar rats, icv. Aβ1-42 | 5 μg/kg ip., daily for 14 days | MWM: ↓ escape latency, ↑ time/distance in target quadrant Y-maze: ↑ spontaneous alternation behavior | ↑ ACh, ChAT activity, ↓ Aβ, AChE activity in hippocampus and prefrontal cortex | n.d. |
| [169] | APPswe/Tau mice ± ip. STZ | 10 μg/kg sc., twice a day, 6 weeks | Barnes circular maze: no diff. in escape latency and error | ↓ soluble Aβ oligomer, no diff. in Thr231-p-Tau | ↑ NeuN+ neurons in the hippocampus (APP/Tau+STZ only) |
| [165] | 5xFAD mice | 100 μg/kg sc., twice a day, 16 weeks | MWM: ↓ escape latency, ↑ number of platform crossings | ↓ TNFα, IL-1β, GFAP and NLRP2 expression in the cortex | ↓ Aβ1-42 load, GFAP+ astrocyte and NLRP2 in the cortex ↑ NeuN+ neurons in the cortex |
| [178] | Wistar rats, icv. STZ | 10 μg/kg ip., daily for 21 days | Y-maze: ↑ frequency and time in the novel arm ORT and ODT: ↑ recognition index | n.d. | ↑ NeuN+ neurons and ↓ cleaved caspase-3 immunoreactivity in the CA1 and CA3 regions and dentate gyrus |
6.2. Liraglutide
| Study | Animal Model | Route of Administration and Dosage | Results | ||
|---|---|---|---|---|---|
| Behavior | Biochemistry | Histology | |||
| [194] | APP/PS1xdb/db crossbreed mice | 500 μg/kg sc., daily for 20 weeks | MWM: ↓ escape latency, ↑ time in target quadrant NOD: ↑ “what” and “where” paradigms Rotarod: no difference in time and speed | no difference in soluble and insoluble Aβ (cortex and hippocampus) ↓ p-Tau (cortex) no difference in IR-A, IR-B, IGF-1R mRNA | ↑ brain weight and cortex size ↑ neuronal density in cortex and hippocampus ↓ Aβ plaque burden in cortex (APP/PS1), no difference in APP/PS1xdb/db animals ↓ Iba-1 + microglia in cortex |
| [227] | 5xFAD mice | 25 nmol/kg sc., daily for 8 weeks | MWM: ↓ escape latency, ↑ number of target crossings | ↑ PSD95 and SYN in cortex ↑ glycolysis, ATP production, p-PI3K, p-Akt, ↓ ROS | ↑ neuronal density in cortex |
| [190] | Sprague-Dawley rats, intravenous homocysteine | 150 (L), 300 (M) and 450 μg/kg (H), twice a day, 2 weeks | MWM: ↓ escape latency, ↑ distance in target quadrant (L, M) | ↓ plasma homocysteine (L, M) ↓ Thr231-, Ser404-, Thr205-p-Tau, PSEN1 (L, M), Aβ, Ser668-p-APP, BACE1, demethylated and p-PP2A (all), ↑ ADAM10 (L, M) ↓ Ser312-p-IRS-1, ↑ GLP-1R (all) | ↓ Thr231-p-Tau |
| [193] | 5xFAD and wild-type mice, icv. STZ | 25 nmol/kg ip., daily for 30 days | OF, NOR, PAL: no statistically significant difference | ↓ Aβ only in 5xFAD-nonSTZ mice ↑ IDE (both), ↑ p-IR in cortex, ↑ p-GSK-3β in hippocampus and cortex (5xFAD, 5xFAD-STZ) | no diff. in the number of degenerating neurons ↓ GFAP+ astrocytes and Iba-1 + microglia (cortex, CA1, CA3) (both) |
| [216] | APPswe/PS1dE9 mice | 25 nmol/kg ip., daily for 8 weeks | n.d. | n.d. | ↓ Aβ plaque number, IRβ expression, S616-p-IRS-1, GFAP+ astrocytes and Iba-1+ microglia in frontal cortex |
| [212] | APPswe/PS1dE9 mice | 25 nmol/kg ip., daily for 8 weeks | NOR: ↑ recognition index MWM: ↑ time and distance in target quadrant | ↓ soluble Aβ oligomers and APP no difference in IR, BDNF, Ngfr mRNA ↑ IDE mRNA | ↓ Aβ plaque load, ↓ Iba-1 + microglia, ↑ Doublecortin+ neurons, ↑ synapse number in the hippocampus |
| [213] | APPswe/PS1dE9 mice | 25 nmol/kg ip., daily for 8 weeks | NOR: ↑ recognition index MWM: ↓ escape latency, ↑ time and distance in target quadrant | ↓ soluble Aβ oligomers and APP | ↓ Aβ plaque load, ↓ Iba-1 + microglia in the cortex ↑ Doublecortin+ neurons ↑ synapse number in the hippocampus |
| [215] | APPswe/PS1dE9 mice | 25 nmol/kg ip., daily for 8 months | OF: no diff. NOR: ↑ recognition index MWM: ↑ time in target quadrant | n.d. | ↓ Aβ plaque load, ↓ Iba-1 + microglia, ↑ Doublecortin + neurons, ↑ synapse number in the cortex and CA1 area |
| [232] | Sprague–Dawley rats, intrahippocampal Aβ25-35 | single dose of 0,05, 0,5, and 5 nmol intrahippocampal drug 30 min prior to Aβ25-35 | MWM: ↓ escape latency (0,5 and 5 nmol), ↓ escape distance (0,5 and 5 nmol), ↑ time and distance in target quadrant | n.d. | n.d. |
| [188] | SAMP8 mice | 100 or 500 μg/kg sc., daily for 4 months | active avoidance T-maze: ↑ memory retention NOR: no improvement | n.d. | ↑ total CA1 pyramidal neuron number and density (100 μg/kg) |
| [207] | C57/BL6 mice, icv. Aβ1-42 | 25 nmol/kg sc., daily for 8 weeks | Y-maze: ↑ spontaneous alternation MWM: ↓ escape latency, ↑ time in target quadrant, and number of platform crosses | ↑ GLP-1R, ↓ Ser396- and Ser202/199-p-Tau, but not total Tau in the hippocampus ↑ Ser473-p-Akt and Ser9-p-GSK-3β in the hippocampus | ↓ Tau phosphorylation in the cortex |
| [220] | hAPPLon/PS1A246E mice hAPPswe/hPS1dE9 mice | 100 or 500 μg/kg sc., daily for 3 months 500 μg/kg sc., daily for 5 months | MWM: no diff. NOR: no diff. active avoidance T-maze: no diff. | n.d. | no atrophy and no diff. in Aβ plaque load in cortex, hippocampus, and striatum (both models) |
| [208] | PLB4 mice | 25 nmol/kg five times a week for 10 weeks | No difference in habituation activity | ↑ serum DPP-4, IR mRNA ↓ Rbp4, Dpp4, Chrebp, Srebp1c mRNA | n.d. |
| [206] | Kunning mice, icv. STZ | 300 μg/kg sc., daily for 30 days | MWM: ↓ escape latency, escape distance, ↑ time in target quadrant, and hidden platform crossings | ↓ p-NF-M/H, ↑ glycated-NF-M/H ↓ Ser199/202-, Ser396/404-, Ser214-, Thr212-, and Thr231-p-Tau ↑ microtubule binding of Tau ↑ p-ERK1, ↓ p-JNK1/2 | ↓ p-NF-M/H, ↑ glycated-NF-M in cortex ↓ Ser199/202-p-Tau in cortex and CA1 ↓ degenerated neurons in CA1, CA4 and cortex |
| [205] | APPswe/PS1dE9 mice | 0.2 mg/kg sc., daily for 2 months | n.d. | No changes on plasma leptin ↓ Thr231-p-Tau, caspase-3 in the hippocampus | ↓ Aβ plaque load in the CA1 region ↓ GFAP+ astrocyte in cortex, no diff. in Iba-1+ microglia |
| [211] | 3xTg-AD mice | 300 μg/kg sc., daily for 8 weeks | MWM: ↓ escape latency, escape distance, ↑ time in target quadrant, and number of platform crossings | ↓ Thr231-, Ser214-, Ser396-, S199/202-p-Tau, but not total Tau ↓ NF-H and NF-M ↑ p-ERK1/2, ↓ p-JNK1/2 | ↓ degenerated neurons in CA3, CA4 and cortex |
| [195] | Male Swiss mice, icv. AβOs | 25 nmol/kg ip., daily for 7 days | NOR, OLM: ↑ novel/displaced object exploration time Contextual fear conditioning: ↑ freezing behavior | ↑ PKA in the hippocampus ↑ IRα mRNA in the hippocampus | n.d. |
| Non-human primates (macaques), icv. AβOs | 0.006 mg/kg for 7 days, then 0.012 mg/kg for 24 days | n.d. | ↓ Ser396-p-Tau (frontal cortex) | ↓ p-Tau in the frontal cortex ↑ IRα and IRβ in the cortex, and IRα in the hippocampus | |
| [229] | APPswe/PS1dE9 mice | 25 nmol/kg ip., daily for 7 and 37 days | n.d. | n.d. | ↑ cell proliferation, Doublecortin+ neurons (acute and chronic), and NeuN+ neurons (chronic) |
| [217] | APPswe/PS1dE9 mice | 25 nmol/kg or 2,5 nmol/kg ip., daily for 10 weeks | OF: no difference NOR: ↑ recognition index (25 nmol/kg only) | n.d. | ↓ Aβ plaque load, Iba-1+ microglia in the cortex (both doses) ↑ synaptic density in the hippocampus and cortex (both doses) |
| [224] | hTauP301L mice | 500 μg/kg sc., daily for 22 weeks | n.d. | n.d. | ↓ total neuronal p-Tau |
| [218] | APPswe/PS1dE9 mice | 25 nmol/kg ip. daily, or sc. implant | n.d. | n.d. | ↓ Aβ plaque load in the cortex, CA1, and dentate gyrus ↓ Iba-1 + microglia and GFAP + astrocyte in the hippocampus |
| [209] | 3xTg-AD mice | 0.2 mg/kg sc., daily for 28 days | OF: no difference Y-maze: no difference MWM: no difference | ↓ Aβ1-42, CRP, pyruvate, reactive carbonyl groups and nitrite level, ↑ G6PDH activity | n.d. |
| [214] | APP/PS1 mice | 25 nmol/kg ip., daily for three months | MWM: ↑ number of platform crossings Y-maze: ↑ frequency in the novel arm | ↓ Aβ1-42, GSK-3β ↑ SYP, GLP-1R, PI3K and Akt | ↓ Aβ plaque load, GSK-3β and ↑ neurons, SYP, PSD95, GLP-1R, p-PI3K and Akt immunoreactivity in the hippocampus |
| [154] | 5xFAD mice | 25 nmol/kg sc., daily for 8 weeks | n.d. | ↑ cAMP, p-PKA, ATP production, and ↓ ROS | ↑ neurons in the cortex |
| [210] | AppNL-G-F mice | 200 μg/kg sc., daily for 4 and 20 weeks | Y-maze: ↑ spontaneous alternation | ↑ PKA activity in the cortex (4 weeks) | ↓ Aβ1-42 positive area and reactive astrocytes, ↑ Ser-p-AQP4 in the cortex (4 weeks) |
| [219] | 3xTg-AD mice | 0.2 mg/kg sc., daily for four months | n.d. | ↓ NF-κB, JNK no diff. in Ser396-p-Tau, GSK-3β, PP2A | ↓ Aβ plaque load in the CA1 region and GFAP+ astrocytes in the amygdala |
6.3. Lixisenatide
6.4. Semaglutide and Other GLP-1RAs
6.5. Dual GLP-1/GIP Receptor Agonists and Other Incretin-Based Multi-Agonists
| Drug and Study | Animal Model | Route of Administration and Dosage | Results | ||
|---|---|---|---|---|---|
| Behavior | Biochemistry | Histology | |||
| DA-JC1 [136] | APPswe/PS1dE9 mice | 50 nmol/kg ip., daily for 4 weeks | n.d. | n.d. | ↑ doublecortin+ neurons in the subventricular zone ↓ Aβ plaque load in the cortex, CA1, and dentate gyrus ↓ GFAP + astrocytes and Iba-1 + microglia in the hippocampus |
| DA-CH3 [252] | APPswe/PS1dE9 mice | 25 nmol/kg ip., daily for 8 weeks | MWM: ↓ escape latency and distance moved, ↑ time in target quadrant reversal MWM: ↑ time in target quadrant | ↑ PSD95 and SYP ↑ Thr308-p-Akt and Ser9-p-GSK-3β | ↓ Aβ plaque load, ↓ GFAP + astrocytes, and Iba-1 + microglia in the cortex and hippocampus |
| DA4-JC [253] | 3xTg-AD mice | 10 nmol/kg ip., daily for 46 days | ORT: ↑ recognition index Y-maze: ↑ spontaneous alternation MWM and reversal MWM: ↑ time in target quadrant and platform crossings Conditional fear memory test: ↑ freezing behavior | ↑ PSD95 and SYP ↑ PINK1/Parkin, ↓ P62 in the hippocampus | ↑ number of synapses and dendritic spines ↑ volume and ↓ number of mitochondria in the hippocampus (TEM) ↓ Aβ- and tau-area in the hippocampus |
