
Chemical Variations on the p53 Reactivation Theme




Abstract
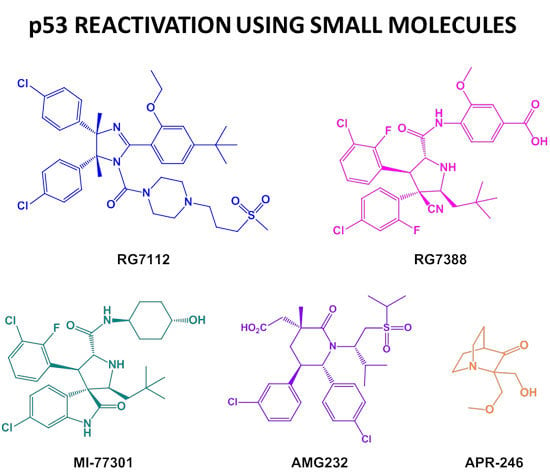
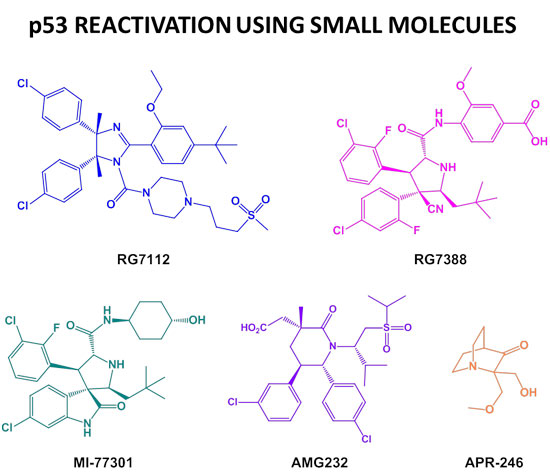
:
1. Introduction
2. Reactivation of p53 as a Therapeutic Strategy
