
Development of a Population Pharmacokinetic Model of Levofloxacin in Healthy Adults and Identification of Optimal Dosing Regimens


Abstract
1. Introduction
2. Results
2.1. Participants
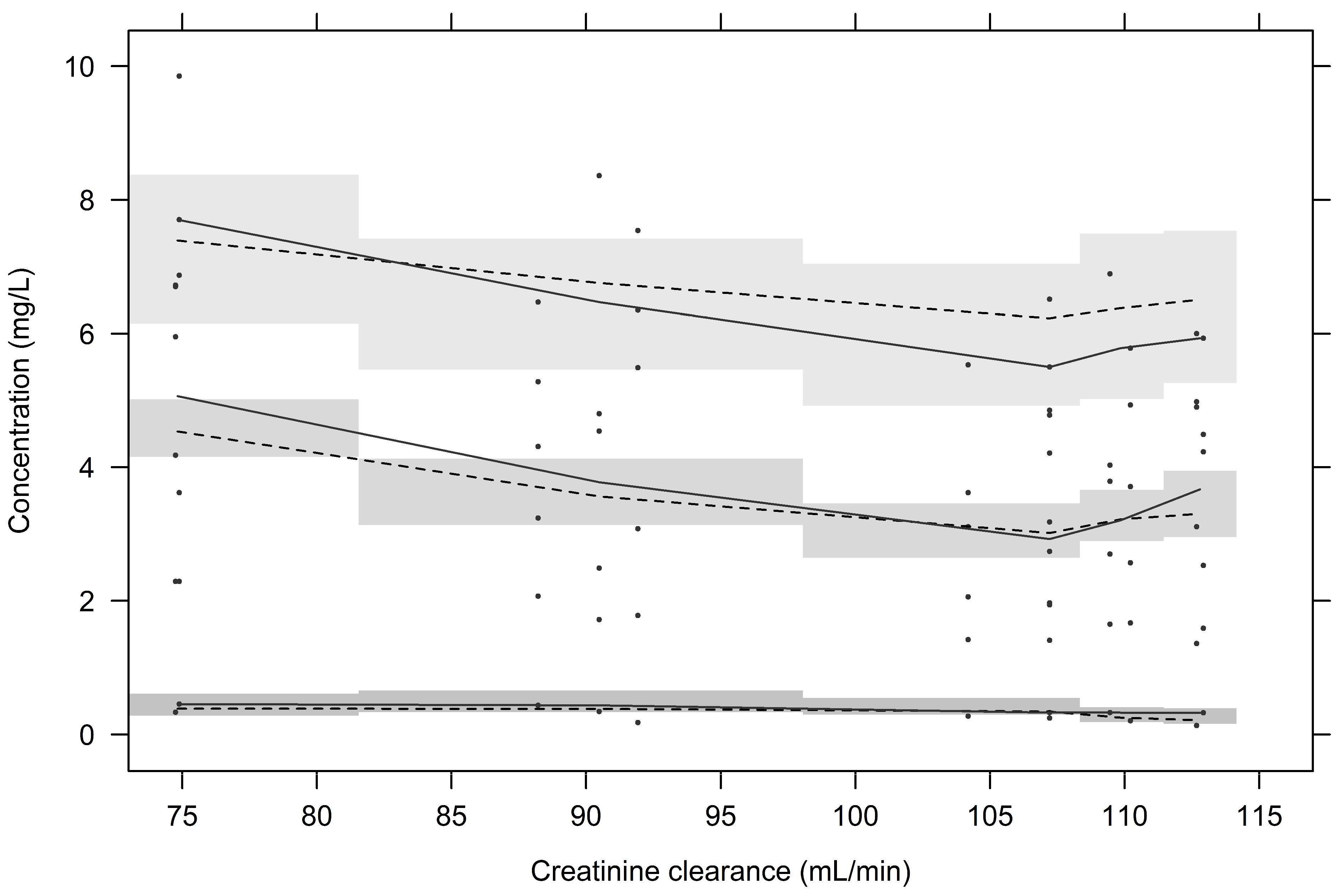
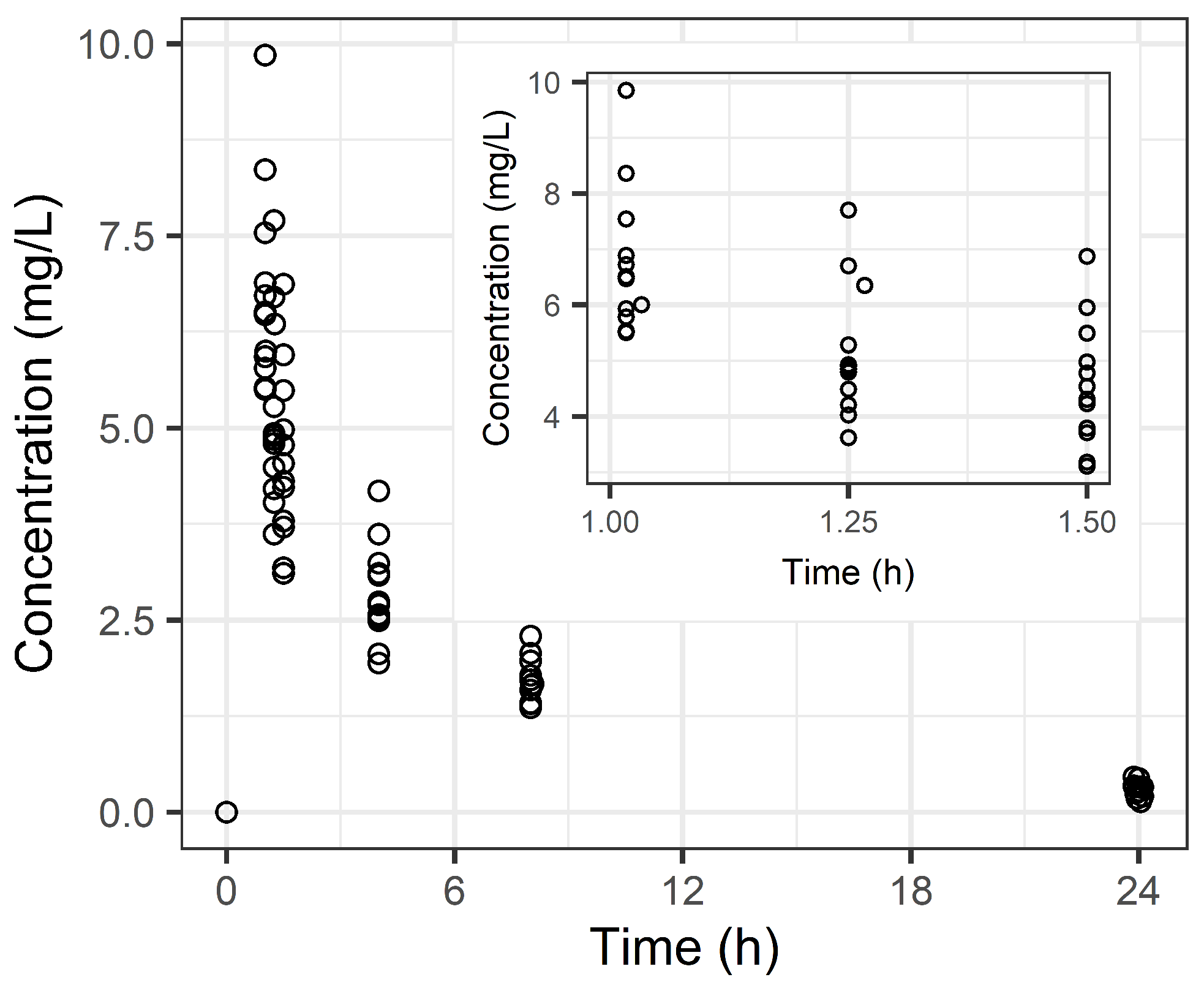
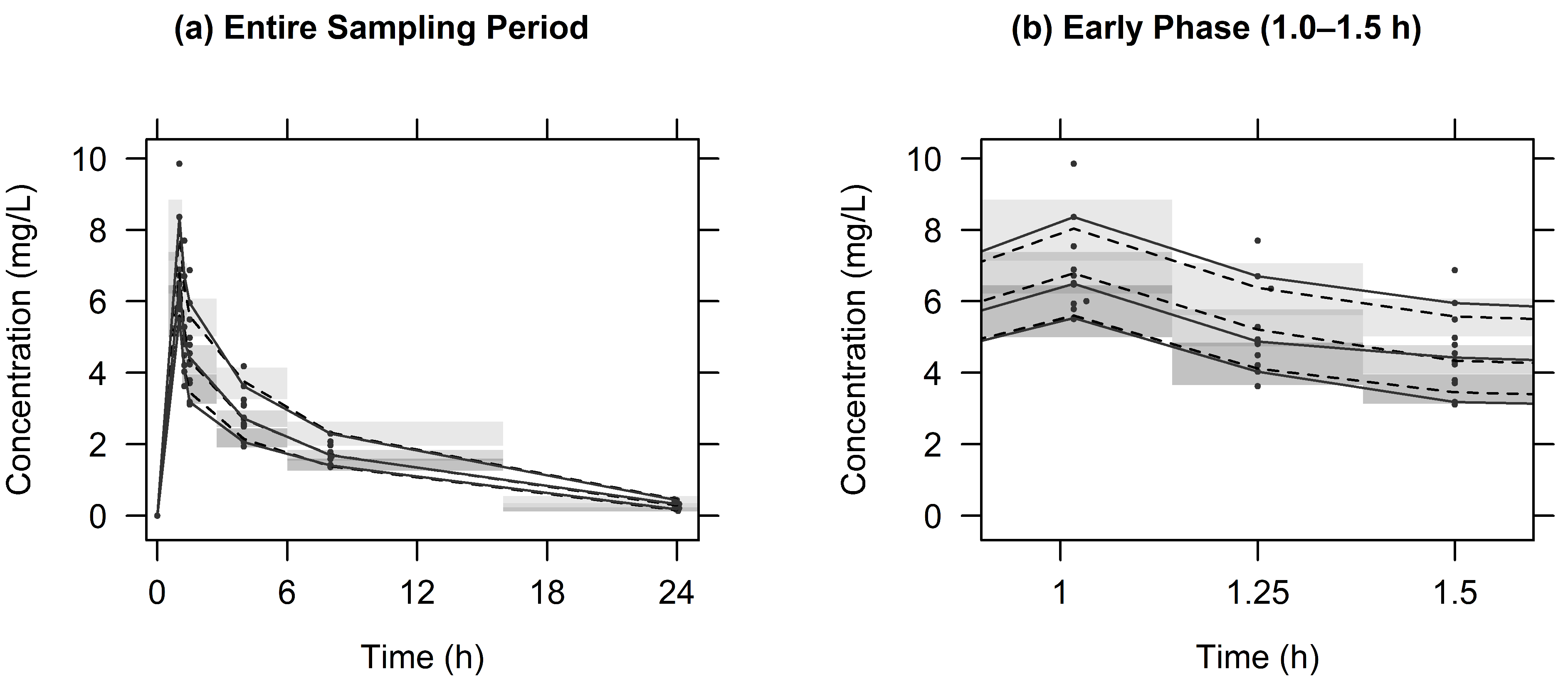
2.2. Population PK Analysis
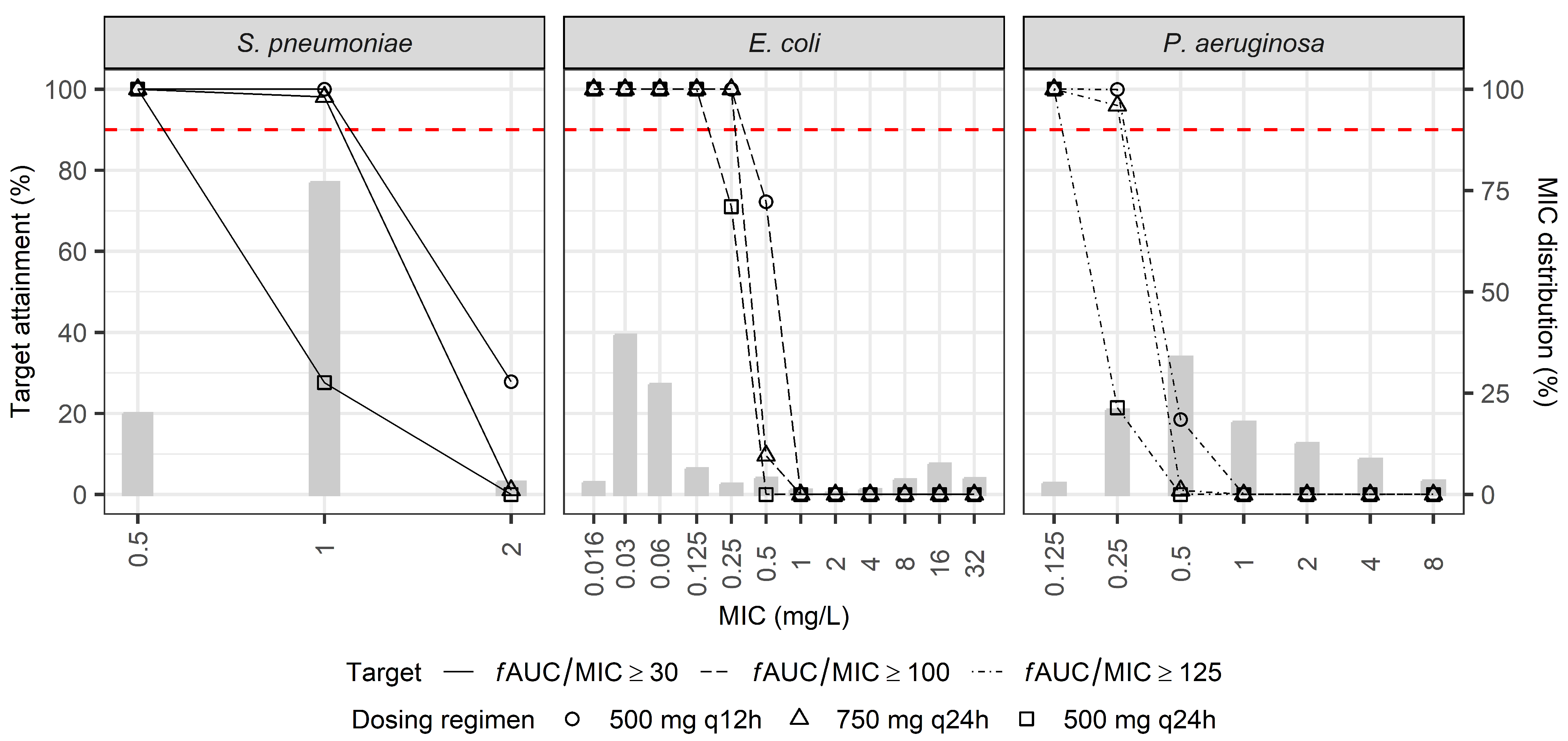
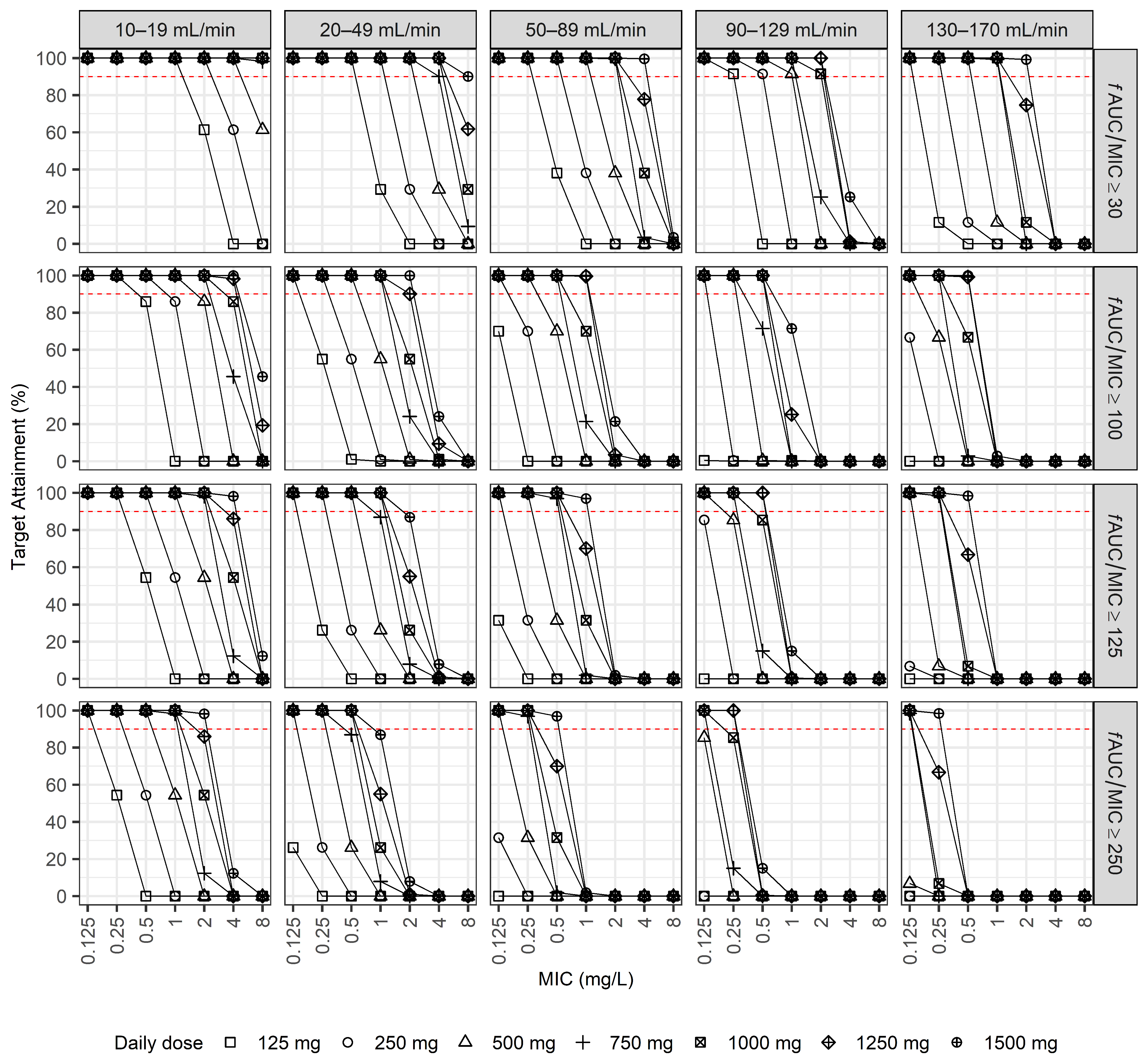
2.3. Dosage Simulations
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Study Design
4.3. Drug Assay
4.4. Population PK Analysis
4.5. Dosage Simulations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LVX | Levofloxacin |
| PK/PD | pharmacokinetic/pharmacodynamic |