| DA4-JC [255] | APPswe/PS1dE9 mice | 10 nmol/kg ip., daily for 6 weeks | MWM: ↑ time in target quadrant | ↓ IL-1β and TNF-α | ↓ Aβ plaque load, ↓ GFAP + astrocytes and Iba-1 + microglia in the cortex |
| DA4-JC [256] | Sprague-Dawley rats, icv. STZ | 10 nmol/kg ip., daily for 2 weeks | Y-maze: ↑ spontaneous alternation MWM: ↓ escape latency, ↑ time in target quadrant | ↓ Bax/Bcl-2 ratio ↑ p-Akt, ↓ p-IRS-1 in the cortex and hippocampus | ↓ Ser396-p-tau, ↓ GFAP + astrocytes and Iba-1 + microglia in the cortex and hippocampus |
| DA5-CH [254] | APPswe/PS1dE9 mice | 10 nmol/kg ip., daily for 4 weeks | Y-maze: ↑ spontaneous alternation MWM and reversal MWM: ↓ escape latency, ↑ time in target quadrant, and platform crossings | ↑ p-PI3K, p-Akt, ↓ p-GSK-3β | ↓ Aβ- and p-tau-immunoreactivity in the hippocampus |
| DA5-CH [135] | Sprague-Dawley rats, icv. STZ | 10 nmol/kg ip., daily for 2 weeks | Y-maze: ↑ spontaneous alternation MWM: ↑ time in target quadrant and platform crossings | ↓ Ser396-p-tau, ↓ Bax/Bcl-2 ratio, ↑ PSD95 and SYN, ↑ p-CREB in the hippocampus | ↓ Ser396-p-tau, ↑ PSD95 and SYN in the hippocampus |
| Tirzepatide [251] | APP/PS1 | 10 nmol/kg ip., daily for 8 weeks | NOR: no difference in recognition index | ↓ GLP-1R, GFAP, BACE1, ↑ GLUT1, HK, G6PDH, PFK mRNA in the cortex | ↓ Aβ plaque number in the cortex, ↓ GFAP + astrocytes in the cortex and hippocampus |
| Tirzepatide [241] | 5xFAD mice | 10 nmol/kg sc., daily for 7 weeks | NOR: ↑ discrimination index (female mice) MWM: no difference | no difference in inflammation-related gene expression | no difference in Aβ plaque load, microglia and astrocyte activation |
7. GLP-1RAs in Clinical Trials of AD
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Lopez, O.L.; Kuller, L.H. Epidemiology of Aging and Associated Cognitive Disorders: Prevalence and Incidence of Alzheimer’s Disease and Other Dementias. Handb. Clin. Neurol. 2019, 167, 139–148. [Google Scholar] [CrossRef] [PubMed]
- 2024 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2024, 20, 3708–3821. [CrossRef] [PubMed]
- Alzheimer’s association report 2022 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2022, 18, 700–789. [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s Disease Drug-Development Pipeline: Few Candidates, Frequent Failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- Tatulian, S.A. Challenges and Hopes for Alzheimer’s Disease. Drug Discov. Today 2022, 27, 1027–1043. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval for Alzheimer’s Drug|FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug (accessed on 22 January 2023).
- Haeberlein, S.B.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef]
- Kang, C. Donanemab: First Approval. Drugs 2024, 84, 1313–1318. [Google Scholar] [CrossRef]
- Hoy, S.M. Lecanemab: First Approval. Drugs 2023, 83, 359–365. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s Disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Sheppard, O.; Coleman, M. Alzheimer’s Disease: Etiology, Neuropathology and Pathogenesis. In Alzheimer’s Disease: Drug Discovery; Huang, X., Ed.; Exon Publications: Brisbane, Australia, 2020; pp. 1–22. [Google Scholar]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
- Chen, M.K.; Mecca, A.P.; Naganawa, M.; Finnema, S.J.; Toyonaga, T.; Lin, S.F.; Najafzadeh, S.; Ropchan, J.; Lu, Y.; McDonald, J.W.; et al. Assessing Synaptic Density in Alzheimer Disease with Synaptic Vesicle Glycoprotein 2A Positron Emission Tomographic Imaging. JAMA Neurol. 2018, 75, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. N. Am. 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S.; Sabbagh, M.N.; Rubino, I. Diagnosis of Early Alzheimer’s Disease: Clinical Practice in 2021. J. Prev. Alzheimer’s Dis. 2021, 8, 371–386. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s Disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s Disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Gunes, S.; Aizawa, Y.; Sugashi, T.; Sugimoto, M.; Rodrigues, P.P. Biomarkers for Alzheimer’s Disease in the Current State: A Narrative Review. Int. J. Mol. Sci. 2022, 23, 4962. [Google Scholar] [CrossRef]
- Gao, Y.; Tan, L.; Yu, J.-T.; Tan, L. Tau in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H.K. Recent Advances in Molecular Pathways and Therapeutic Implications Targeting Neuroinflammation for Alzheimer’s Disease. Inflammopharmacology 2021, 29, 1669. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 Inflammasome Activation Drives Tau Pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Kepp, K.P.; Robakis, N.K.; Høilund-Carlsen, P.F.; Sensi, S.L.; Vissel, B. The Amyloid Cascade Hypothesis: An Updated Critical Review. Brain 2023, 146, 3969–3990. [Google Scholar] [CrossRef]
- Nowell, J.; Blunt, E.; Edison, P. Incretin and Insulin Signaling as Novel Therapeutic Targets for Alzheimer’s and Parkinson’s Disease. Mol. Psychiatry 2023, 28, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; Hofman, A.; Van Harskamp, F.; Grobbee, D.E.; Breteler, M.M.B. Association of Diabetes Mellitus and Dementia: The Rotterdam Study. Diabetologia 1996, 39, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Hyperinsulinemia and Risk of Alzheimer Disease. Neurology 2004, 63, 1187–1192. [Google Scholar] [CrossRef]
- Noguchi-Shinohara, M.; Yuki-Nozaki, S.; Abe, C.; Mori, A.; Horimoto, M.; Yokogawa, M.; Ishida, N.; Suga, Y.; Ishizaki, J.; Ishimiya, M.; et al. Diabetes Mellitus, Elevated Hemoglobin A1c, and Glycated Albumin Are Associated with the Presence of All-Cause Dementia and Alzheimer’s Disease: The JPSC-AD Study. J. Alzheimer’s Dis. 2022, 85, 235–247. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yamagishi, S.I. Involvement of Toxic AGEs (TAGE) in the Pathogenesis of Diabetic Vascular Complications and Alzheimer’s Disease. J. Alzheimer’s Dis. 2009, 16, 845–858. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Silva, M.V.F.; Loures, C.M.G.; Alves, L.C.V.; de Souza, L.C.; Borges, K.B.G.; Carvalho, M.D.G. Alzheimer’s Disease: Risk Factors and Potentially Protective Measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes Mellitus and the Risk of Dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef]
- Akomolafe, A.; Beiser, A.; Meigs, J.B.; Au, R.; Green, R.C.; Farrer, L.A.; Wolf, P.A.; Seshadri, S. Diabetes Mellitus and Risk of Developing Alzheimer Disease: Results from the Framingham Study. Arch. Neurol. 2006, 63, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 2687. [Google Scholar] [CrossRef]
- Chauhan, N.; Paliwal, S.; Jain, S.; Verma, K.; Paliwal, S.; Sharma, S. GSK-3β and Its Inhibitors in Alzheimer’s Disease: A Recent Update. Mini Rev. Med. Chem. 2022, 22, 2881–2895. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Dubey, S.; Jain, S. Association Between Type 2 Diabetes Mellitus and Alzheimer’s Disease: Common Molecular Mechanism and Therapeutic Targets. Cell Biochem. Funct. 2024, 42, e4111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, D.; Zhu, X.; Wang, Y.; Zhu, P. Structural Characterization and Cryo-Electron Tomography Analysis of Human Islet Amyloid Polypeptide Suggest a Synchronous Process of the HIAPP1-37 Amyloid Fibrillation. Biochem. Biophys. Res. Commun. 2020, 533, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, M.; Dominguez, L.J. Type 2 Diabetes Mellitus and Alzheimer’s Disease. World J. Diabetes 2014, 5, 889–893. [Google Scholar] [CrossRef]
- Kang, P.; Wang, Z.; Qiao, D.; Zhang, B.; Mu, C.; Cui, H.; Li, S. Dissecting Genetic Links between Alzheimer’s Disease and Type 2 Diabetes Mellitus in a Systems Biology Way. Front. Genet. 2022, 13, 1019860. [Google Scholar] [CrossRef]
- Chung, Y.; Lee, H. Correlation between Alzheimer’s Disease and Type 2 Diabetes Using Non-Negative Matrix Factorization. Sci. Rep. 2021, 11, 15265. [Google Scholar] [CrossRef]
- Hao, K.; Di Narzo, A.F.; Ho, L.; Luo, W.; Li, S.; Chen, R.; Li, T.; Dubner, L.; Pasinetti, G.M. Shared Genetic Etiology Underlying Alzheimer’s Disease and Type 2 Diabetes. Mol. Asp. Med. 2015, 43–44, 66–76. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated Brain Insulin Resistance in Alzheimer’s Disease Patients Is Associated with IGF-1 Resistance, IRS-1 Dysregulation, and Cognitive Decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Burillo, J.; Marqués, P.; Jiménez, B.; González-Blanco, C.; Benito, M.; Guillén, C. Insulin Resistance and Diabetes Mellitus in Alzheimer’s Disease. Cells 2021, 10, 1236. [Google Scholar] [CrossRef]
- Rhea, E.M.; Banks, W.A. Insulin and the Blood–Brain Barrier. Vitam. Horm. 2024, 126, 169–190. [Google Scholar] [CrossRef]
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in Central Nervous System: More than Just a Peripheral Hormone. J. Aging Res. 2012, 2012, 384017. [Google Scholar] [CrossRef] [PubMed]