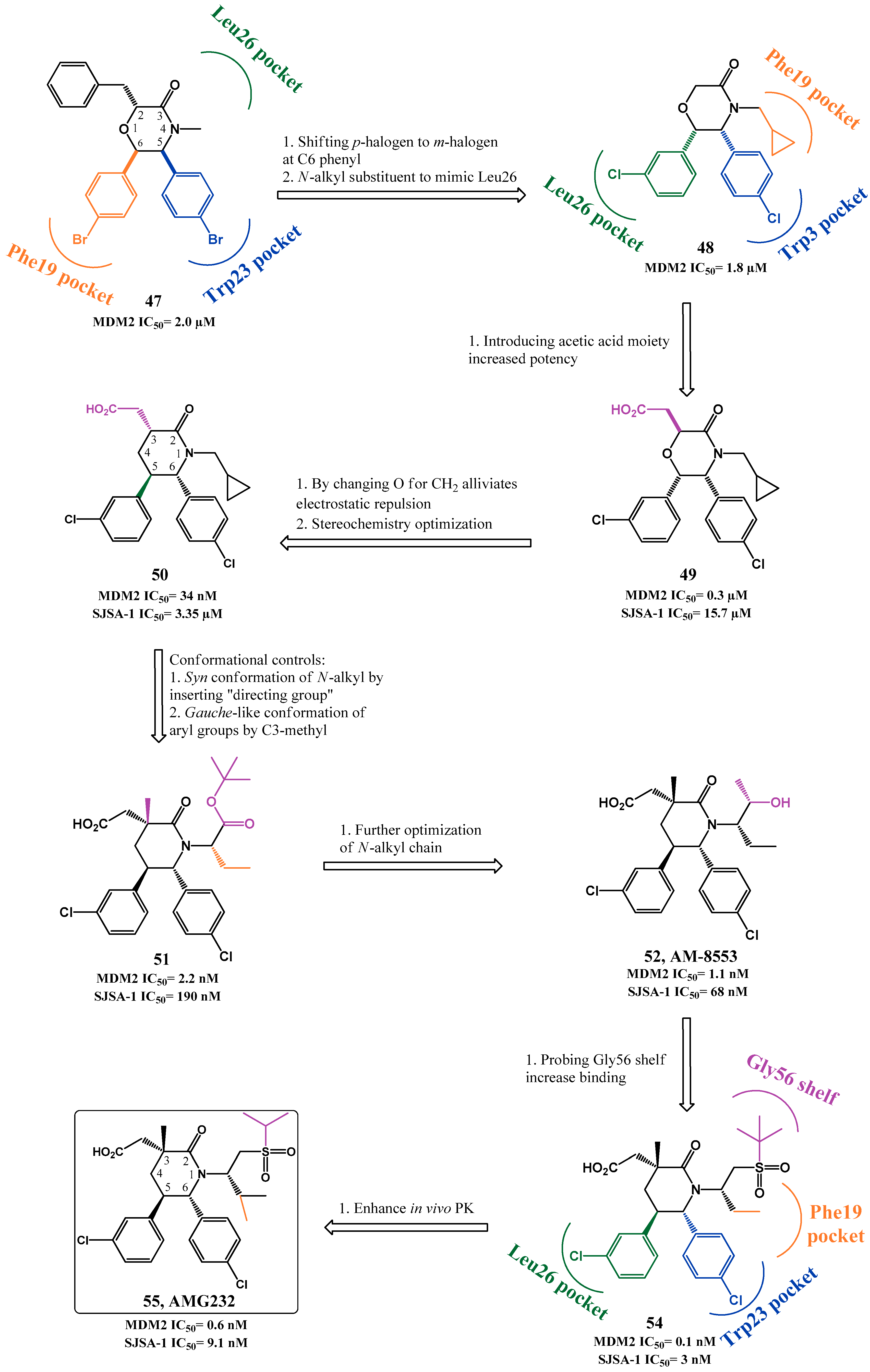
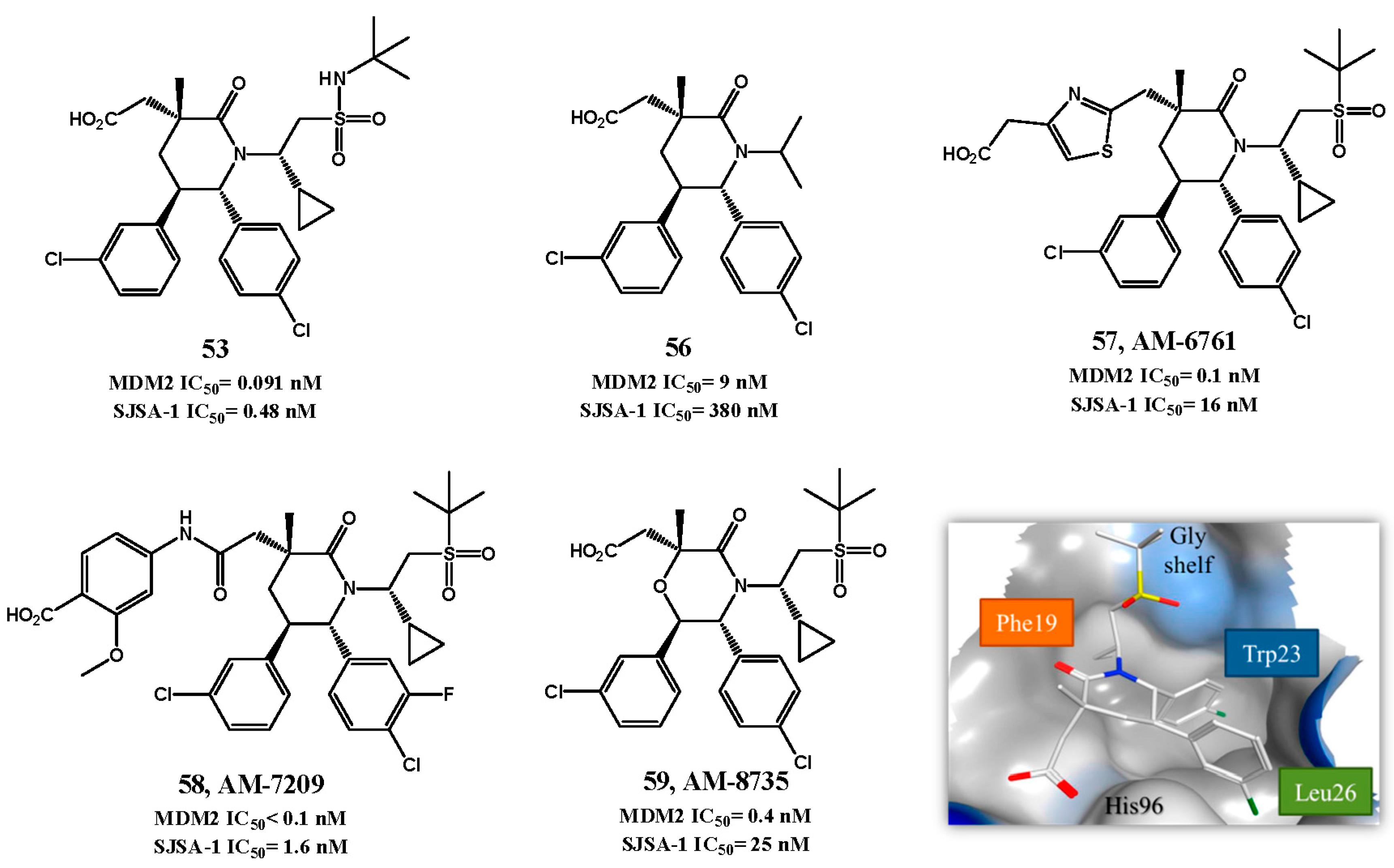