| fAUC/MIC | the ratio of the 24-h area under the free drug concentration–time curve to the minimum inhibitory concentration |
| FDA | United States Food and Drug Administration |
| EMA | European Medicines Agency |
| CrCl | creatinine clearance |
| PTA | probability of target attainment |
| LC-MS/MS | liquid chromatography–tandem mass spectrometry |
| OFV | objective function value |
| VPC | visual predictive check |
| CWRES | conditional weighted residuals |
| PRED | population predictions |
| EUCAST | European Committee on Antimicrobial Susceptibility Testing |
| TBW | total body weight |
| LBM | lean body mass |
| eGFR | estimated glomerular filtration rate |
| MDRD | modification of diet in renal disease |
| CKD-EPI | chronic kidney disease epidemiology collaboration |
| CR | creatinine |
| CC | cystatin C |
| CL | total clearance |
| V1 | volume of distribution in the central compartment |
| V2 | volume of distribution for the peripheral compartments |
| Q | intercompartmental clearance between V1 and V2 |
| RSE | relative standard error |
| CI | confidence interval |
| Vd | volume of distribution |
Appendix A



Appendix B. Monte Carlo Simulation
References
- Fish, D.N.; Chow, A.T. The clinical pharmacokinetics of levofloxacin. Clin. Pharmacokinet. 1997, 32, 101–119. [Google Scholar] [CrossRef]
- Wimer, S.M.; Schoonover, L.; Garrison, M.W. Levofloxacin: A therapeutic review. Clin. Ther. 1998, 20, 1049–1070. [Google Scholar] [CrossRef]
- Langtry, H.D.; Lamb, H.M. Levofloxacin. Its use in infections of the respiratory tract, skin, soft tissues and urinary tract. Drugs 1998, 56, 487–515. [Google Scholar] [CrossRef] [PubMed]
- Croom, K.F.; Goa, K.L. Levofloxacin: A review of its use in the treatment of bacterial infections in the United States. Drugs 2003, 63, 2769–2802. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Levofloxacin Injection. Available online: https://nctr-crs.fda.gov/fdalabel/services/spl/set-ids/699fbdb8-a6fd-476a-9b80-08197d869534/spl-doc?hl=levofloxacin (accessed on 2 January 2025).