- Pomytkin, I.; Pinelis, V. Brain Insulin Resistance: Focus on Insulin Receptor-Mitochondria Interactions. Life 2021, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Hwang, J.H.; Son, S.U.; Choi, J.; You, S.W.; Park, H.; Cha, S.Y.; Maeng, S. How Can Insulin Resistance Cause Alzheimer’s Disease? Int. J. Mol. Sci. 2023, 24, 3506. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Sah, S.P. Insulin Signaling Pathway and Related Molecules: Role in Neurodegeneration and Alzheimer’s Disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Hernandez-Garzón, E.; Perez-Domper, P.; Perez-Alvarez, A.; Mederos, S.; Matsui, T.; Santi, A.; Trueba-Saiz, A.; García-Guerra, L.; Pose-Utrilla, J.; et al. Insulin Regulates Astrocytic Glucose Handling Through Cooperation With IGF-I. Diabetes 2017, 66, 64–74. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Kuga, G.K.; Muñoz, V.R.; Gaspar, R.C.; Nakandakari, S.C.B.R.; da Silva, A.S.R.; Botezelli, J.D.; de Almeida Leme, J.A.C.; Gomes, R.J.; de Moura, L.P.; Cintra, D.E.; et al. Impaired Insulin Signaling and Spatial Learning in Middle-Aged Rats: The Role of PTP1B. Exp. Gerontol. 2018, 104, 66–71. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Dodd, G.T.; Tiganis, T. Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharmacol. Sci. 2015, 36, 661–674. [Google Scholar] [CrossRef]
- Qiu, J.; Bosch, M.A.; Zhang, C.; Rønnekleiv, O.K.; Kelly, M.J. Estradiol Protects Neuropeptide Y/Agouti-Related Peptide Neurons against Insulin Resistance in Females. Neuroendocrinology 2019, 110, 105–118. [Google Scholar] [CrossRef]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 Receptor, Insulin Receptor and IRS-1/2 in Alzheimer’s Disease Indicate Possible Resistance to IGF-1 and Insulin Signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Martínez Báez, A.; Ayala, G.; Pedroza-Saavedra, A.; González-Sánchez, H.M.; Chihu Amparan, L. Phosphorylation Codes in IRS-1 and IRS-2 Are Associated with the Activation/Inhibition of Insulin Canonical Signaling Pathways. Curr. Issues Mol. Biol. 2024, 46, 634–649. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-α Mediates PKR-Dependent Memory Impairment and Brain IRS-1 Inhibition Induced by Alzheimer’s β-Amyloid Oligomers in Mice and Monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In Vivo Seeding and Cross-Seeding of Localized Amyloidosis: A Molecular Link between Type 2 Diabetes and Alzheimer Disease. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Bové, J.; Martínez-Vicente, M.; Vila, M. Fighting Neurodegeneration with Rapamycin: Mechanistic Insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Alldred, M.J.; Ginsberg, S.D.; Ohno, M. Mechanisms Underlying Insulin Deficiency-Induced Acceleration of β-Amyloidosis in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2012, 7, e32792. [Google Scholar] [CrossRef]
- Zuo, X.; Jia, J. Promoter Polymorphisms Which Modulate Insulin Degrading Enzyme Expression May Increase Susceptibility to Alzheimer’s Disease. Brain Res. 2009, 1249, 1–8. [Google Scholar] [CrossRef]
- Chow, H.M.; Shi, M.; Cheng, A.; Gao, Y.; Chen, G.; Song, X.; So, R.W.L.; Zhang, J.; Herrup, K. Age-Related Hyperinsulinemia Leads to Insulin Resistance in Neurons and Cell-Cycle-Induced Senescence. Nat. Neurosci. 2019, 22, 1806–1819. [Google Scholar] [CrossRef]
- Sayas, C.L.; Ávila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like Peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef]
- Trapp, S.; Richards, J.E. The Gut Hormone Glucagon-like Peptide-1 Produced in Brain: Is This Physiologically Relevant? Curr. Opin. Pharmacol. 2013, 13, 964–969. [Google Scholar] [CrossRef] [PubMed]
- McLean, B.A.; Wong, C.K.; Campbell, J.E.; Hodson, D.J.; Trapp, S.; Drucker, D.J. Revisiting the Complexity of GLP-1 Action from Sites of Synthesis to Receptor Activation. Endocr. Rev. 2021, 42, 101–132. [Google Scholar] [CrossRef] [PubMed]
- Holz, G.G. Epac: A New CAMP-Binding Protein in Support of Glucagon-like Peptide-1 Receptor-Mediated Signal Transduction in the Pancreatic Beta-Cell. Diabetes 2004, 53, 5–13. [Google Scholar] [CrossRef]
- Kang, G.; Chepurny, O.G.; Holz, G.G. CAMP-Regulated Guanine Nucleotide Exchange Factor II (Epac2) Mediates Ca2+-Induced Ca2+ Release in INS-1 Pancreatic Beta-Cells. J. Physiol. 2001, 536, 375–385. [Google Scholar] [CrossRef]
- Kimura, R.; Okouchi, M.; Kato, T.; Imaeda, K.; Okayama, N.; Asai, K.; Joh, T. Epidermal Growth Factor Receptor Transactivation Is Necessary for Glucagon-like Peptide-1 to Protect PC12 Cells from Apoptosis. Neuroendocrinology 2013, 97, 300–308. [Google Scholar] [CrossRef]
- Buteau, J.; Foisy, S.; Joly, E.; Prentki, M. Glucagon-like Peptide 1 Induces Pancreatic Beta-Cell Proliferation via Transactivation of the Epidermal Growth Factor Receptor. Diabetes 2003, 52, 124–132. [Google Scholar] [CrossRef]
- Marzook, A.; Tomas, A.; Jones, B. The Interplay of Glucagon-Like Peptide-1 Receptor Trafficking and Signalling in Pancreatic Beta Cells. Front. Endocrinol. 2021, 12, 678055. [Google Scholar] [CrossRef]
- Guan, G.; Zhang, J.; Liu, S.; Huang, W.; Gong, Y.; Gu, X. Glucagon-like Peptide-1 Attenuates Endoplasmic Reticulum Stress-Induced Apoptosis in H9c2 Cardiomyocytes during Hypoxia/Reoxygenation through the GLP-1R/PI3K/Akt Pathways. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 715–722. [Google Scholar] [CrossRef]
- Buteau, J.; Spatz, M.L.; Accili, D. Transcription Factor FoxO1 Mediates Glucagon-Like Peptide-1 Effects on Pancreatic β-Cell Mass. Diabetes 2006, 55, 1190–1196. [Google Scholar] [CrossRef]
- Li, Y.; Perry, T.A.; Kindy, M.S.; Harvey, B.K.; Tweedie, D.; Holloway, H.W.; Powers, K.; Shen, H.; Egan, J.M.; Sambamurti, K.; et al. GLP-1 Receptor Stimulation Preserves Primary Cortical and Dopaminergic Neurons in Cellular and Rodent Models of Stroke and Parkinsonism. Proc. Natl. Acad. Sci. USA 2009, 106, 1285–1290. [Google Scholar] [CrossRef]
- Yang, J.L.; Chen, W.Y.; Chen, Y.P.; Kuo, C.Y.; Chen, S. Der Activation of GLP-1 Receptor Enhances Neuronal Base Excision Repair via PI3K-AKT-Induced Expression of Apurinic/Apyrimidinic Endonuclease 1. Theranostics 2016, 6, 2015–2027. [Google Scholar] [CrossRef] [PubMed]
- Buteau, J.; Foisy, S.; Rhodes, C.J.; Carpenter, L.; Biden, T.J.; Prentki, M. Protein Kinase Cζ Activation Mediates Glucagon-Like Peptide-1–Induced Pancreatic β-Cell Proliferation. Diabetes 2001, 50, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Dalle, S.; Ravier, M.A.; Bertrand, G. Emerging Roles for β-Arrestin-1 in the Control of the Pancreatic β-Cell Function and Mass: New Therapeutic Strategies and Consequences for Drug Screening. Cell. Signal. 2011, 23, 522–528. [Google Scholar] [CrossRef]
- Sonoda, N.; Imamura, T.; Yoshizaki, T.; Babendure, J.L.; Lu, J.C.; Olefsky, J.M. β-Arrestin-1 Mediates Glucagon-like Peptide-1 Signaling to Insulin Secretion in Cultured Pancreatic β Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 6614. [Google Scholar] [CrossRef]
- Jones, B. The Therapeutic Potential of GLP-1 Receptor Biased Agonism. Br. J. Pharmacol. 2022, 179, 492–510. [Google Scholar] [CrossRef]
- Quoyer, J.; Longuet, C.; Broca, C.; Linck, N.; Costes, S.; Varin, E.; Bockaert, J.L.; Bertrand, G.; Dalle, S. GLP-1 Mediates Antiapoptotic Effect by Phosphorylating Bad through a β-Arrestin 1-Mediated ERK1/2 Activation in Pancreatic β-Cells. J. Biol. Chem. 2010, 285, 1989–2002. [Google Scholar] [CrossRef]
- Benchoula, K.; Parhar, I.S.; Madhavan, P.; Hwa, W.E. CREB Nuclear Transcription Activity as a Targeting Factor in the Treatment of Diabetes and Diabetes Complications. Biochem. Pharmacol. 2021, 188, 114531. [Google Scholar] [CrossRef]
- Campbell, J.E.; Drucker, D.J. Pharmacology, Physiology, and Mechanisms of Incretin Hormone Action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef]
- Reich, N.; Hölscher, C. The Neuroprotective Effects of Glucagon-like Peptide 1 in Alzheimer’s and Parkinson’s Disease: An in-Depth Review. Front. Neurosci. 2022, 16, 970925. [Google Scholar] [CrossRef]
- Hayes, M.R.; Leichner, T.M.; Zhao, S.; Lee, G.S.; Chowansky, A.; Zimmer, D.; De Jonghe, B.C.; Kanoski, S.E.; Grill, H.J.; Bence, K.K. Intracellular Signals Mediating the Food Intake-Suppressive Effects of Hindbrain Glucagon-like Peptide-1 Receptor Activation. Cell Metab. 2011, 13, 320–330. [Google Scholar] [CrossRef]
- Boer, G.A.; Hay, D.L.; Tups, A. Obesity Pharmacotherapy: Incretin Action in the Central Nervous System. Trends Pharmacol. Sci. 2023, 44, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.E.; Egan, J.M. Mechanisms of Action of Glucagon-like Peptide 1 in the Pancreas. Pharmacol. Ther. 2007, 113, 546–593. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidis, L.A.; Mankad, S.; Sokos, G.G.; Miske, G.; Shah, A.; Elahi, D.; Shannon, R.P. Effects of Glucagon-like Peptide-1 in Patients with Acute Myocardial Infarction and Left Ventricular Dysfunction after Successful Reperfusion. Circulation 2004, 109, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Ong, Z.Y.; Liu, J.J.; Pang, Z.P.; Grill, H.J. Paraventricular Thalamic Control of Food Intake and Reward: Role of Glucagon-Like Peptide-1 Receptor Signaling. Neuropsychopharmacology 2017, 42, 2387–2397. [Google Scholar] [CrossRef]
- Trujillo, J.M.; Nuffer, W.; Smith, B.A. GLP-1 Receptor Agonists: An Updated Review of Head-to-Head Clinical Studies. Ther. Adv. Endocrinol. Metab. 2021, 12, 2042018821997320. [Google Scholar] [CrossRef]
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Front. Endocrinol. 2019, 10, 155. [Google Scholar] [CrossRef]
- Yu, M.; Benjamin, M.M.; Srinivasan, S.; Morin, E.E.; Shishatskaya, E.I.; Schwendeman, S.P.; Schwendeman, A. Battle of GLP-1 Delivery Technologies. Adv. Drug Deliv. Rev. 2018, 130, 113–130. [Google Scholar] [CrossRef]
- Lyxumia|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/lyxumia (accessed on 5 January 2025).
- Bydureon|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/bydureon (accessed on 6 January 2025).