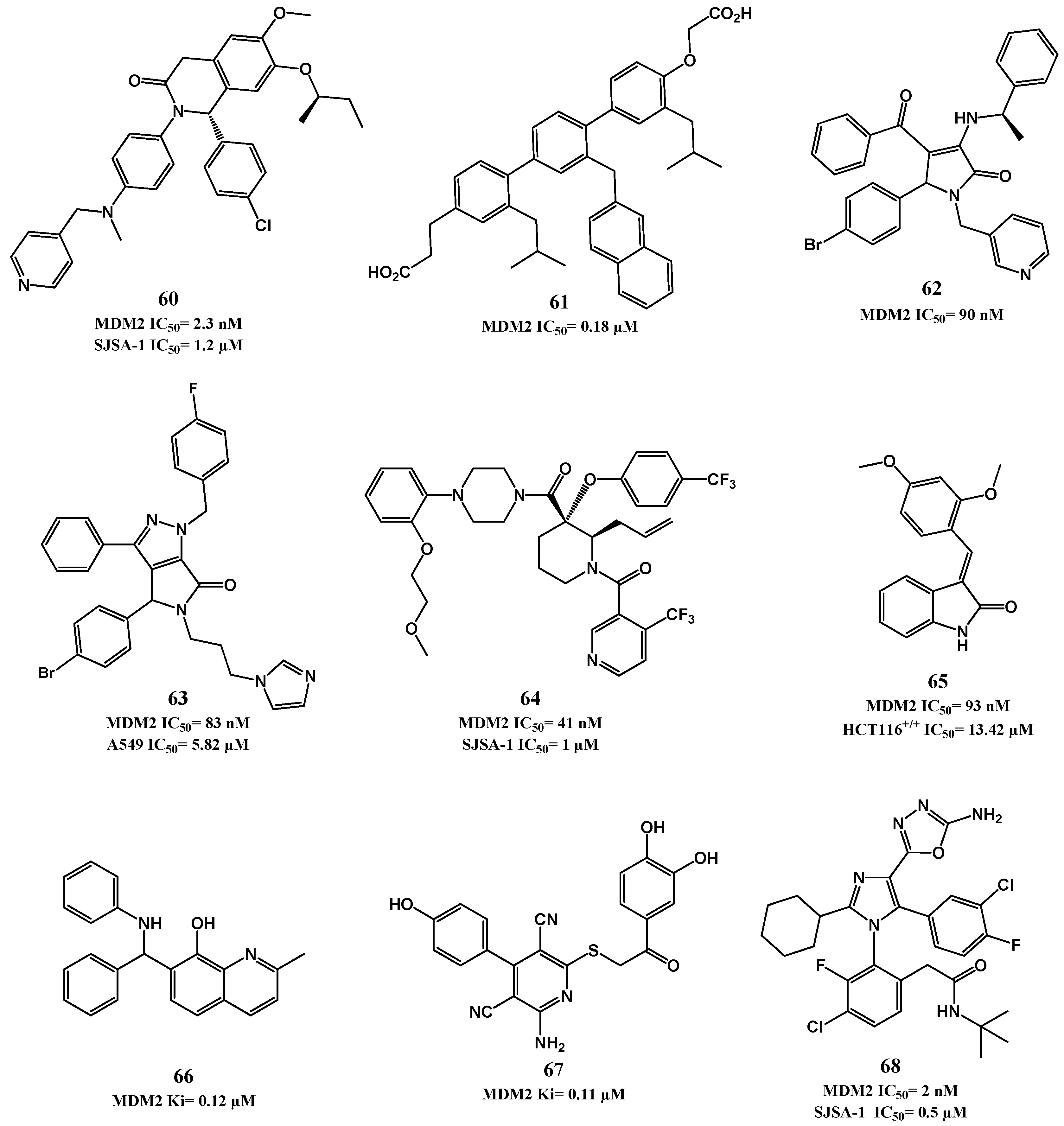
2.1. Targeting p53-MDM2 Interaction
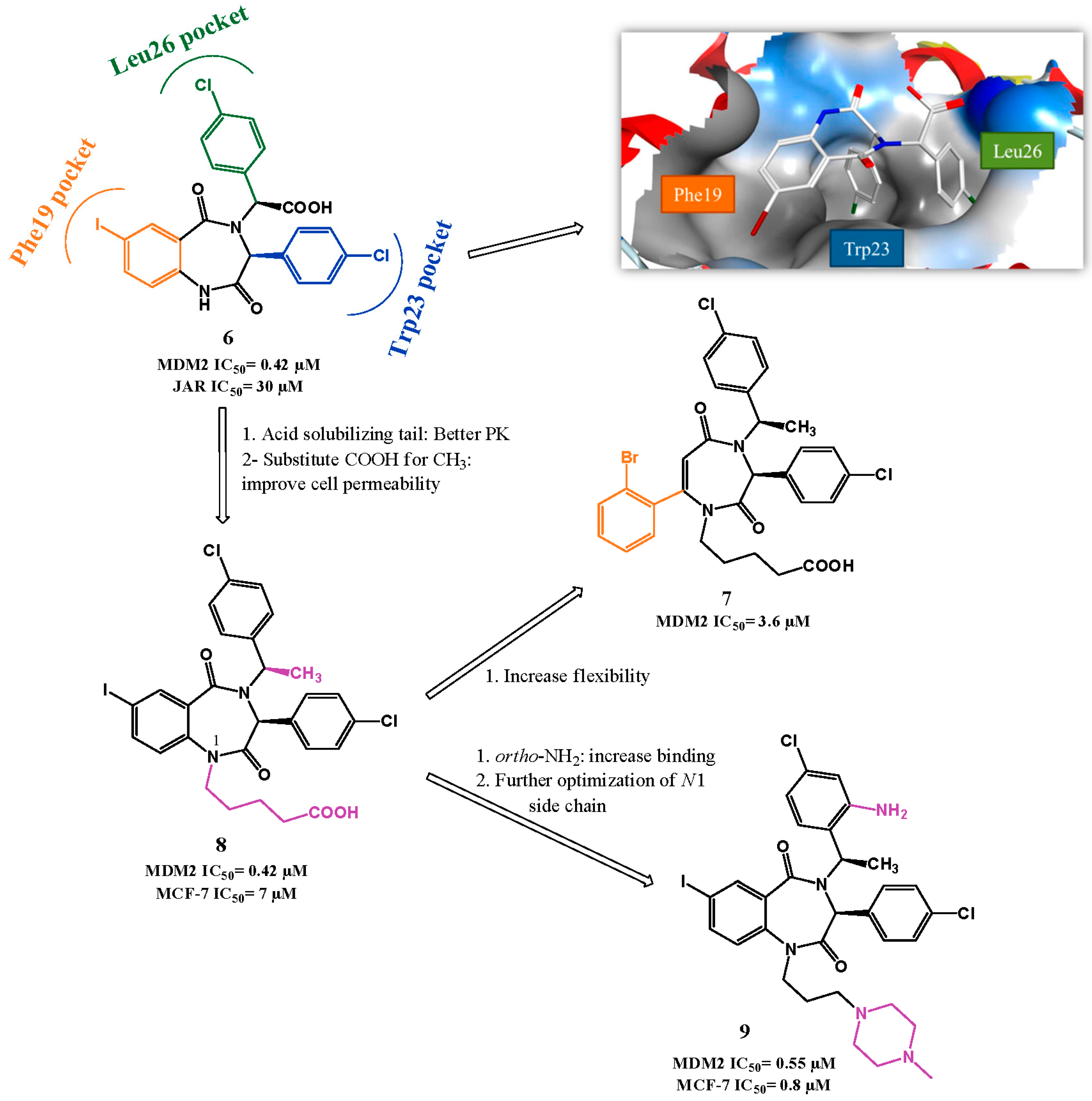