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. Medchemcomm 2019, 10, 1719–1739. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.H.; Brown, G.H.; Peterson, M.L.; Rotschafer, J.C. Application of fluoroquinolone pharmacodynamics. J. Antimicrob. Chemother. 2000, 46, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Craig, W.A. Pharmacokinetic/pharmacodynamic parameters: Rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 1998, 26, 1–10, quiz 11–12. [Google Scholar] [CrossRef]
- European Medicines Agency. Tavanic. Available online: https://www.ema.europa.eu/en/medicines/human/referrals/tavanic (accessed on 2 January 2025).
- Udy, A.A.; Baptista, J.P.; Lim, N.L.; Joynt, G.M.; Jarrett, P.; Wockner, L.; Boots, R.J.; Lipman, J. Augmented renal clearance in the ICU: Results of a multicenter observational study of renal function in critically ill patients with normal plasma creatinine concentrations*. Crit. Care Med. 2014, 42, 520–527. [Google Scholar] [CrossRef]
- Bilbao-Meseguer, I.; Rodriguez-Gascon, A.; Barrasa, H.; Isla, A.; Solinis, M.A. Augmented Renal Clearance in Critically Ill Patients: A Systematic Review. Clin. Pharmacokinet. 2018, 57, 1107–1121. [Google Scholar] [CrossRef]
- Chen, I.H.; Nicolau, D.P. Augmented Renal Clearance and How to Augment Antibiotic Dosing. Antibiotics 2020, 9, 393. [Google Scholar] [CrossRef]
- Sistanizad, M.; Hassanpour, R.; Pourheidar, E. Are Antibiotics Appropriately Dosed in Critically Ill Patients with Augmented Renal Clearance? A Narrative Review. Int. J. Clin. Pract. 2022, 2022, 1867674. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.X.; Qu, Q.; Zhuang, H.H.; Teng, X.Q.; Xu, W.X.; Liu, Y.P.; Xiao, Y.W.; Qu, J. Individualized antibiotic dosage regimens for patients with augmented renal clearance. Front. Pharmacol. 2023, 14, 1137975. [Google Scholar] [CrossRef]
- Lacy, M.K.; Lu, W.; Xu, X.; Tessier, P.R.; Nicolau, D.P.; Quintiliani, R.; Nightingale, C.H. Pharmacodynamic comparisons of levofloxacin, ciprofloxacin, and ampicillin against Streptococcus pneumoniae in an in vitro model of infection. Antimicrob. Agents Chemother. 1999, 43, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Bhavnani, S.M.; Forrest, A.; Hammel, J.P.; Drusano, G.L.; Rubino, C.M.; Ambrose, P.G. Pharmacokinetics-pharmacodynamics of quinolones against Streptococcus pneumoniae in patients with community-acquired pneumonia. Diagn. Microbiol. Infect. Dis. 2008, 62, 99–101. [Google Scholar] [CrossRef]
- Pai, M.P.; Cojutti, P.; Pea, F. Levofloxacin dosing regimen in severely morbidly obese patients (BMI ≥ 40 kg/m2) should be guided by creatinine clearance estimates based on ideal body weight and optimized by therapeutic drug monitoring. Clin. Pharmacokinet. 2014, 53, 753–762. [Google Scholar] [CrossRef]
- Boonpeng, A.; Jaruratanasirikul, S.; Wattanavijitkul, T.; Nawakitrangsan, M.; Samaeng, M. Population pharmacokinetics of oral levofloxacin in healthy volunteers and dosing optimization for multidrug-resistant tuberculosis therapy. Biopharm. Drug Dispos. 2021, 42, 329–337. [Google Scholar] [CrossRef]
- Canoui, E.; Kerneis, S.; Morand, P.; Enser, M.; Gauzit, R.; Eyrolle, L.; Leclerc, P.; Contejean, A.; Zheng, Y.; Anract, P.; et al. Oral levofloxacin: Population pharmacokinetics model and pharmacodynamics study in bone and joint infections. J. Antimicrob. Chemother. 2022, 77, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, E.; Abdul-Aziz, M.H.; Cotta, M.O.; Susaniwati, S.; Cahjono, H.; Sari, I.Y.; Wibowo, T.; Marpaung, F.R.; Roberts, J.A. Population pharmacokinetics and dose optimization of intravenous levofloxacin in hospitalized adult patients. Sci. Rep. 2022, 12, 8930. [Google Scholar] [CrossRef]