- Skovbjerg, G.; Roostalu, U.; Salinas, C.G.; Skytte, J.L.; Perens, J.; Clemmensen, C.; Elster, L.; Frich, C.K.; Hansen, H.H.; Hecksher-Sørensen, J. Uncovering CNS Access of Lipidated Exendin-4 Analogues by Quantitative Whole-Brain 3D Light Sheet Imaging. Neuropharmacology 2023, 238, 109637. [Google Scholar] [CrossRef]
- Kastin, A.J.; Akerstrom, V. Entry of Exendin-4 into Brain Is Rapid but May Be Limited at High Doses. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 313–318. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Xu, Y.; Yu, Q.; Li, L.; Guo, Y. Intranasal Administration of Exendin-4 Antagonizes Aβ31–35-Induced Disruption of Circadian Rhythm and Impairment of Learning and Memory. Aging Clin. Exp. Res. 2016, 28, 1259–1266. [Google Scholar] [CrossRef]
- Hunter, K.; Hölscher, C. Drugs Developed to Treat Diabetes, Liraglutide and Lixisenatide, Cross the Blood Brain Barrier and Enhance Neurogenesis. BMC Neurosci. 2012, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Secher, A.; Jelsing, J.; Baquero, A.F.; Hecksher-Sørensen, J.; Cowley, M.A.; Dalbøge, L.S.; Hansen, G.; Grove, K.L.; Pyke, C.; Raun, K.; et al. The Arcuate Nucleus Mediates GLP-1 Receptor Agonist Liraglutide-Dependent Weight Loss. J. Clin. Investig. 2014, 124, 4473–4488. [Google Scholar] [CrossRef] [PubMed]
- Ast, J.; Arvaniti, A.; Fine, N.H.F.; Nasteska, D.; Ashford, F.B.; Stamataki, Z.; Koszegi, Z.; Bacon, A.; Jones, B.J.; Lucey, M.A.; et al. Super-Resolution Microscopy Compatible Fluorescent Probes Reveal Endogenous Glucagon-like Peptide-1 Receptor Distribution and Dynamics. Nat. Commun. 2020, 11, 467. [Google Scholar] [CrossRef]
- Fortin, S.M.; Lipsky, R.K.; Lhamo, R.; Chen, J.; Kim, E.; Borner, T.; Schmidt, H.D.; Hayes, M.R. GABA Neurons in the Nucleus Tractus Solitarius Express GLP-1 Receptors and Mediate Anorectic Effects of Liraglutide in Rats. Sci. Transl. Med. 2020, 12, eaay8071. [Google Scholar] [CrossRef]
- Imbernon, M.; Saponaro, C.; Helms, H.C.C.; Duquenne, M.; Fernandois, D.; Deligia, E.; Denis, R.G.P.; Chao, D.H.M.; Rasika, S.; Staels, B.; et al. Tanycytes Control Hypothalamic Liraglutide Uptake and Its Anti-Obesity Actions. Cell Metab. 2022, 34, 1054–1063.e7. [Google Scholar] [CrossRef]
- Salameh, T.S.; Rhea, E.M.; Talbot, K.; Banks, W.A. Brain Uptake Pharmacokinetics of Incretin Receptor Agonists Showing Promise as Alzheimer’s and Parkinson’s Disease Therapeutics. Biochem. Pharmacol. 2020, 180, 114187. [Google Scholar] [CrossRef]
- Lee, T.S.; Park, E.J.; Choi, M.; Oh, H.S.; An, Y.; Kim, T.; Kim, T.H.; Shin, B.S.; Shin, S. Novel LC-MS/MS Analysis of the GLP-1 Analog Semaglutide with Its Application to Pharmacokinetics and Brain Distribution Studies in Rats. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2023, 1221, 123688. [Google Scholar] [CrossRef]
- Gabery, S.; Salinas, C.G.; Paulsen, S.J.; Ahnfelt-Rønne, J.; Alanentalo, T.; Baquero, A.F.; Buckley, S.T.; Farkas, E.; Fekete, C.; Frederiksen, K.S.; et al. Semaglutide Lowers Body Weight in Rodents via Distributed Neural Pathways. JCI Insight 2020, 5, e133429. [Google Scholar] [CrossRef]
- Abdulhameed, N.; Babin, A.; Hansen, K.; Weaver, R.; Banks, W.A.; Talbot, K.; Rhea, E.M. Comparing Regional Brain Uptake of Incretin Receptor Agonists after Intranasal Delivery in CD-1 Mice and the APP/PS1 Mouse Model of Alzheimer’s Disease. Alzheimer’s Res. Ther. 2024, 16, 173. [Google Scholar] [CrossRef]
- Rhea, E.M.; Babin, A.; Thomas, P.; Omer, M.; Weaver, R.; Hansen, K.; Banks, W.A.; Talbot, K. Brain Uptake Pharmacokinetics of Albiglutide, Dulaglutide, Tirzepatide, and DA5-CH in the Search for New Treatments of Alzheimer’s and Parkinson’s Diseases. Tissue Barriers 2023, 12, 2292461. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, M.; Wen, Z.; Lu, Z.; Cui, L.; Fu, C.; Xue, H.; Liu, Y.; Zhang, Y. GLP-1 Receptor Agonists: Beyond Their Pancreatic Effects. Front. Endocrinol. 2021, 12, 721135. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Zhang, A.; Li, D.; Wu, Y.; Wang, C.Z.; Wan, J.Y.; Yuan, C.S. Comparative Effectiveness of GLP-1 Receptor Agonists on Glycaemic Control, Body Weight, and Lipid Profile for Type 2 Diabetes: Systematic Review and Network Meta-Analysis. BMJ 2024, 384, e076410. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Y.; Poudel, A.; Welchko, R.; Mekala, N.; Chandramani-Shivalingappa, P.; Rosca, M.G.; Li, L. Liraglutide Improves Insulin Sensitivity in High Fat Diet Induced Diabetic Mice through Multiple Pathways. Eur. J. Pharmacol. 2019, 861, 172594. [Google Scholar] [CrossRef]
- Elsayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Care in Diabetes-2023. Diabetes Care 2023, 46, S140–S157. [Google Scholar] [CrossRef]
- Ussher, J.R.; Drucker, D.J. Glucagon-like Peptide 1 Receptor Agonists: Cardiovascular Benefits and Mechanisms of Action. Nat. Rev. Cardiol. 2023, 20, 463–474. [Google Scholar] [CrossRef]
- Zhang, F.; Tong, Y.; Su, N.; Li, Y.; Tang, L.; Huang, L.; Tong, N. Weight Loss Effect of Glucagon-like Peptide-1 Mimetics on Obese/Overweight Adults without Diabetes: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Diabetes 2015, 7, 329–339. [Google Scholar] [CrossRef]
- Xu, F.; Lin, B.; Zheng, X.; Chen, Z.; Cao, H.; Xu, H.; Liang, H.; Weng, J. GLP-1 Receptor Agonist Promotes Brown Remodelling in Mouse White Adipose Tissue through SIRT1. Diabetologia 2016, 59, 1059–1069. [Google Scholar] [CrossRef]
- Maselli, D.B.; Camilleri, M. Effects of GLP-1 and Its Analogs on Gastric Physiology in Diabetes Mellitus and Obesity. Adv. Exp. Med. Biol. 2021, 1307, 171–192. [Google Scholar] [CrossRef]
- Brierley, D.I.; Holt, M.K.; Singh, A.; de Araujo, A.; McDougle, M.; Vergara, M.; Afaghani, M.H.; Lee, S.J.; Scott, K.; Maske, C.; et al. Central and Peripheral GLP-1 Systems Independently Suppress Eating. Nat. Metab. 2021, 3, 258–273. [Google Scholar] [CrossRef]
- Ard, J.; Fitch, A.; Fruh, S.; Herman, L. Weight Loss and Maintenance Related to the Mechanism of Action of Glucagon-Like Peptide 1 Receptor Agonists. Adv. Ther. 2021, 38, 2821–2839. [Google Scholar] [CrossRef]
- Drucker, D.J. GLP-1 Physiology Informs the Pharmacotherapy of Obesity. Mol. Metab. 2021, 57, 101351. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, D.; Prasad-Reddy, L.; Srivastava, S.B. Role of Glucagon-like Peptide 1 Receptor Agonists in Management of Obesity. Am. J. Health Syst. Pharm. 2016, 73, 1493–1507. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves New Drug Treatment for Chronic Weight Management, First Since 2014. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-treatment-chronic-weight-management-first-2014 (accessed on 26 February 2023).
- Chao, A.M.; Tronieri, J.S.; Amaro, A.; Wadden, T.A. Semaglutide for the Treatment of Obesity. Trends Cardiovasc. Med. 2021, 33, 159–166. [Google Scholar] [CrossRef]
- Ceriello, A.; Esposito, K.; Testa, R.; Bonfigli, A.R.; Marra, M.; Giugliano, D. The Possible Protective Role of Glucagon-like Peptide 1 on Endothelium during the Meal and Evidence for an “Endothelial Resistance” to Glucagon-like Peptide 1 in Diabetes. Diabetes Care 2011, 34, 697–702. [Google Scholar] [CrossRef]
- Drucker, D.J. Efficacy and Safety of GLP-1 Medicines for Type 2 Diabetes and Obesity. Diabetes Care 2024, 47, 1873–1888. [Google Scholar] [CrossRef]
- Dong, S.; Sun, C. Can Glucagon-like Peptide-1 Receptor Agonists Cause Acute Kidney Injury? An Analytical Study Based on Post-Marketing Approval Pharmacovigilance Data. Front. Endocrinol. 2022, 13, 1032199. [Google Scholar] [CrossRef]
- He, L.; Wang, J.; Ping, F.; Yang, N.; Huang, J.; Li, Y.; Xu, L.; Li, W.; Zhang, H. Association of Glucagon-Like Peptide-1 Receptor Agonist Use with Risk of Gallbladder and Biliary Diseases: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. JAMA Intern. Med. 2022, 182, 513–519. [Google Scholar] [CrossRef]
- Samms, R.J.; Coghlan, M.P.; Sloop, K.W. How May GIP Enhance the Therapeutic Efficacy of GLP-1? Trends Endocrinol. Metab. 2020, 31, 410–421. [Google Scholar] [CrossRef]
- Frias, J.P.; Bastyr, E.J.; Vignati, L.; Tschöp, M.H.; Schmitt, C.; Owen, K.; Christensen, R.H.; DiMarchi, R.D. The Sustained Effects of a Dual GIP/GLP-1 Receptor Agonist, NNC0090-2746, in Patients with Type 2 Diabetes. Cell Metab. 2017, 26, 343–352.e2. [Google Scholar] [CrossRef]
- Liu, Q.K. Mechanisms of Action and Therapeutic Applications of GLP-1 and Dual GIP/GLP-1 Receptor Agonists. Front. Endocrinol. 2024, 15, 1431292. [Google Scholar] [CrossRef]
- Frías, J.P.; Davies, M.J.; Rosenstock, J.; Pérez Manghi, F.C.; Fernández Landó, L.; Bergman, B.K.; Liu, B.; Cui, X.; Brown, K. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Melson, E.; Ashraf, U.; Papamargaritis, D.; Davies, M.J. What Is the Pipeline for Future Medications for Obesity? Int. J. Obes. 2024, 49, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Khoo, B.; Tan, T. Targeting the Incretin System in Obesity and Type 2 Diabetes Mellitus. Nat. Rev. Endocrinol. 2024, 20, 447–459. [Google Scholar] [CrossRef]
- Nogueiras, R.; Nauck, M.A.; Tschöp, M.H. Gut Hormone Co-Agonists for the Treatment of Obesity: From Bench to Bedside. Nat. Metab. 2023, 5, 933–944. [Google Scholar] [CrossRef]
- Adriaenssens, A.E.; Biggs, E.K.; Darwish, T.; Tadross, J.; Sukthankar, T.; Girish, M.; Polex-Wolf, J.; Lam, B.Y.; Zvetkova, I.; Pan, W.; et al. Glucose-Dependent Insulinotropic Polypeptide Receptor-Expressing Cells in the Hypothalamus Regulate Food Intake. Cell Metab. 2019, 30, 987–996.e6. [Google Scholar] [CrossRef]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the Two Incretin Hormones: Similarities and Differences. J. Diabetes Investig. 2010, 1, 8. [Google Scholar] [CrossRef]
- Mayendraraj, A.; Rosenkilde, M.M.; Gasbjerg, L.S. GLP-1 and GIP Receptor Signaling in Beta Cells—A Review of Receptor Interactions and Co-Stimulation. Peptides 2022, 151, 170749. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M. The Role of GIP Receptor in the CNS for the Pathogenesis of Obesity. Diabetes 2021, 70, 1929. [Google Scholar] [CrossRef]
- Liskiewicz, A.; Khalil, A.; Liskiewicz, D.; Novikoff, A.; Grandl, G.; Maity-Kumar, G.; Gutgesell, R.M.; Bakhti, M.; Bastidas-Ponce, A.; Czarnecki, O.; et al. Glucose-Dependent Insulinotropic Polypeptide Regulates Body Weight and Food Intake via GABAergic Neurons in Mice. Nat. Metab. 2023, 5, 2075–2085. [Google Scholar] [CrossRef]
- Li, C.; Liu, W.; Li, X.; Zhang, Z.; Qi, H.; Liu, S.; Yan, N.; Xing, Y.; Hölscher, C.; Wang, Z. The Novel GLP-1/GIP Analogue DA5-CH Reduces Tau Phosphorylation and Normalizes Theta Rhythm in the Icv. STZ Rat Model of AD. Brain Behav. 2020, 10, e01505. [Google Scholar] [CrossRef]
- Salles, G.N.; Calió, M.L.; Hölscher, C.; Pacheco-Soares, C.; Porcionatto, M.; Lobo, A.O. Neuroprotective and Restorative Properties of the GLP-1/GIP Dual Agonist DA-JC1 Compared with a GLP-1 Single Agonist in Alzheimer’s Disease. Neuropharmacology 2020, 162, 107813. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.J.; Xue, G.F.; Cheng, H.F.; Meng, P.F.; Lian, X.; Hölscher, C.; Li, D.F. The GLP-1/GIP Dual-Receptor Agonist DA5-CH Inhibits the NF-ΚB Inflammatory Pathway in the MPTP Mouse Model of Parkinson’s Disease More Effectively than the GLP-1 Single-Receptor Agonist NLY01. Brain Behav. 2021, 11, e2231. [Google Scholar] [CrossRef]
- Duffy, A.M.; Hölscher, C. The Incretin Analogue D-Ala2GIP Reduces Plaque Load, Astrogliosis and Oxidative Stress in an APP/PS1 Mouse Model of Alzheimer’s Disease. Neuroscience 2013, 228, 294–300. [Google Scholar] [CrossRef]