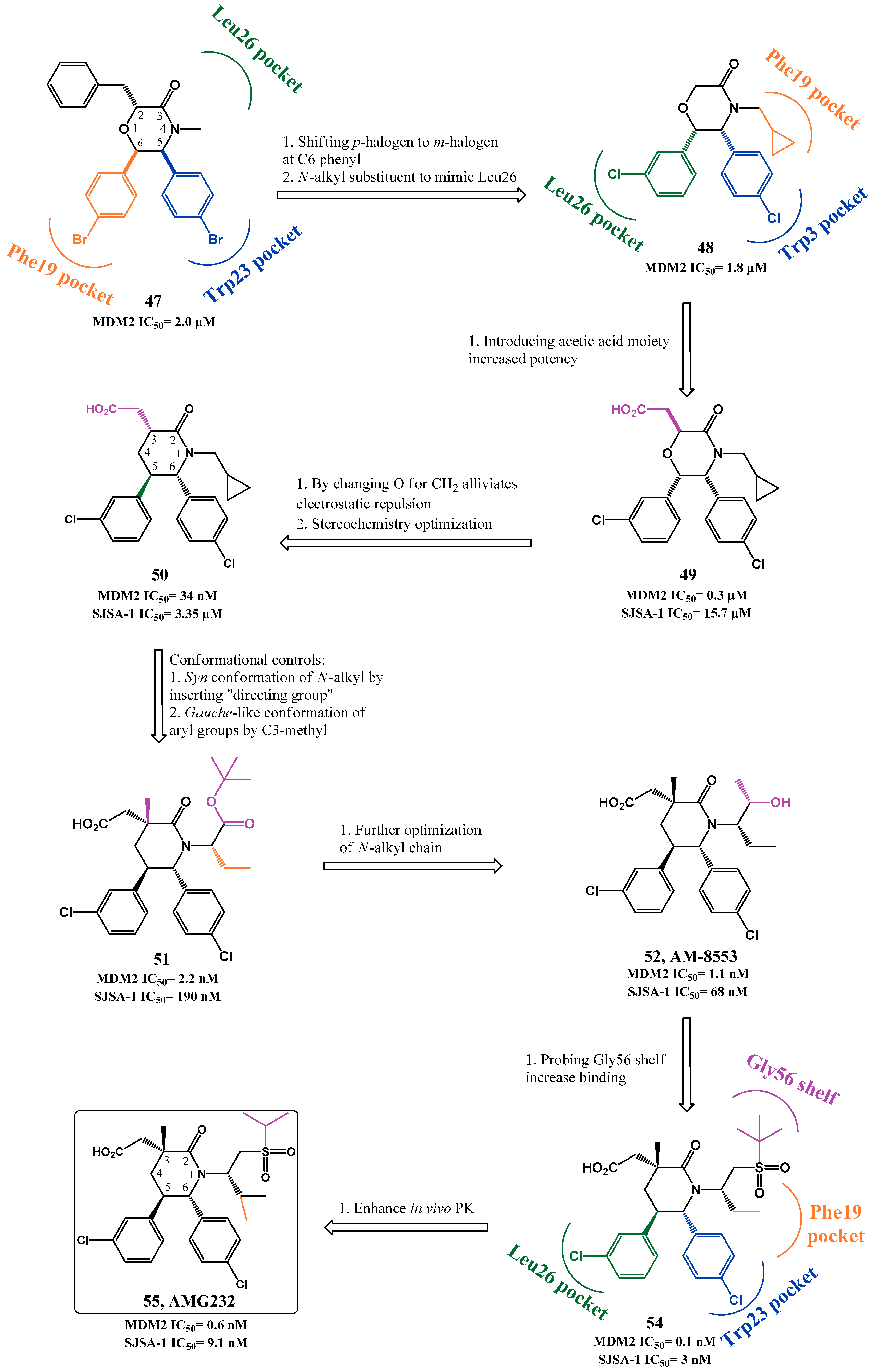
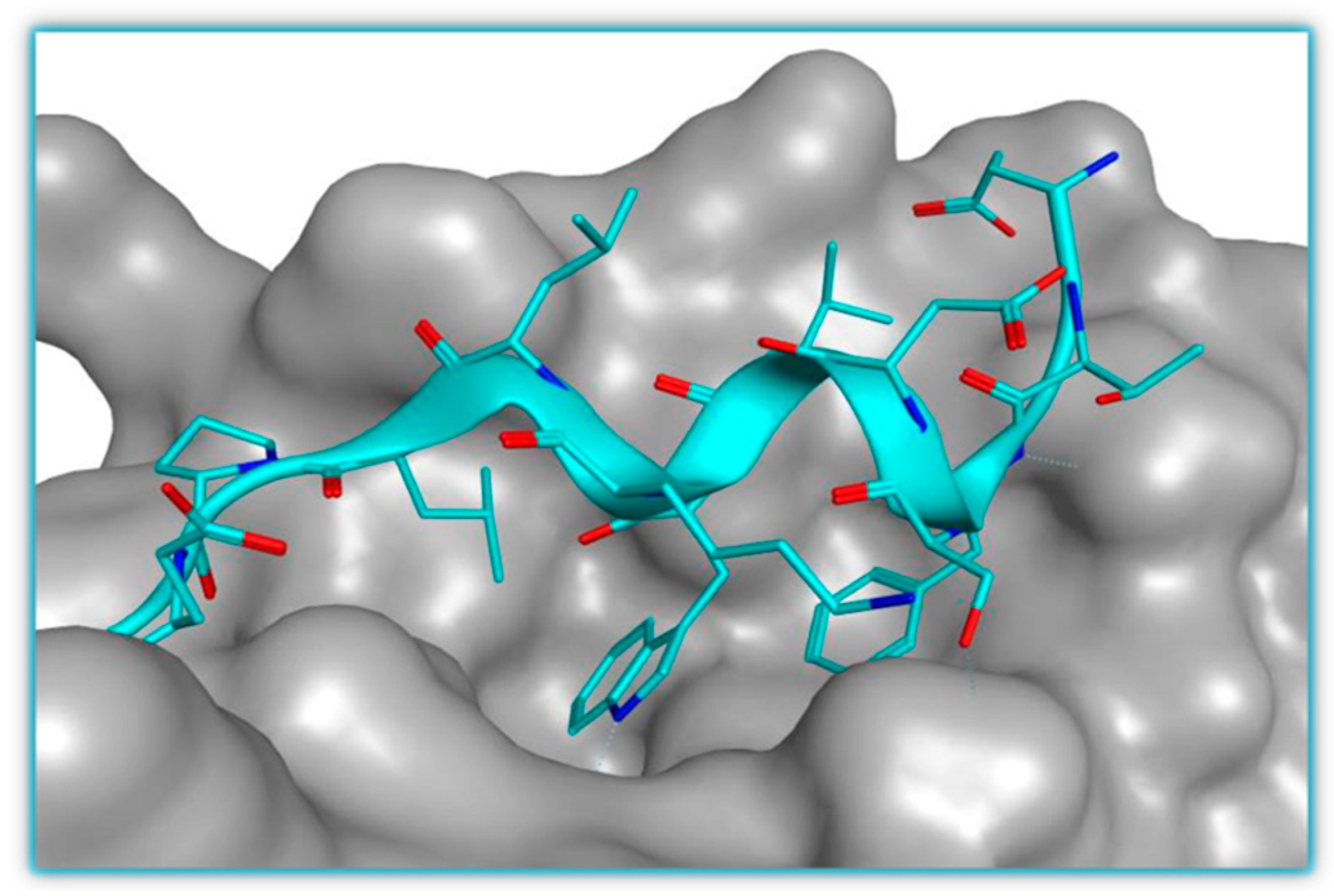