- He, Y.Y.; Sun, J.; Wu, Y.E.; Wang, Y.B.; van den Anker, J.; Hao, G.X.; Sun, D.Q.; Zhao, W. Population pharmacokinetics and dose optimization of levofloxacin in elderly patients with pneumonia. Br. J. Clin. Pharmacol. 2024, 90, 1213–1221. [Google Scholar] [CrossRef]
- James, W.P.T. Research on obesity. Nutr. Bull. 1977, 4, 187–190. [Google Scholar] [CrossRef]
- Du Bois, D.; Du Bois, E.F. A formula to estimate the approximate surface area if height and weight be known. 1916. Nutrition 1989, 5, 303–311; discussion 312–313. [Google Scholar]
- Matthews, I.; Kirkpatrick, C.; Holford, N. Quantitative justification for target concentration intervention—Parameter variability and predictive performance using population pharmacokinetic models for aminoglycosides. Br. J. Clin. Pharmacol. 2004, 58, 8–19. [Google Scholar] [CrossRef]
- Holford, N.; O’Hanlon, C.J.; Allegaert, K.; Anderson, B.; Falcao, A.; Simon, N.; Lo, Y.L.; Thomson, A.H.; Sherwin, C.M.; Jacqz-Aigrain, E.; et al. A physiological approach to renal clearance: From premature neonates to adults. Br. J. Clin. Pharmacol. 2024, 90, 1066–1080. [Google Scholar] [CrossRef]
- Overholser, B.R.; Kays, M.B.; Lagvankar, S.; Goldman, M.; Mueller, B.A.; Sowinski, K.M. Pharmacokinetics of intravenously administered levofloxacin in men and women. Pharmacotherapy 2005, 25, 1310–1318. [Google Scholar] [CrossRef]
- Cao, G.; Zhang, J.; Wu, X.; Yu, J.; Chen, Y.; Ye, X.; Zhu, D.; Zhang, Y.; Guo, B.; Shi, Y. Pharmacokinetics and pharmacodynamics of levofloxacin injection in healthy Chinese volunteers and dosing regimen optimization. J. Clin. Pharm. Ther. 2013, 38, 394–400. [Google Scholar] [CrossRef]
- Cook, A.M.; Martin, C.; Adams, V.R.; Morehead, R.S. Pharmacokinetics of intravenous levofloxacin administered at 750 milligrams in obese adults. Antimicrob. Agents Chemother. 2011, 55, 3240–3243. [Google Scholar] [CrossRef]
- Eloy, G.; Lebeaux, D.; Launay, M.; Fernandez-Gerlinger, M.P.; Billaud, E.; Douez, E.; Mainardi, J.L.; Bouyer, B.; Jullien, V. Influence of Renal Function and Age on the Pharmacokinetics of Levofloxacin in Patients with Bone and Joint Infections. Antibiotics 2020, 9, 401. [Google Scholar] [CrossRef]
- Cojutti, P.G.; Ramos-Martin, V.; Schiavon, I.; Rossi, P.; Baraldo, M.; Hope, W.; Pea, F. Population Pharmacokinetics and Pharmacodynamics of Levofloxacin in Acutely Hospitalized Older Patients with Various Degrees of Renal Function. Antimicrob. Agents Chemother. 2017, 61, 10–128. [Google Scholar] [CrossRef]
- Roberts, J.A.; Cotta, M.O.; Cojutti, P.; Lugano, M.; Della Rocca, G.; Pea, F. Does Critical Illness Change Levofloxacin Pharmacokinetics? Antimicrob. Agents Chemother. 2015, 60, 1459–1463. [Google Scholar] [CrossRef]
- Thomas, J.K.; Forrest, A.; Bhavnani, S.M.; Hyatt, J.M.; Cheng, A.; Ballow, C.H.; Schentag, J.J. Pharmacodynamic evaluation of factors associated with the development of bacterial resistance in acutely ill patients during therapy. Antimicrob. Agents Chemother. 1998, 42, 521–527. [Google Scholar] [CrossRef]
- Watkins, R.R. Antibiotic stewardship in the era of precision medicine. JAC Antimicrob. Resist. 2022, 4, dlac066. [Google Scholar] [CrossRef]
- Rybak, M.J.; Le, J.; Lodise, T.P.; Levine, D.P.; Bradley, J.S.; Liu, C.; Mueller, B.A.; Pai, M.P.; Wong-Beringer, A.; Rotschafer, J.C.; et al. Therapeutic Monitoring of Vancomycin for Serious Methicillin-resistant Staphylococcus aureus Infections: A Revised Consensus Guideline and Review by the American Society of Health-system Pharmacists, the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists. Clin. Infect. Dis. 2020, 71, 1361–1364. [Google Scholar] [CrossRef]
- Mahmoud, S.H.; Shen, C. Augmented Renal Clearance in Critical Illness: An Important Consideration in Drug Dosing. Pharmaceutics 2017, 9, 36. [Google Scholar] [CrossRef]