- Faivre, E.; Hölscher, C. D-Ala2GIP Facilitated Synaptic Plasticity and Reduces Plaque Load in Aged Wild Type Mice and in an Alzheimer’s Disease Mouse Model. J. Alzheimer’s Dis. 2013, 35, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Kopp, K.O.; Li, Y.; Glotfelty, E.J.; Tweedie, D.; Greig, N.H. Incretin-Based Multi-Agonist Peptides Are Neuroprotective and Anti-Inflammatory in Cellular Models of Neurodegeneration. Biomolecules 2024, 14, 872. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in Neurodegenerative Disorders: The Roles of Microglia and Astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Zheng, Z.; Zong, Y.; Ma, Y.; Tian, Y.; Pang, Y.; Zhang, C.; Gao, J. Glucagon-like Peptide-1 Receptor: Mechanisms and Advances in Therapy. Signal Transduct. Target. Ther. 2024, 9, 234. [Google Scholar] [CrossRef]
- During, M.J.; Cao, L.; Zuzga, D.S.; Francis, J.S.; Fitzsimons, H.L.; Jiao, X.; Bland, R.J.; Klugmann, M.; Banks, W.A.; Drucker, D.J.; et al. Glucagon-like Peptide-1 Receptor Is Involved in Learning and Neuroprotection. Nat. Med. 2003, 9, 1173–1179. [Google Scholar] [CrossRef]
- Hsu, T.M.; Hahn, J.D.; Konanur, V.R.; Lam, A.; Kanoski, S.E. Hippocampal GLP-1 Receptors Influence Food Intake, Meal Size, and Effort-Based Responding for Food through Volume Transmission. Neuropsychopharmacology 2015, 40, 327–337. [Google Scholar] [CrossRef]
- Perry, T.; Haughey, N.J.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Protection and Reversal of Excitotoxic Neuronal Damage by Glucagon-like Peptide-1 and Exendin-4. J. Pharmacol. Exp. Ther. 2002, 302, 881–888. [Google Scholar] [CrossRef]
- Ghosh, P.; Fontanella, R.A.; Scisciola, L.; Pesapane, A.; Taktaz, F.; Franzese, M.; Puocci, A.; Ceriello, A.; Prattichizzo, F.; Rizzo, M.R.; et al. Targeting Redox Imbalance in Neurodegeneration: Characterizing the Role of GLP-1 Receptor Agonists. Theranostics 2023, 13, 4872–4884. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative Toxicity in Diabetes and Alzheimer’s Disease: Mechanisms behind ROS/RNS Generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef] [PubMed]
- Chidambaram, S.B.; Anand, N.; Varma, S.R.; Ramamurthy, S.; Vichitra, C.; Sharma, A.; Mahalakshmi, A.M.; Essa, M.M. Superoxide Dismutase and Neurological Disorders. IBRO Neurosci. Rep. 2024, 16, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Sun, X.; Xu, Y.; Wu, T.; Tao, L. Superoxide Dismutase Activity and Risk of Cognitive Decline in Older Adults: Findings from the Chinese Longitudinal Healthy Longevity Survey. Exp. Gerontol. 2019, 118, 72–77. [Google Scholar] [CrossRef]
- Park, K.W.; Baik, H.H.; Jin, B.K. IL-13-Induced Oxidative Stress via Microglial NADPH Oxidase Contributes to Death of Hippocampal Neurons in Vivo. J. Immunol. 2009, 183, 4666–4674. [Google Scholar] [CrossRef]
- Mangmool, S.; Hemplueksa, P.; Parichatikanond, W.; Chattipakorn, N. Epac Is Required for GLP-1R-Mediated Inhibition of Oxidative Stress and Apoptosis in Cardiomyocytes. Mol. Endocrinol. 2015, 29, 583–596. [Google Scholar] [CrossRef]
- Robichaux, W.G.; Cheng, X. Intracellular CAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol. Rev. 2018, 98, 919–1053. [Google Scholar] [CrossRef]
- Li, Y.; Glotfelty, E.J.; Karlsson, T.; Fortuno, L.V.; Harvey, B.K.; Greig, N.H. The Metabolite GLP-1 (9-36) Is Neuroprotective and Anti-Inflammatory in Cellular Models of Neurodegeneration. J. Neurochem. 2021, 159, 867–886. [Google Scholar] [CrossRef]
- Xie, Y.; Zheng, J.; Li, S.; Li, H.; Zhou, Y.; Zheng, W.; Zhang, M.; Liu, L.; Chen, Z. GLP-1 Improves the Neuronal Supportive Ability of Astrocytes in Alzheimer’s Disease by Regulating Mitochondrial Dysfunction via the CAMP/PKA Pathway. Biochem. Pharmacol. 2021, 188, 114578. [Google Scholar] [CrossRef]
- Oh, Y.S.; Jun, H.S. Effects of Glucagon-Like Peptide-1 on Oxidative Stress and Nrf2 Signaling. Int. J. Mol. Sci. 2017, 19, 26. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Basic Principles and Emerging Concepts in the Redox Control of Transcription Factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; DeFronzo, R.A.; Gastaldelli, A.; Holst, J.J. Glucagon-like Peptide-1 and the Central/Peripheral Nervous System: Crosstalk in Diabetes. Trends Endocrinol. Metab. 2017, 28, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, Y.; Shi, Z.; Lu, D.; Li, T.; Ding, Y.; Ruan, Y.; Xu, A. The Neuroprotection of Liraglutide Against Ischaemia-Induced Apoptosis through the Activation of the PI3K/AKT and MAPK Pathways. Sci. Rep. 2016, 6, 268559. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Wódkiewicz, E.; Słupski, M.; Walczak, M.; Socha, M.; Malinowski, B.; Pawlak-Osińska, K. Neuroprotective Activity of Sitagliptin via Reduction of Neuroinflammation beyond the Incretin Effect: Focus on Alzheimer’s Disease. Biomed. Res. Int. 2018, 2018, 6091014. [Google Scholar] [CrossRef]
- Laurijssens, B.; Aujard, F.; Rahman, A. Animal Models of Alzheimer’s Disease and Drug Development. Drug Discov. Today Technol. 2013, 10, e319–e327. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s Disease: Experimental Models and Reality. Acta Neuropathol. 2016, 133, 155–175. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.; Xu, Z.; Chen, S.; Yao, W.; Gao, X. GLP-1 Receptor Agonists Downregulate Aberrant GnT-III Expression in Alzheimer’s Disease Models through the Akt/GSK-3β/β-Catenin Signaling. Neuropharmacology 2018, 131, 190–199. [Google Scholar] [CrossRef]
- An, J.; Zhou, Y.; Zhang, M.; Xie, Y.; Ke, S.; Liu, L.; Pan, X.; Chen, Z. Exenatide Alleviates Mitochondrial Dysfunction and Cognitive Impairment in the 5xFAD Mouse Model of Alzheimer’s Disease. Behav. Brain Res. 2019, 370, 111932. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An Anti-Diabetes Agent Protects the Mouse Brain from Defective Insulin Signaling Caused by Alzheimer’s Disease–Associated Aβ Oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, Y.; Gao, R.; Chen, X.; Chen, R.; Chen, Z. Glucagon-like Peptide-1 Analogs Mitigate Neuroinflammation in Alzheimer’s Disease by Suppressing NLRP2 Activation in Astrocytes. Mol. Cell. Endocrinol. 2022, 542, 111529. [Google Scholar] [CrossRef]
- Bomba, M.; Granzotto, A.; Castelli, V.; Onofrj, M.; Lattanzio, R.; Cimini, A.; Sensi, S.L. Exenatide Reverts the High-Fat-Diet-Induced Impairment of BDNF Signaling and Inflammatory Response in an Animal Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 70, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Bomba, M.; Ciavardelli, D.; Silvestri, E.; Canzoniero, L.M.T.; Lattanzio, R.; Chiappini, P.; Piantelli, M.; Di Ilio, C.; Consoli, A.; Sensi, S.L. Exenatide Promotes Cognitive Enhancement and Positive Brain Metabolic Changes in PS1-KI Mice but Has No Effects in 3xTg-AD Animals. Cell Death Dis. 2013, 4, e612. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Duffy, K.B.; Ottinger, M.A.; Ray, B.; Bailey, J.A.; Holloway, H.W.; Tweedie, D.; Perry, T.; Mattson, M.P.; Kapogiannis, D.; et al. GLP-1 Receptor Stimulation Reduces Amyloid-β Peptide Accumulation and Cytotoxicity in Cellular and Animal Models of Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 19, 1205–1219. [Google Scholar] [CrossRef]
- King, M.R.; Anderson, N.J.; Deciu, M.; Guernsey, L.S.; Cundiff, M.; Hajizadeh, S.; Jolivalt, C.G. Insulin Deficiency, but Not Resistance, Exaggerates Cognitive Deficits in Transgenic Mice Expressing Human Amyloid and Tau Proteins. Reversal by Exendin-4 Treatment. J. Neurosci. Res. 2020, 98, 2357–2369. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Liu, S.; Sun, H.M.; Ma, M.D.; Gao, Y.J.; Qi, C.C.; Xia, Q.R.; Ge, J.F. Bilateral Intracerebroventricular Injection of Streptozotocin Induces AD-like Behavioral Impairments and Neuropathological Features in Mice: Involved with the Fundamental Role of Neuroinflammation. Biomed. Pharmacother. 2022, 153, 113375. [Google Scholar] [CrossRef]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. 2021, 1, e78. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Morris Water Maze: Procedures for Assessing Spatial and Related Forms of Learning and Memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef]
- Pitts, M. Barnes Maze Procedure for Spatial Learning and Memory in Mice. Bio Protoc. 2018, 8, e2744. [Google Scholar] [CrossRef]
- Ohtake, N.; Saito, M.; Eto, M.; Seki, K. Exendin-4 Promotes the Membrane Trafficking of the AMPA Receptor GluR1 Subunit and ADAM10 in the Mouse Neocortex. Regul. Pept. 2014, 190–191, 1–11. [Google Scholar] [CrossRef]
- Huang, H.J.; Chen, Y.H.; Liang, K.C.; Jheng, Y.S.; Jhao, J.J.; Su, M.T.; Lee-Chen, G.J.; Hsieh-Li, H.M. Exendin-4 Protected against Cognitive Dysfunction in Hyperglycemic Mice Receiving an Intrahippocampal Lipopolysaccharide Injection. PLoS ONE 2012, 7, e39656. [Google Scholar] [CrossRef]
- Solmaz, V.; Çınar, B.P.; Yiğittürk, G.; Çavuşoğlu, T.; Taşkiran, D.; Erbaş, O. Exenatide Reduces TNF-α Expression and Improves Hippocampal Neuron Numbers and Memory in Streptozotocin Treated Rats. Eur. J. Pharmacol. 2015, 765, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Garabadu, D.; Verma, J. Exendin-4 Attenuates Brain Mitochondrial Toxicity through PI3K/Akt-Dependent Pathway in Amyloid Beta (1-42)-Induced Cognitive Deficit Rats. Neurochem. Int. 2019, 128, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Zago, A.M.; Carvalho, F.B.; Rahmeier, F.L.; Santin, M.; Guimarães, G.R.; Gutierres, J.M.; da C. Fernandes, M. Exendin-4 Prevents Memory Loss and Neuronal Death in Rats with Sporadic Alzheimer-Like Disease. Mol. Neurobiol. 2024, 61, 2631–2652. [Google Scholar] [CrossRef]
- Jia, X.T.; Tian, Y.; Li, Y.; Zhang, G.J.; Liu, Z.Q.; Di, Z.L.; Ying, X.P.; Fang, Y.; Song, E.F.; Qi, J.S.; et al. Exendin-4, a Glucagon-like Peptide 1 Receptor Agonist, Protects against Amyloid-β Peptide-Induced Impairment of Spatial Learning and Memory in Rats. Physiol. Behav. 2016, 159, 72–79. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Jiang, R.; Yuan, Y.; Yu, Q.; Li, Y. Exendin-4 Antagonizes Aβ1-42-Induced Suppression of Long-Term Potentiation by Regulating Intracellular Calcium Homeostasis in Rat Hippocampal Neurons. Brain Res. 2015, 1627, 101–108. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen Synthase Kinase-3 Signaling in Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Chen, S.; Liu, A.R.; An, F.M.; Yao, W.B.; Gao, X.D. Amelioration of Neurodegenerative Changes in Cellular and Rat Models of Diabetes-Related Alzheimer’s Disease by Exendin-4. Age 2012, 34, 1211–1224. [Google Scholar] [CrossRef]
- Robinson, A.; Lubitz, I.; Atrakchi-Baranes, D.; Licht-Murava, A.; Katsel, P.; Leroith, D.; Liraz-Zaltsman, S.; Haroutunian, V.; Beeri, M.S. Combination of Insulin with a GLP1 Agonist Is Associated with Better Memory and Normal Expression of Insulin Receptor Pathway Genes in a Mouse Model of Alzheimer’s Disease. J. Mol. Neurosci. 2019, 67, 504–510. [Google Scholar] [CrossRef]
- Park, J.S.; Kam, T.I.; Lee, S.; Park, H.; Oh, Y.; Kwon, S.H.; Song, J.J.; Kim, D.; Kim, H.; Jhaldiyal, A.; et al. Blocking Microglial Activation of Reactive Astrocytes Is Neuroprotective in Models of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 78. [Google Scholar] [CrossRef]
- Duda, P.; Hajka, D.; Wójcicka, O.; Rakus, D.; Gizak, A. GSK3β: A Master Player in Depressive Disorder Pathogenesis and Treatment Responsiveness. Cells 2020, 9, 727. [Google Scholar] [CrossRef]
- APPswe/PSEN1dE9 (Line 85)|ALZFORUM. Available online: https://www.alzforum.org/research-models/appswepsen1de9-line-85 (accessed on 11 January 2025).