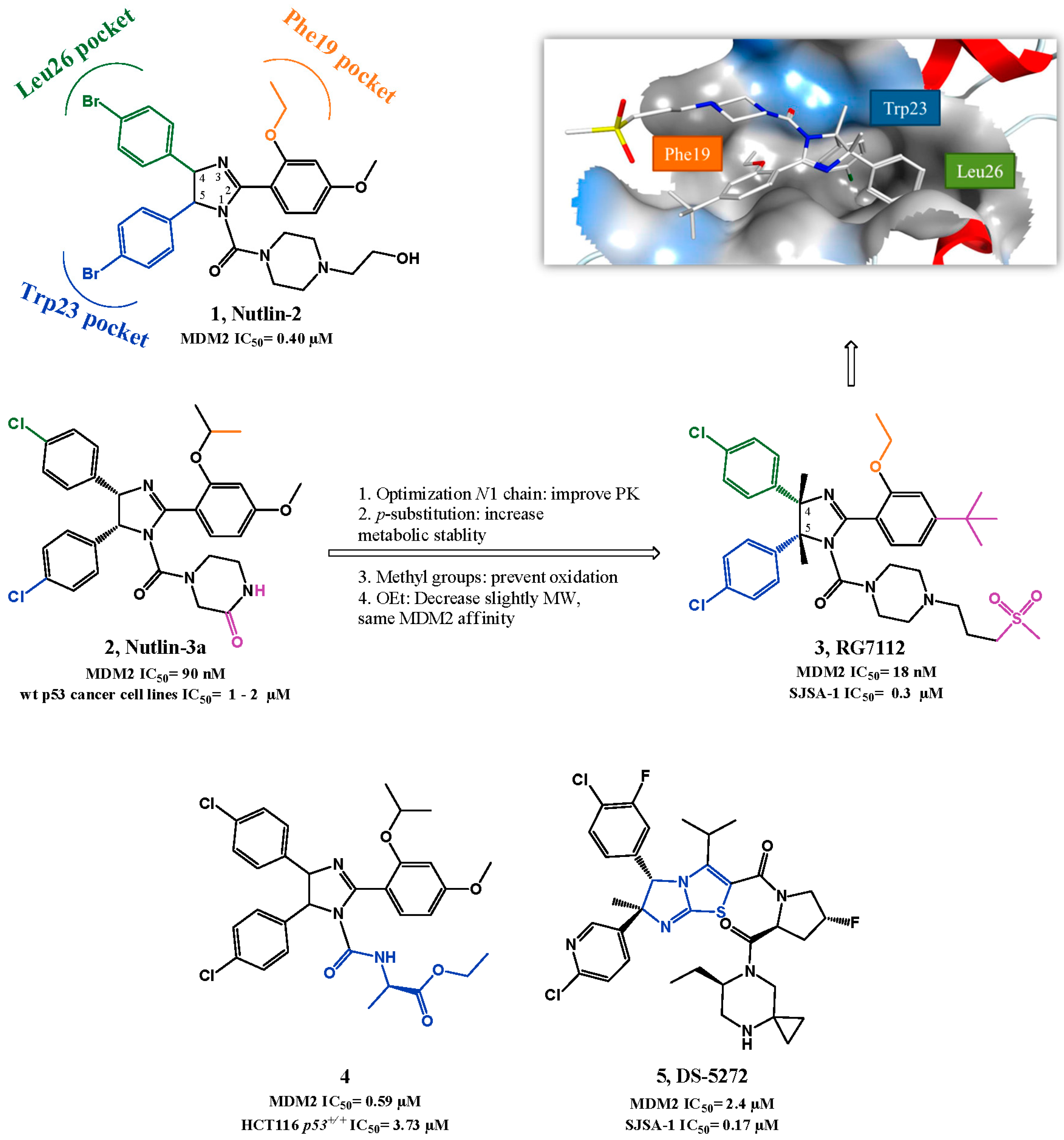
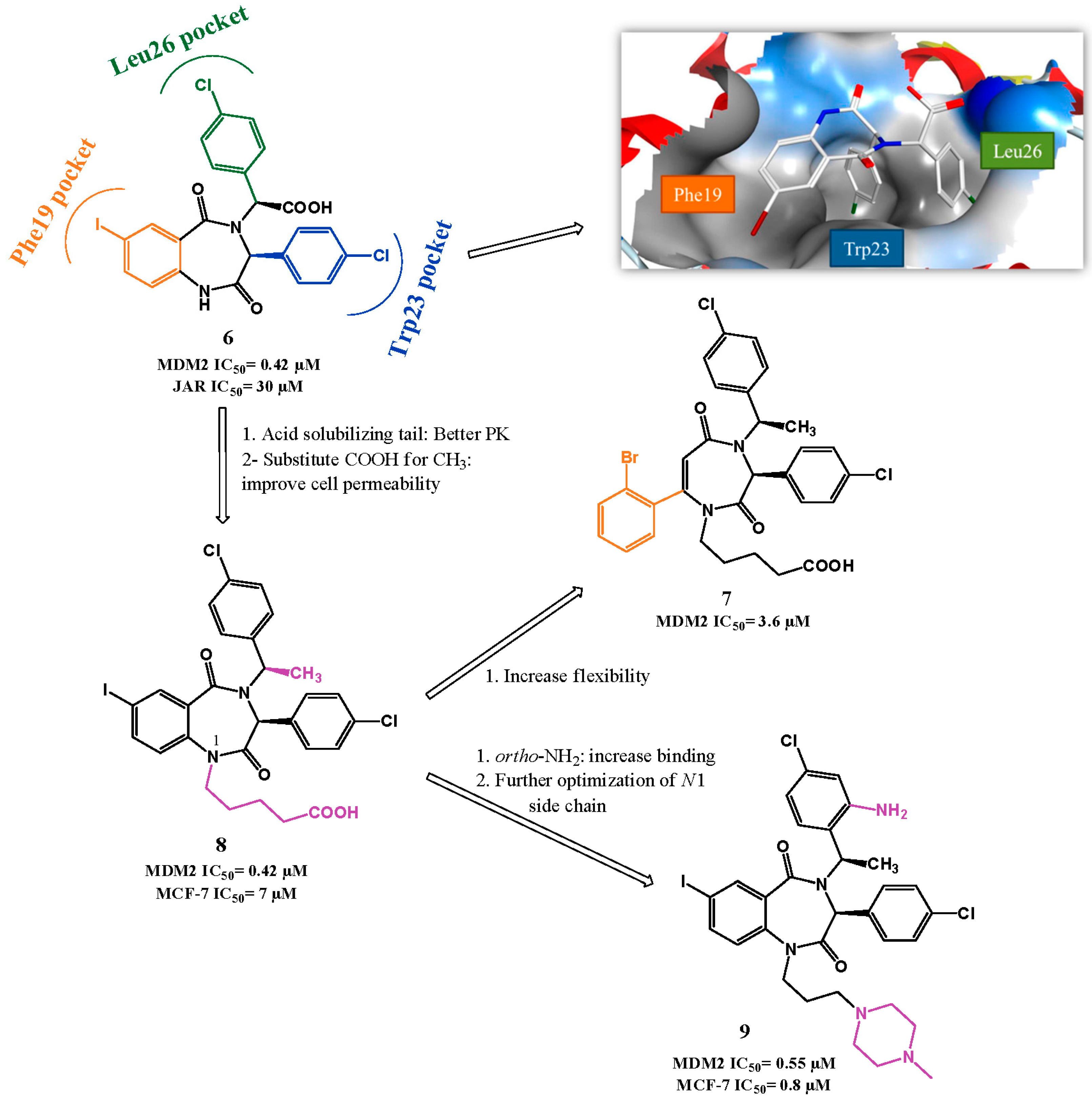
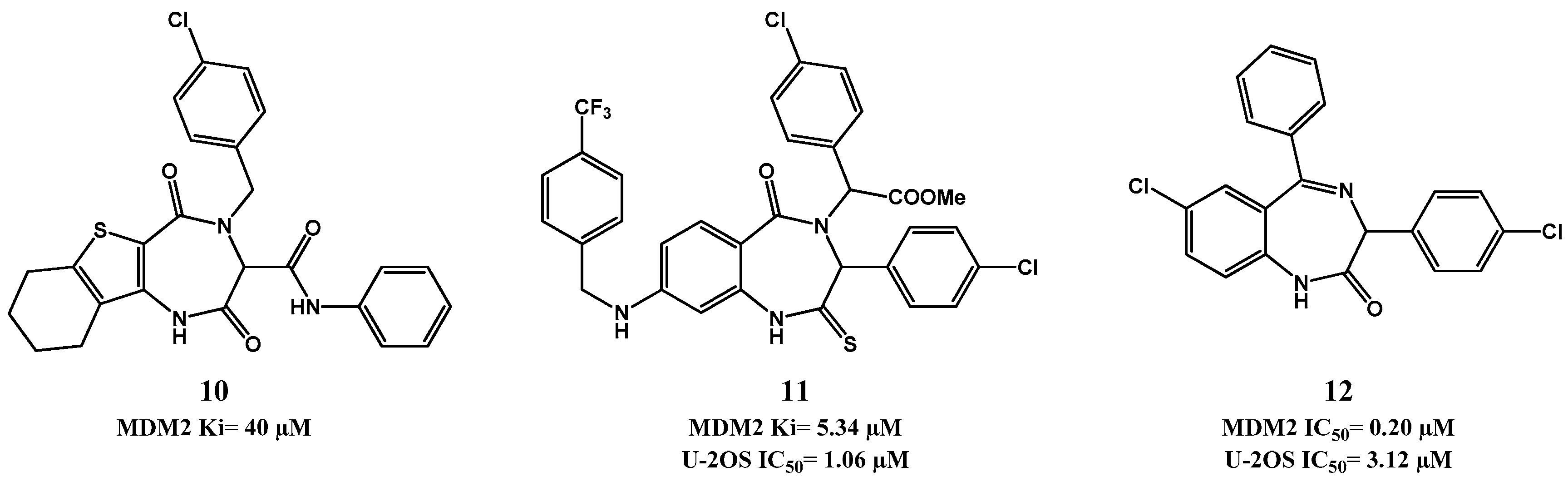