- Sanghavi, K.; Ribbing, J.; Rogers, J.A.; Ahmed, M.A.; Karlsson, M.O.; Holford, N.; Chasseloup, E.; Ahamadi, M.; Kowalski, K.G.; Cole, S.; et al. Covariate modeling in pharmacometrics: General points for consideration. CPT Pharmacomet. Syst. Pharmacol. 2024, 13, 710–728. [Google Scholar] [CrossRef]
- Gonzalez-Sales, M.; Holford, N.; Bonnefois, G.; Desrochers, J. Wide size dispersion and use of body composition and maturation improves the reliability of allometric exponent estimates. J. Pharmacokinet. Pharmacodyn. 2022, 49, 151–165. [Google Scholar] [CrossRef]
- Janmahasatian, S.; Duffull, S.B.; Ash, S.; Ward, L.C.; Byrne, N.M.; Green, B. Quantification of lean bodyweight. Clin. Pharmacokinet. 2005, 44, 1051–1065. [Google Scholar] [CrossRef]
- Anderson, B.J.; Holford, N.H. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab. Pharmacokinet. 2009, 24, 25–36. [Google Scholar] [CrossRef]
- Rhodin, M.M.; Anderson, B.J.; Peters, A.M.; Coulthard, M.G.; Wilkins, B.; Cole, M.; Chatelut, E.; Grubb, A.; Veal, G.J.; Keir, M.J.; et al. Human renal function maturation: A quantitative description using weight and postmenstrual age. Pediatr. Nephrol. 2009, 24, 67–76. [Google Scholar] [CrossRef]
- Cockcroft, D.W.; Gault, M.H. Prediction of creatinine clearance from serum creatinine. Nephron 1976, 16, 31–41. [Google Scholar] [CrossRef]
- Levey, A.S.; Coresh, J.; Greene, T.; Stevens, L.A.; Zhang, Y.L.; Hendriksen, S.; Kusek, J.W.; Van Lente, F.; Chronic Kidney Disease Epidemiology, C. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann. Intern. Med. 2006, 145, 247–254. [Google Scholar] [CrossRef]
- Inker, L.A.; Eneanya, N.D.; Coresh, J.; Tighiouart, H.; Wang, D.; Sang, Y.; Crews, D.C.; Doria, A.; Estrella, M.M.; Froissart, M.; et al. New Creatinine- and Cystatin C-Based Equations to Estimate GFR without Race. N. Engl. J. Med. 2021, 385, 1737–1749. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Median (Min–Max) | ||
|---|---|---|---|
| Total (n = 12) | Female (n = 8) | Male (n = 4) | |
| Demographic characteristics | |||
| Age, years | 35.5 (29.0–44.0) | 37.5 (29.0–44.0) | 33.0 (30.0–44.0) |
| TBW, kg | 68.0 (47.3–77.2) | 58.4 (47.3–75.7) | 72.8 (67.5–77.2) |
| LBM, kg a | 47.9 (37.7–60.3) | 43.6 (37.7–51.3) | 58.1 (53.6–60.3) |
| Height, cm | 165 (152–181) | 161 (152–171) | 174 (168–181) |
| Body surface area, m2 b | 1.75 (1.45–1.94) | 1.63 (1.45–1.86) | 1.89 (1.77–1.94) |
| Body mass index, kg/m2 | 23.5 (18.3–28.9) | 22.5 (18.3–28.9) | 23.8 (21.3–26.8) |
| Laboratory characteristics | |||
| Protein, g/dL | 7.45 (6.80–8.00) | 7.50 (6.80–8.00) | 7.20 (6.90–7.50) |
| Albumin, g/dL | 4.75 (4.40–5.20) | 4.75 (4.40–5.20) | 4.80 (4.60–4.90) |
| Creatinine, mg/dL | 0.850 (0.560–1.11) | 0.780 (0.560–0.900) | 1.06 (0.880–1.11) |
| Cystatin C, mg/dL | 0.880 (0.780–1.06) | 0.880 (0.780–1.06) | 0.885 (0.850–0.930) |
| Renal functions | |||
| CrCl, mL/min c | 106 (74.8–113) | 99.6 (74.8–113) | 106 (90.5–113) |
| eGFRMDRD, mL/min/1.73 m2 d | 80.1 (69.8–121) | 81.6 (69.8–121) | 78.8 (77.8–99.1) |
| eGFRCKD-EPI_CR, mL/min/1.73 m2 e | 95.8 (83.1–120) | 98.1 (83.1–120) | 92.7 (91.6–116) |
| eGFRCKD-EPI_CRCC, mL/min/1.73 m2 f | 98.2 (81.3–119) | 97.2 (81.3–119) | 99.4 (94.0–113) |
| Parameter | Estimates | RSE (%) [Shrinkage, %] | Bootstrap Median (95% CI) |
|---|---|---|---|
| Structural model | |||
| CL = θ1 × (CrCl/105.71) θ2 | |||
| θ1 (L/h) | 13.4 | 3.36 | 13.4 (12.6–14.5) |
| θ2 | 0.901 | 16.8 | 0.900 (0.392–1.26) |
| V1 | 34.3 | 8.93 | 34.5 (29.6–41.3) |
| Q | 72.8 | 10.9 | 72.4 (56.6–86.2) |
| V2 = θ3 × (LBM/47.91) θ4 | |||
| θ3 (L) | 67.7 | 3.42 | 67.4 (62.1–71.9) |
| θ4 | 1.75 | 12.5 | 1.76 (1.32–2.27) |
| Interindividual variability | |||
| CL (%) | 8.99 | 15.3 [3.58] | 8.23 (4.65–11.1) |
| Q (%) | 36.0 | 30.6 [10.2] | 35.3 (0.000–53.0) |