- Morley, J.E.; Farr, S.A.; Kumar, V.B.; Armbrecht, H.J. The SAMP8 Mouse: A Model to Develop Therapeutic Interventions for Alzheimer’s Disease. Curr. Pharm. Des. 2012, 18, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Fabricius, K.; Barkholt, P.; Niehoff, M.L.; Morley, J.E.; Jelsing, J.; Pyke, C.; Knudsen, L.B.; Farr, S.A.; Vrang, N. The GLP-1 Receptor Agonist Liraglutide Improves Memory Function and Increases Hippocampal CA1 Neuronal Numbers in a Senescence-Accelerated Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 46, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Plucińska, K.; Crouch, B.; Koss, D.; Robinson, L.; Siebrecht, M.; Riedel, G.; Platt, B. Knock-in of Human BACE1 Cleaves Murine APP and Reiterates Alzheimer-like Phenotypes. J. Neurosci. 2014, 34, 10710–10728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, J.Z.; Xu, X.Y.; Hu, J.; Xu, T.; Jin, S.; Yang, S.J.; Wang, J.Z. Liraglutide Ameliorates Hyperhomocysteinemia-Induced Alzheimer-Like Pathology and Memory Deficits in Rats via Multi-Molecular Targeting. Neurosci. Bull. 2019, 35, 724–734. [Google Scholar] [CrossRef]
- Zhang, C.E.; Wei, W.; Liu, Y.H.; Peng, J.H.; Tian, Q.; Liu, G.P.; Zhang, Y.; Wang, J.Z. Hyperhomocysteinemia Increases Beta-Amyloid by Enhancing Expression of Gamma-Secretase and Phosphorylation of Amyloid Precursor Protein in Rat Brain. Am. J. Pathol. 2009, 174, 1481–1491. [Google Scholar] [CrossRef]
- Chai, G.S.; Jiang, X.; Ni, Z.F.; Ma, Z.W.; Xie, A.J.; Cheng, X.S.; Wang, Q.; Wang, J.Z.; Liu, G.P. Betaine Attenuates Alzheimer-like Pathological Changes and Memory Deficits Induced by Homocysteine. J. Neurochem. 2013, 124, 388–396. [Google Scholar] [CrossRef]
- Paladugu, L.; Gharaibeh, A.; Kolli, N.; Learman, C.; Hall, T.C.; Li, L.; Rossignol, J.; Maiti, P.; Dunbar, G.L. Liraglutide Has Anti-Inflammatory and Anti-Amyloid Properties in Streptozotocin-Induced and 5xFAD Mouse Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 860. [Google Scholar] [CrossRef]
- Carranza-Naval, M.J.; del Marco, A.; Hierro-Bujalance, C.; Alves-Martinez, P.; Infante-Garcia, C.; Vargas-Soria, M.; Herrera, M.; Barba-Cordoba, B.; Atienza-Navarro, I.; Lubian-Lopez, S.; et al. Liraglutide Reduces Vascular Damage, Neuronal Loss, and Cognitive Impairment in a Mixed Murine Model of Alzheimer’s Disease and Type 2 Diabetes. Front. Aging Neurosci. 2021, 13, 741923. [Google Scholar] [CrossRef]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra e Silva, N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al. The Diabetes Drug Liraglutide Reverses Cognitive Impairment in Mice and Attenuates Insulin Receptor and Synaptic Pathology in a Non-Human Primate Model of Alzheimer’s Disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef]
- Forny-Germano, L.; Lyra E Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.B.; et al. Alzheimer’s Disease-like Pathology Induced by Amyloid-β Oligomers in Nonhuman Primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef]
- Denninger, J.K.; Smith, B.M.; Kirby, E.D. Novel Object Recognition and Object Location Behavioral Testing in Mice on a Budget. J. Vis. Exp. 2018, 2018, e58593. [Google Scholar] [CrossRef]
- Barker, G.R.I.; Warburton, E.C. When Is the Hippocampus Involved in Recognition Memory? J. Neurosci. 2011, 31, 10721–10731. [Google Scholar] [CrossRef] [PubMed]
- Vogel-Ciernia, A.; Wood, M.A. Examining Object Location and Object Recognition Memory in Mice. Curr. Protoc. Neurosci. 2014, 69, 8.31.1–8.31.17. [Google Scholar] [CrossRef] [PubMed]
- Dere, E.; Huston, J.P.; De Souza Silva, M.A. Episodic-like Memory in Mice: Simultaneous Assessment of Object, Place and Temporal Order Memory. Brain Res. Protoc. 2005, 16, 10–19. [Google Scholar] [CrossRef]
- Kraeuter, A.K.; Guest, P.C.; Sarnyai, Z. The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol. Biol. 2019, 1916, 105–111. [Google Scholar] [CrossRef]
- Kim, J.; Kang, H.; Lee, Y.B.; Lee, B.; Lee, D. A Quantitative Analysis of Spontaneous Alternation Behaviors on a Y-Maze Reveals Adverse Effects of Acute Social Isolation on Spatial Working Memory. Sci. Rep. 2023, 13, 14722. [Google Scholar] [CrossRef]
- Kim, W.B.; Cho, J.H. Encoding of Contextual Fear Memory in Hippocampal–Amygdala Circuit. Nat. Commun. 2020, 11, 1382. [Google Scholar] [CrossRef]
- Curzon, P.; Rustay, N.R.; Browman, K.E. Cued and Contextual Fear Conditioning for Rodents. In Methods of Behavior Analysis in Neuroscience; Buccafusco, J.J., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009; pp. 19–37. ISBN 9781420052343. [Google Scholar]
- Holubová, M.; Hrubá, L.; Popelová, A.; Bencze, M.; Pražienková, V.; Gengler, S.; Kratochvílová, H.; Haluzík, M.; Železná, B.; Kuneš, J.; et al. Liraglutide and a Lipidized Analog of Prolactin-Releasing Peptide Show Neuroprotective Effects in a Mouse Model of β-Amyloid Pathology. Neuropharmacology 2019, 144, 377–387. [Google Scholar] [CrossRef]
- Xiong, H.; Zheng, C.; Wang, J.; Song, J.; Zhao, G.; Shen, H.; Deng, Y. The Neuroprotection of Liraglutide on Alzheimer-like Learning and Memory Impairment by Modulating the Hyperphosphorylation of Tau and Neurofilament Proteins and Insulin Signaling Pathways in Mice. J. Alzheimer’s Dis. 2013, 37, 623–635. [Google Scholar] [CrossRef]
- Qi, L.; Ke, L.; Liu, X.; Liao, L.; Ke, S.; Liu, X.; Wang, Y.; Lin, X.; Zhou, Y.; Wu, L.; et al. Subcutaneous Administration of Liraglutide Ameliorates Learning and Memory Impairment by Modulating Tau Hyperphosphorylation via the Glycogen Synthase Kinase-3β Pathway in an Amyloid β Protein Induced Alzheimer Disease Mouse Model. Eur. J. Pharmacol. 2016, 783, 23–32. [Google Scholar] [CrossRef]
- Dekeryte, R.; Hull, C.; Plucińska, K.; Khan, S.; Kamli-Salino, S.; Mody, N.; Morrice, N.; McLaughlin, C.; Gault, V.; Platt, B.; et al. Effects of Liraglutide and Fenretinide Treatments on the Diabetic Phenotype of Neuronal Human BACE1 Knock-in Mice. Biochem. Pharmacol. 2019, 166, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.I.; Candeias, E.; Alves, I.N.; Mena, D.; Silva, D.F.; Machado, N.J.; Campos, E.J.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Liraglutide Protects Against Brain Amyloid-Β1-42 Accumulation in Female Mice with Early Alzheimer’s Disease-Like Pathology by Partially Rescuing Oxidative/Nitrosative Stress and Inflammation. Int. J. Mol. Sci. 2020, 21, 1746. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Fujita, H.; Sato, T.; Kato, S.; Takahashi, Y.; Takeshita, Y.; Kanda, T.; Saito, T.; Saido, T.C.; Hattori, S.; et al. GLP-1 Receptor Signaling Restores Aquaporin 4 Subcellular Polarization in Reactive Astrocytes and Promotes Amyloid β Clearance in a Mouse Model of Alzheimer’s Disease. Biochem. Biophys. Res. Commun. 2024, 741, 151016. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, J.; Zhao, G.; Guo, A.; Chen, Y.; Fu, R.; Deng, Y. Liraglutide Improves Water Maze Learning and Memory Performance While Reduces Hyperphosphorylation of Tau and Neurofilaments in APP/PS1/Tau Triple Transgenic Mice. Neurochem. Res. 2017, 42, 2326–2335. [Google Scholar] [CrossRef]
- McClean, P.L.; Hölscher, C. Liraglutide Can Reverse Memory Impairment, Synaptic Loss and Reduce Plaque Load in Aged APP/PS1 Mice, a Model of Alzheimer’s Disease. Neuropharmacology 2014, 76 Pt A, 57–67. [Google Scholar] [CrossRef]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Holscher, C. The Diabetes Drug Liraglutide Prevents Degenerative Processes in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef]
- Gao, C.Y.; Qin, G.F.; Zheng, M.C.; Tian, M.J.; He, Y.N.; Wang, P.W. Banxia Xiexin Decoction Alleviated Cerebral Glucose Metabolism Disorder by Regulating Intestinal Microbiota in APP/PS1 Mice. Chin. J. Integr. Med. 2024, 30, 701–712. [Google Scholar] [CrossRef]
- McClean, P.L.; Jalewa, J.; Hölscher, C. Prophylactic Liraglutide Treatment Prevents Amyloid Plaque Deposition, Chronic Inflammation and Memory Impairment in APP/PS1 Mice. Behav. Brain Res. 2015, 293, 96–106. [Google Scholar] [CrossRef]
- Long-Smith, C.M.; Manning, S.; McClean, P.L.; Coakley, M.F.; O’Halloran, D.J.; Holscher, C.; O’Neill, C. The Diabetes Drug Liraglutide Ameliorates Aberrant Insulin Receptor Localisation and Signalling in Parallel with Decreasing Both Amyloid-β Plaque and Glial Pathology in a Mouse Model of Alzheimer’s Disease. Neuromol. Med. 2013, 15, 102–114. [Google Scholar] [CrossRef]