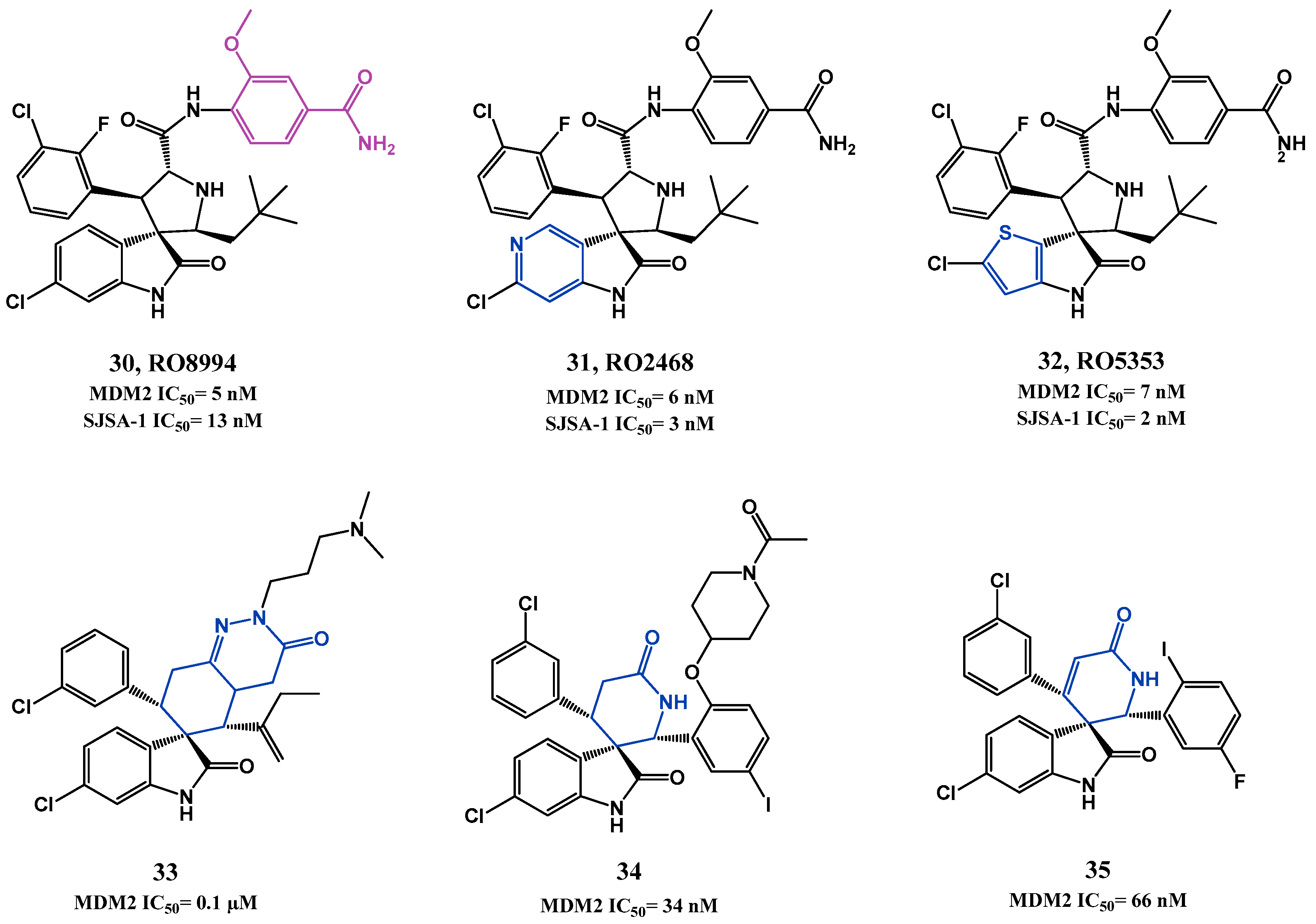
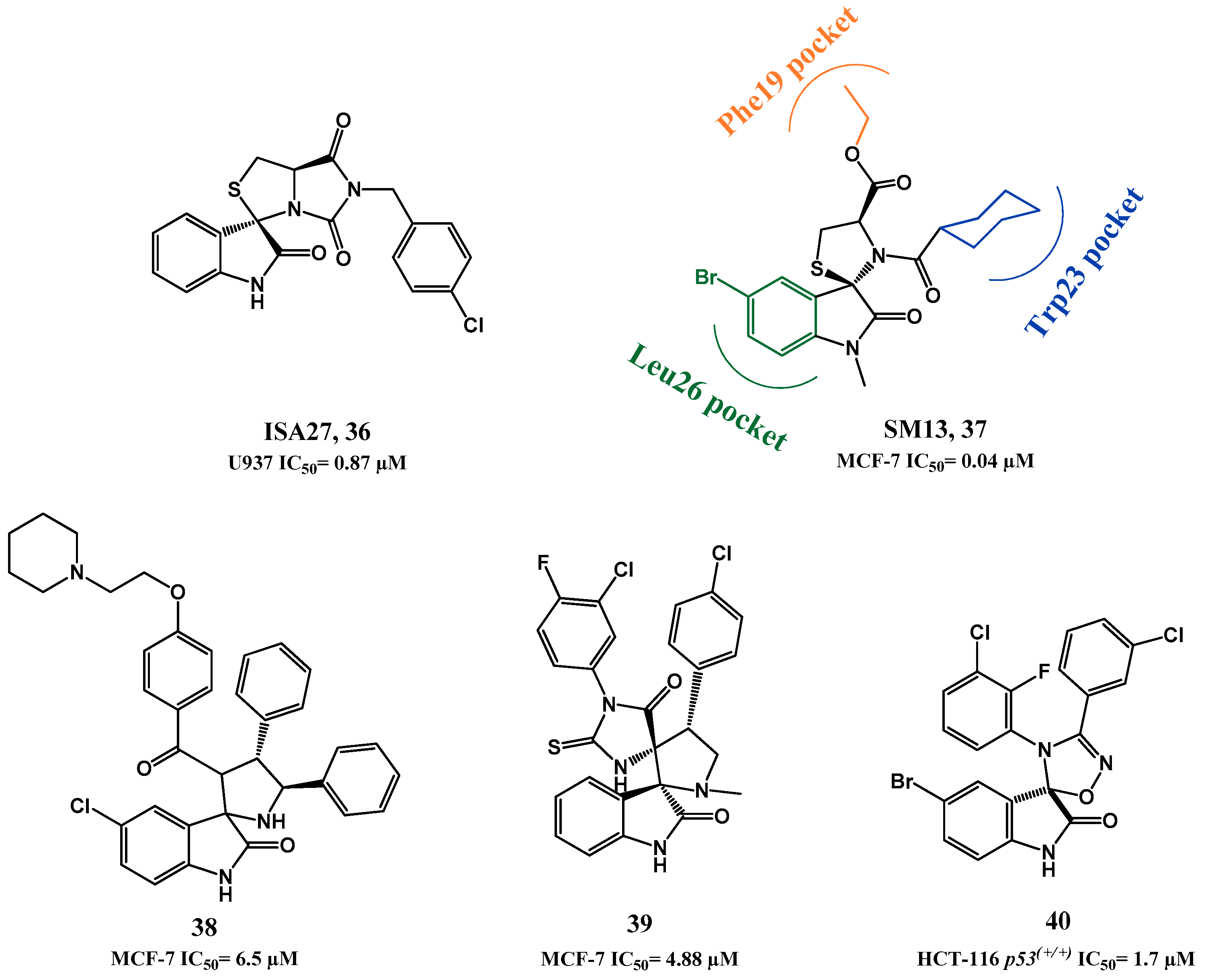