| Residual variability | |||
| Proportional error (%) | 6.99 | 13.8 [7.09] | 6.72 (4.68–8.24) |
| CrCl (mL/min) | |||||
|---|---|---|---|---|---|
| MIC (mg/L) | 10–19 | 20–49 | 50–89 | 90–129 | 130–170 |
| fAUC/MIC ≥ 30 | |||||
| 0.125 | 125 (100) | 125 (100) | 125 (100) | 125 (100) | 125 (100) |
| 0.25 | 125 (100) | 125 (100) | 125 (100) | 125 (91.5) | 250 (100) |
| 0.5 | 125 (100) | 125 (100) | 250 (100) | 250 (91.5) | 500 (100) |
| 1 | 125 (100) | 250 (100) | 500 (100) | 500 (91.5) | 750 (99.2) |
| 2 | 250 (100) | 500 (100) | 750 (99.6) | 1000 (91.5) | 1500 (99.2) |
| 4 | 500 (100) | 750 (90.1) | 1500 (99.6) | 1500 (25.2) | – |
| 8 | 750 (98.2) | 1500 (90.1) | – | – | – |
| fAUC/MIC ≥ 100 | |||||
| 0.125 | 125 (100) | 125 (100) | 250 (100) | 250 (100) | 500 (100) |
| 0.25 | 125 (100) | 250 (100) | 500 (100) | 500 (100) | 750 (100) |
| 0.5 | 250 (100) | 500 (100) | 750 (100) | 1000 (100) | 1250 (99.2) |
| 1 | 500 (100) | 750 (100) | 1250 (99.6) | 1500 (71.5) | – |
| 2 | 750 (100) | 1250 (90.1) | 1500 (21.4) | – | – |
| 4 | 1250 (98.2) | 1500 (24.1) | – | – | – |
| 8 | 1500 (45.6) | – | – | – | – |
| fAUC/MIC ≥ 125 | |||||
| 0.125 | 125 (100) | 125 (100) | 250 (100) | 500 (100) | 500 (100) |
| 0.25 | 125 (100) | 250 (100) | 500 (100) | 750 (100) | 750 (98.4) |
| 0.5 | 250 (100) | 500 (100) | 750 (96.9) | 1250 (100) | 1500 (98.4) |
| 1 | 500 (100) | 1000 (100) | 1500 (96.9) | 1500 (15.0) | – |
| 2 | 750 (98.2) | 1500 (86.9) | – | – | – |
| 4 | 1500 (98.2) | – | – | – | – |
| 8 | 1500 (12.3) | – | – | – | – |
| fAUC/MIC ≥ 250 | |||||
| 0.125 | 125 (100) | 250 (100) | 500 (100) | 750 (100) | 750 (98.4) |
| 0.25 | 250 (100) | 500 (100) | 750 (96.9) | 1250 (100) | 1500 (98.4) |
| 0.5 | 500 (100) | 1000 (100) | 1500 (96.9) | 1500 (15.0) | – |
| 1 | 750 (98.2) | 1500 (86.9) | – | – | – |
| 2 | 1500 (98.2) | – | – | – | – |
| 4 | 1500 (12.3) | – | – | – | – |
| 8 | – | – | – | – | – |
| Study [Reference] | Population | CL (L/h) (70 kg) | Vss (L) (70 kg) | CL (L/h) (53 kg LBM) | Vss (L) (53 kg LBM) | CrCl (mL/min) |
|---|---|---|---|---|---|---|
| Our Study | Healthy Korean adults | M: 11.9 F: 11.1 | M: 123 F: 93.0 | M: 13.2 F: 13.7 | M: 118 F: 112 | M: 106 F: 99.6 |
| Overholser et al. [26] | Healthy U.S. adults | M: 11.2 F: 10.5 | M: 113 F: 91.7 | M: 10.6 F: 10.6 | M: 106 F: 92.2 | M: 105.7 F: 97.0 |
| Cao et al. [27] | Healthy Chinese adults | SDS: 9.32 MDS: 7.79 | SDS: 103 MDS: 96.9 | NR | NR | SDS: 138.0 MDS: 125.9 |
| Cook et al. [28] | Obese patients | AMB: 11.1 HOS: 3.11 | AMB: 49.1 HOS: 35.8 | NR | NR | AMB: 87.2 HOS: 82.2 |
| Eloy et al. [29] | Patients with bone and joint infections | 5.08 | 88.0 | NR | NR | 120.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-J.; Kang, G.; Zang, D.-Y.; Lee, D.-H. Development of a Population Pharmacokinetic Model of Levofloxacin in Healthy Adults and Identification of Optimal Dosing Regimens. Pharmaceuticals 2025, 18, 621. https://doi.org/10.3390/ph18050621
Lee Y-J, Kang G, Zang D-Y, Lee D-H. Development of a Population Pharmacokinetic Model of Levofloxacin in Healthy Adults and Identification of Optimal Dosing Regimens. Pharmaceuticals. 2025; 18(5):621. https://doi.org/10.3390/ph18050621
Chicago/Turabian StyleLee, Yun-Jung, Gaeun Kang, Dae-Young Zang, and Dong-Hwan Lee. 2025. "Development of a Population Pharmacokinetic Model of Levofloxacin in Healthy Adults and Identification of Optimal Dosing Regimens" Pharmaceuticals 18, no. 5: 621. https://doi.org/10.3390/ph18050621
APA StyleLee, Y.-J., Kang, G., Zang, D.-Y., & Lee, D.-H. (2025). Development of a Population Pharmacokinetic Model of Levofloxacin in Healthy Adults and Identification of Optimal Dosing Regimens. Pharmaceuticals, 18(5), 621. https://doi.org/10.3390/ph18050621