- McClean, P.L.; Hölscher, C. Lixisenatide, a Drug Developed to Treat Type 2 Diabetes, Shows Neuroprotective Effects in a Mouse Model of Alzheimer’s Disease. Neuropharmacology 2014, 86, 241–258. [Google Scholar] [CrossRef]
- Salles, G.N.; Calió, M.L.; Afewerki, S.; Pacheco-Soares, C.; Porcionatto, M.; Hölscher, C.; Lobo, A.O. Prolonged Drug-Releasing Fibers Attenuate Alzheimer’s Disease-like Pathogenesis. ACS Appl. Mater. Interfaces 2018, 10, 36693–36702. [Google Scholar] [CrossRef] [PubMed]
- Mengr, A.; Šmotková, Z.; Pačesová, A.; Železná, B.; Kuneš, J.; Maletínská, L. Reduction of Neuroinflammation as a Common Mechanism of Action of Anorexigenic and Orexigenic Peptide Analogues in the Triple Transgenic Mouse Model of Alzheimer’s Disease. J. Neuroimmune Pharmacol. 2025, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Fabricius, K.; Barkholt, P.; Kongsbak-Wismann, P.; Schlumberger, C.; Jelsing, J.; Terwel, D.; Termont, A.; Pyke, C.; Knudsen, L.B.; et al. Long-Term Treatment with Liraglutide, a Glucagon-Like Peptide-1 (GLP-1) Receptor Agonist, Has No Effect on β-Amyloid Plaque Load in Two Transgenic APP/PS1 Mouse Models of Alzheimer’s Disease. PLoS ONE 2016, 11, e0158205. [Google Scholar] [CrossRef]
- Tian, Y.; Jing, G.; Zhang, M. Insulin-Degrading Enzyme: Roles and Pathways in Ameliorating Cognitive Impairment Associated with Alzheimer’s Disease and Diabetes. Ageing Res. Rev. 2023, 90, 101999. [Google Scholar] [CrossRef] [PubMed]
- Kurochkin, I.V.; Guarnera, E.; Berezovsky, I.N. Insulin-Degrading Enzyme in the Fight against Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 49–58. [Google Scholar] [CrossRef]
- Mestre, H.; Mori, Y.; Nedergaard, M. The Brain’s Glymphatic System: Current Controversies. Trends Neurosci. 2020, 43, 466. [Google Scholar] [CrossRef]
- Hansen, H.H.; Barkholt, P.; Fabricius, K.; Jelsing, J.; Terwel, D.; Pyke, C.; Knudsen, L.B.; Vrang, N. The GLP-1 Receptor Agonist Liraglutide Reduces Pathology-Specific Tau Phosphorylation and Improves Motor Function in a Transgenic HTauP301L Mouse Model of Tauopathy. Brain Res. 2016, 1634, 158–170. [Google Scholar] [CrossRef]
- Sontag, J.M.; Sontag, E. Protein Phosphatase 2A Dysfunction in Alzheimer’s Disease. Front. Mol. Neurosci. 2014, 7, 16. [Google Scholar] [CrossRef]
- Zhao, W.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid Beta Oligomers Induce Impairment of Neuronal Insulin Receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef]
- Zheng, J.; Xie, Y.; Ren, L.; Qi, L.; Wu, L.; Pan, X.; Zhou, J.; Chen, Z.; Liu, L. GLP-1 Improves the Supportive Ability of Astrocytes to Neurons by Promoting Aerobic Glycolysis in Alzheimer’s Disease. Mol. Metab. 2021, 47, 101180. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain Insulin Resistance in Type 2 Diabetes and Alzheimer Disease: Concepts and Conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Parthsarathy, V.; Hölscher, C. Chronic Treatment with the GLP1 Analogue Liraglutide Increases Cell Proliferation and Differentiation into Neurons in an AD Mouse Model. PLoS ONE 2013, 8, e58784. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Gault, V.A.; Harriott, P.; Hölscher, C. Glucagon-like Peptide-1 Analogues Enhance Synaptic Plasticity in the Brain: A Link between Diabetes and Alzheimer’s Disease. Eur. J. Pharmacol. 2010, 630, 158–162. [Google Scholar] [CrossRef]
- Thakur, S.; Dhapola, R.; Sarma, P.; Medhi, B.; Reddy, D.H. Neuroinflammation in Alzheimer’s Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation 2023, 46, 1–17. [Google Scholar] [CrossRef]
- Han, W.N.; Hölscher, C.; Yuan, L.; Yang, W.; Wang, X.H.; Wu, M.N.; Qi, J.S. Liraglutide Protects against Amyloid-β Protein-Induced Impairment of Spatial Learning and Memory in Rats. Neurobiol. Aging 2013, 34, 576–588. [Google Scholar] [CrossRef]
- Cai, H.Y.; Hölscher, C.; Yue, X.H.; Zhang, S.X.; Wang, X.H.; Qiao, F.; Yang, W.; Qi, J.S. Lixisenatide Rescues Spatial Memory and Synaptic Plasticity from Amyloid β Protein-Induced Impairments in Rats. Neuroscience 2014, 277, 6–13. [Google Scholar] [CrossRef]
- Cai, H.Y.; Wang, Z.J.; Hölscher, C.; Yuan, L.; Zhang, J.; Sun, P.; Li, J.; Yang, W.; Wu, M.N.; Qi, J.S. Lixisenatide Attenuates the Detrimental Effects of Amyloid β Protein on Spatial Working Memory and Hippocampal Neurons in Rats. Behav. Brain Res. 2017, 318, 28–35. [Google Scholar] [CrossRef]
- Cai, H.Y.; Yang, J.T.; Wang, Z.J.; Zhang, J.; Yang, W.; Wu, M.N.; Qi, J.S. Lixisenatide Reduces Amyloid Plaques, Neurofibrillary Tangles and Neuroinflammation in an APP/PS1/Tau Mouse Model of Alzheimer’s Disease. Biochem. Biophys. Res. Commun. 2018, 495, 1034–1040. [Google Scholar] [CrossRef]
- Meca, A.D.; Boboc, I.K.S.; Mititelu-Tartau, L.; Bogdan, M. Unlocking the Potential: Semaglutide’s Impact on Alzheimer’s and Parkinson’s Disease in Animal Models. Curr. Issues Mol. Biol. 2024, 46, 5929–5949. [Google Scholar] [CrossRef]
- Wang, Z.J.; Li, X.R.; Chai, S.F.; Li, W.R.; Li, S.; Hou, M.; Li, J.L.; Ye, Y.C.; Cai, H.Y.; Hölscher, C.; et al. Semaglutide Ameliorates Cognition and Glucose Metabolism Dysfunction in the 3xTg Mouse Model of Alzheimer’s Disease via the GLP-1R/SIRT1/GLUT4 Pathway. Neuropharmacology 2023, 240, 109716. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, C.; He, Y.; Zhang, Y.; Li, Q.; Zhang, T.; Zhao, B.; Tong, A.; Zhong, Q.; Zhong, Z. Semaglutide Ameliorates Alzheimer’s Disease and Restores Oxytocin in APP/PS1 Mice and Human Brain Organoid Models. Biomed. Pharmacother. 2024, 180, 117540. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Han, W.N.; Chai, S.F.; Li, Y.; Fu, C.J.; Wang, C.F.; Cai, H.Y.; Li, X.Y.; Wang, X.; Hölscher, C.; et al. Semaglutide Promotes the Transition of Microglia from M1 to M2 Type to Reduce Brain Inflammation in APP/PS1/Tau Mice. Neuroscience 2024, 563, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Elbadawy, N.N.; Saad, M.A.; Elfarrash, S.; Ahmed, M.A.E.; Abdelkader, N.F. The GLP-1 Agonist Semaglutide Ameliorates Cognitive Regression in P301S Tauopathy Mice Model via Autophagy/ACE2/SIRT1/FOXO1-Mediated Microglia Polarization. Eur. J. Pharmacol. 2025, 991, 177305. [Google Scholar] [CrossRef]
- Forny Germano, L.; Koehler, J.A.; Baggio, L.L.; Cui, F.; Wong, C.K.; Rittig, N.; Cao, X.; Matthews, D.; Drucker, D.J. The GLP-1 Medicines Semaglutide and Tirzepatide Do Not Alter Disease-Related Pathology, Behaviour or Cognitive Function in 5XFAD and APP/PS1 Mice. Mol. Metab. 2024, 89, 102019. [Google Scholar] [CrossRef]
- Boboc, I.K.S.; Dumitrelea, P.D.; Meca, A.D.; Mititelu-Tartau, L.; Bogdan, M. Exploring the Impact of Semaglutide on Cognitive Function and Anxiety-Related Behaviors in a Murine Model of Alzheimer’s Disease. Biomedicines 2024, 12, 2689. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Li, L.; Hölscher, C. Semaglutide Is Neuroprotective and Reduces α-Synuclein Levels in the Chronic MPTP Mouse Model of Parkinson’s Disease. J. Parkinson’s Dis. 2019, 9, 157–171. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Li, L.; Hölscher, C. Neuroprotective Effects of the Novel GLP-1 Long Acting Analogue Semaglutide in the MPTP Parkinson’s Disease Mouse Model. Neuropeptides 2018, 71, 70–80. [Google Scholar] [CrossRef]
- Tipa, R.O.; Balan, D.G.; Georgescu, M.T.; Ignat, L.A.; Vacaroiu, I.A.; Georgescu, D.E.; Raducu, L.; Mihai, D.A.; Chiperi, L.V.; Balcangiu-Stroescu, A.E. A Systematic Review of Semaglutide’s Influence on Cognitive Function in Preclinical Animal Models and Cell-Line Studies. Int. J. Mol. Sci. 2024, 25, 4972. [Google Scholar] [CrossRef]
- Basalay, M.V.; Davidson, S.M.; Yellon, D.M. Neuroprotection in Rats Following Ischaemia-Reperfusion Injury by GLP-1 Analogues-Liraglutide and Semaglutide. Cardiovasc. Drugs Ther. 2019, 33, 661–667. [Google Scholar] [CrossRef]
- Wang, L.; Ding, J.; Zhu, C.; Guo, B.; Yang, W.; He, W.; Li, X.; Wang, Y.; Li, W.; Wang, F.; et al. Semaglutide Attenuates Seizure Severity and Ameliorates Cognitive Dysfunction by Blocking the NLR Family Pyrin Domain Containing 3 Inflammasome in Pentylenetetrazole-kindled Mice. Int. J. Mol. Med. 2021, 48, 219. [Google Scholar] [CrossRef]
- Sadek, M.A.; Kandil, E.A.; El Sayed, N.S.; Sayed, H.M.; Rabie, M.A. Semaglutide, a Novel Glucagon-like Peptide-1 Agonist, Amends Experimental Autoimmune Encephalomyelitis-Induced Multiple Sclerosis in Mice: Involvement of the PI3K/Akt/GSK-3β Pathway. Int. Immunopharmacol. 2023, 115, 109647. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Chen, S.; Peng, P.; Gu, Z.; Yu, J.; Zhao, G.; Deng, Y. Dulaglutide Ameliorates STZ Induced AD-like Impairment of Learning and Memory Ability by Modulating Hyperphosphorylation of Tau and NFs through GSK3β. Biochem. Biophys. Res. Commun. 2019, 511, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Lei, M.; Zhao, J.; Wu, M.; Ren, Z.; Yang, X.; Ouyang, C.; Liu, X.; Liu, C.; Chen, Q. Tirzepatide Ameliorates Spatial Learning and Memory Impairment through Modulation of Aberrant Insulin Resistance and Inflammation Response in Diabetic Rats. Front. Pharmacol. 2023, 14, 1146960. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, X.; Zhang, Y.; Tang, Q.; Li, Y.; Du, Y.; Yu, P. Tirzepatide Shows Neuroprotective Effects via Regulating Brain Glucose Metabolism in APP/PS1 Mice. Peptides 2024, 179, 171271. [Google Scholar] [CrossRef]
- Panagaki, T.; Gengler, S.; Hölscher, C. The Novel DA-CH3 Dual Incretin Restores Endoplasmic Reticulum Stress and Autophagy Impairments to Attenuate Alzheimer-Like Pathology and Cognitive Decrements in the APPSWE/PS1ΔE9 Mouse Model. J. Alzheimer’s Dis. 2018, 66, 195–218. [Google Scholar] [CrossRef]
- Cai, H.Y.; Yang, D.; Qiao, J.; Yang, J.T.; Wang, Z.J.; Wu, M.N.; Qi, J.S.; Hölscher, C. A GLP-1/GIP Dual Receptor Agonist DA4-JC Effectively Attenuates Cognitive Impairment and Pathology in the APP/PS1/Tau Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 83, 799–818. [Google Scholar] [CrossRef]
- Cao, Y.; Hölscher, C.; Hu, M.M.; Wang, T.; Zhao, F.; Bai, Y.; Zhang, J.; Wu, M.N.; Qi, J.S. DA5-CH, a Novel GLP-1/GIP Dual Agonist, Effectively Ameliorates the Cognitive Impairments and Pathology in the APP/PS1 Mouse Model of Alzheimer’s Disease. Eur. J. Pharmacol. 2018, 827, 215–226. [Google Scholar] [CrossRef]
- Maskery, M.; Goulding, E.M.; Gengler, S.; Melchiorsen, J.U.; Rosenkilde, M.M.; Hölscher, C. The Dual GLP-1/GIP Receptor Agonist DA4-JC Shows Superior Protective Properties Compared to the GLP-1 Analogue Liraglutide in the APP/PS1 Mouse Model of Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Dement. 2020, 35, 1533317520953041. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, Z.; Li, L.; Hölscher, C. A Novel Dual GLP-1/GIP Receptor Agonist Alleviates Cognitive Decline by Re-Sensitizing Insulin Signaling in the Alzheimer Icv. STZ Rat Model. Behav. Brain Res. 2017, 327, 65–74. [Google Scholar] [CrossRef]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN Signalling in Neurodegeneration and Neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef]
- Li, T.; Jiao, J.J.; Su, Q.; Hölscher, C.; Zhang, J.; Yan, X.D.; Zhao, H.M.; Cai, H.Y.; Qi, J.S. A GLP-1/GIP/Gcg Receptor Triagonist Improves Memory Behavior, as Well as Synaptic Transmission, Neuronal Excitability and Ca2+ Homeostasis in 3xTg-AD Mice. Neuropharmacology 2020, 170, 108042. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jiao, J.J.; Hölscher, C.; Wu, M.N.; Zhang, J.; Tong, J.Q.; Dong, X.F.; Qu, X.S.; Cao, Y.; Cai, H.Y.; et al. A Novel GLP-1/GIP/Gcg Triagonist Reduces Cognitive Deficits and Pathology in the 3xTg Mouse Model of Alzheimer’s Disease. Hippocampus 2018, 28, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Tai, J.; Liu, W.; Li, Y.; Li, L.; Hölscher, C. Neuroprotective Effects of a Triple GLP-1/GIP/Glucagon Receptor Agonist in the APP/PS1 Transgenic Mouse Model of Alzheimer’s Disease. Brain Res. 2018, 1678, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Han, Y.F.; Zhao, F.; Yang, G.Z.; Yuan, L.; Cai, H.Y.; Yang, J.T.; Holscher, C.; Qi, J.S.; Wu, M.N. A Dual GLP-1 and Gcg Receptor Agonist Rescues Spatial Memory and Synaptic Plasticity in APP/PS1 Transgenic Mice. Horm. Behav. 2020, 118, 104640. [Google Scholar] [CrossRef]
- Tang, H.; Shao, H.; Shaaban, C.E.; Yang, K.; Brown, J.; Anton, S.; Wu, Y.; Bress, A.; Donahoo, W.T.; DeKosky, S.T.; et al. Newer Glucose-Lowering Drugs and Risk of Dementia: A Systematic Review and Meta-Analysis of Observational Studies. J. Am. Geriatr. Soc. 2023, 71, 2096. [Google Scholar] [CrossRef]
- Li, Z.; Lin, C.; Cai, X.; Lv, F.; Yang, W.; Ji, L. Anti-Diabetic Agents and the Risks of Dementia in Patients with Type 2 Diabetes: A Systematic Review and Network Meta-Analysis of Observational Studies and Randomized Controlled Trials. Alzheimer’s Res. Ther. 2024, 16, 272. [Google Scholar] [CrossRef]
- Zhou, B.; Zissimopoulos, J.; Nadeem, H.; Crane, M.A.; Goldman, D.; Romley, J.A. Association between Exenatide Use and Incidence of Alzheimer’s Disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12139. [Google Scholar] [CrossRef]
- Mullins, R.J.; Mustapic, M.; Chia, C.W.; Carlson, O.; Gulyani, S.; Tran, J.; Li, Y.; Mattson, M.P.; Resnick, S.; Egan, J.M.; et al. A Pilot Study of Exenatide Actions in Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 741. [Google Scholar] [CrossRef]
- Dei Cas, A.; Micheli, M.M.; Aldigeri, R.; Gardini, S.; Ferrari-Pellegrini, F.; Perini, M.; Messa, G.; Antonini, M.; Spigoni, V.; Cinquegrani, G.; et al. Long-Acting Exenatide Does Not Prevent Cognitive Decline in Mild Cognitive Impairment: A Proof-of-Concept Clinical Trial. J. Endocrinol. Investig. 2024, 47, 2339. [Google Scholar] [CrossRef]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Møller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Brændgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef]
- Gejl, M.; Brock, B.; Egefjord, L.; Vang, K.; Rungby, J.; Gjedde, A. Blood-Brain Glucose Transfer in Alzheimer’s Disease: Effect of GLP-1 Analog Treatment. Sci. Rep. 2017, 7, 17490. [Google Scholar] [CrossRef] [PubMed]
- Bi, Z.; Wang, L.; Wang, W. Evaluating the Effects of Glucagon-like Peptide-1 Receptor Agonists on Cognitive Function in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Adv. Clin. Exp. Med. 2023, 32, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Atri, A.; Feldman, H.H.; Hansson, O.; Sano, M.; Knop, F.K.; Johannsen, P.; León, T.; Scheltens, P. Evoke and Evoke+: Design of Two Large-Scale, Double-Blind, Placebo-Controlled, Phase 3 Studies Evaluating Efficacy, Safety, and Tolerability of Semaglutide in Early-Stage Symptomatic Alzheimer’s Disease. Alzheimer’s Res. Ther. 2025, 17, 14. [Google Scholar] [CrossRef] [PubMed]
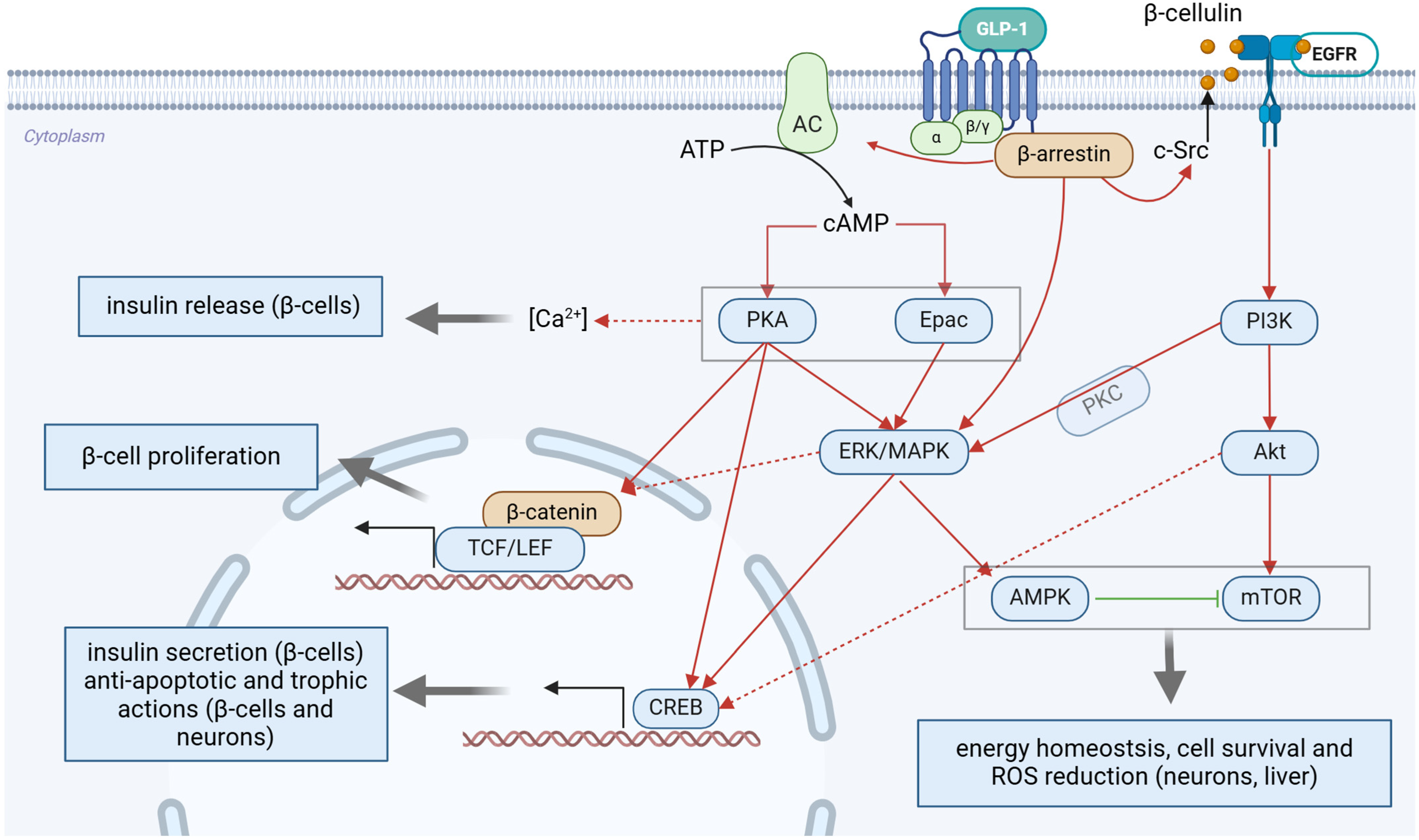
| Study | Animal Model | Route of Administration and Dosage | Results | ||
|---|---|---|---|---|---|
| Behavior | Biochemistry | Histology | |||
| [217] | APPswe/PS1dE9 mice | 1 or 10 nmol/kg ip., daily for 10 weeks | OF: no difference NOR: ↑ recognition index (both doses) | n.d. | ↓ Aβ plaque load, Iba-1+ microglia in the cortex (both doses) ↑ synaptic density in the hippocampus and cortex (both doses) |
| [233] | Spargue-Dawley rats, intrahippocampal Aβ25-35 | 10 nmol intrahippocampal drug 15 min prior to Aβ25-35 | MWM: ↓ escape latency, ↑ time in target quadrant | ↑ Ser9-p-GSK-3β and ↓ Tyr216-p- GSK-3β in the hippocampus | n.d. |
| [234] | Spargue-Dawley rats, intrahippocampal Aβ25-35 | 10 nmol intrahippocampal drug 15 min prior to Aβ25-35 | Y-maze: ↑ spontaneous alternation | n.d. | n.d. |
| [235] | APP/PS1/tau mice | 10 nmol/kg ip., daily for 60 days | n.d. | ↑ p-PKA, p-CREB ↓ p-p38 MAPK in the hippocampus | ↓ Aβ plaque and p-Tau load, ↓ Iba-1+ microglia in the hippocampus |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urkon, M.; Ferencz, E.; Szász, J.A.; Szabo, M.I.M.; Orbán-Kis, K.; Szatmári, S.; Nagy, E.E. Antidiabetic GLP-1 Receptor Agonists Have Neuroprotective Properties in Experimental Animal Models of Alzheimer’s Disease. Pharmaceuticals 2025, 18, 614. https://doi.org/10.3390/ph18050614
Urkon M, Ferencz E, Szász JA, Szabo MIM, Orbán-Kis K, Szatmári S, Nagy EE. Antidiabetic GLP-1 Receptor Agonists Have Neuroprotective Properties in Experimental Animal Models of Alzheimer’s Disease. Pharmaceuticals. 2025; 18(5):614. https://doi.org/10.3390/ph18050614
Chicago/Turabian StyleUrkon, Melinda, Elek Ferencz, József Attila Szász, Monica Iudita Maria Szabo, Károly Orbán-Kis, Szabolcs Szatmári, and Előd Ernő Nagy. 2025. "Antidiabetic GLP-1 Receptor Agonists Have Neuroprotective Properties in Experimental Animal Models of Alzheimer’s Disease" Pharmaceuticals 18, no. 5: 614. https://doi.org/10.3390/ph18050614
APA StyleUrkon, M., Ferencz, E., Szász, J. A., Szabo, M. I. M., Orbán-Kis, K., Szatmári, S., & Nagy, E. E. (2025). Antidiabetic GLP-1 Receptor Agonists Have Neuroprotective Properties in Experimental Animal Models of Alzheimer’s Disease. Pharmaceuticals, 18(5), 614. https://doi.org/10.3390/ph18050614