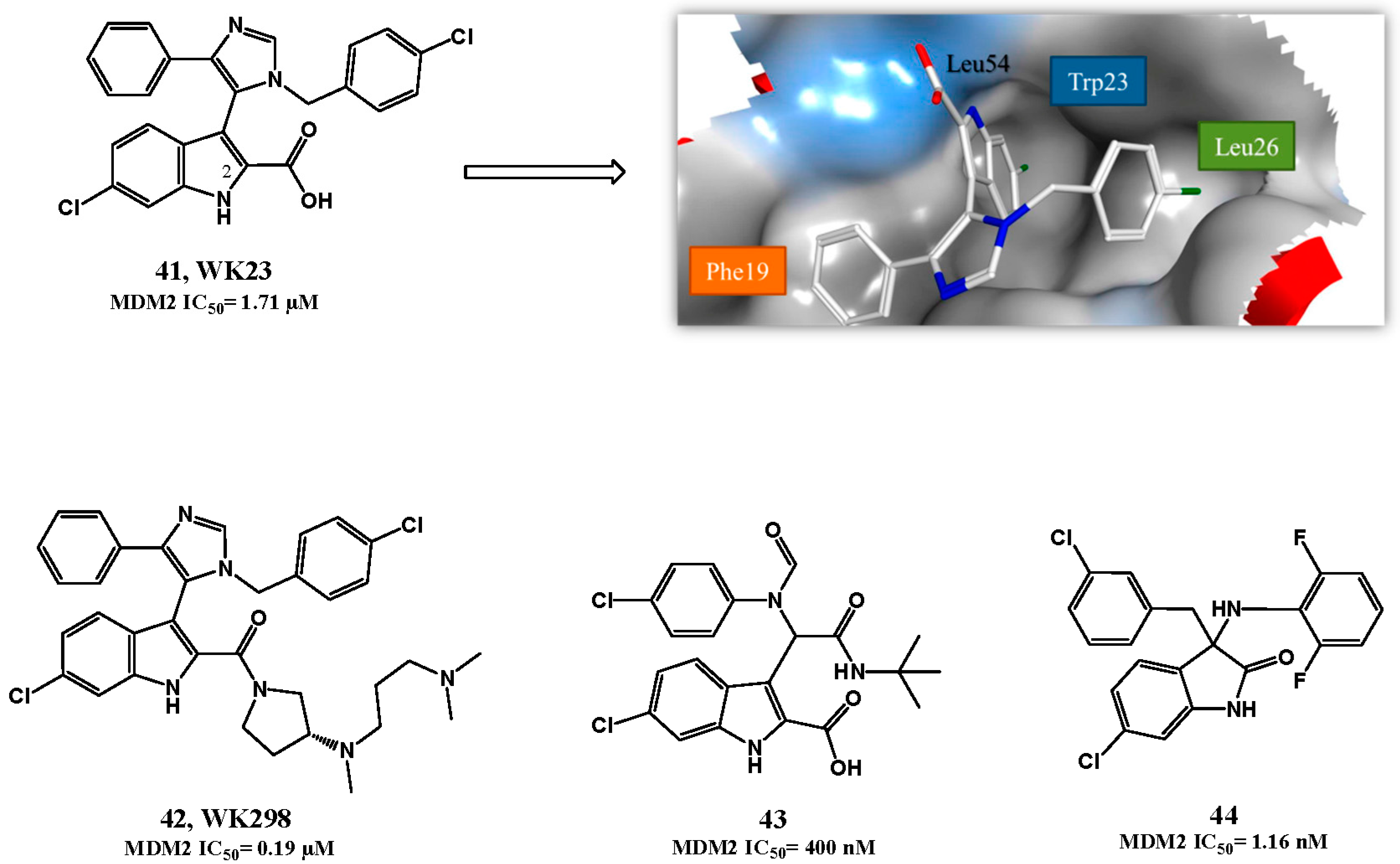
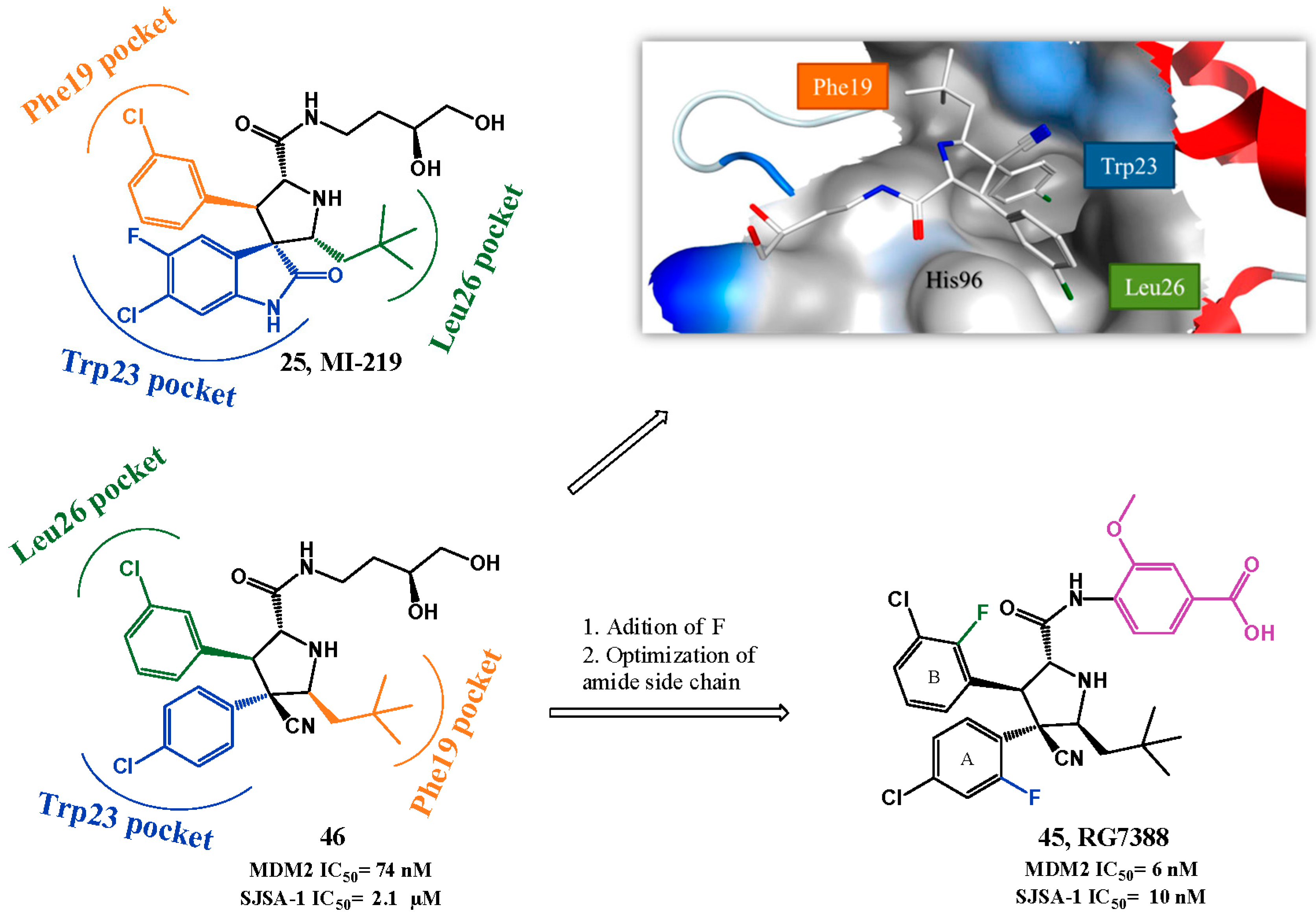
p53-MDM2 Interaction Inhibitors
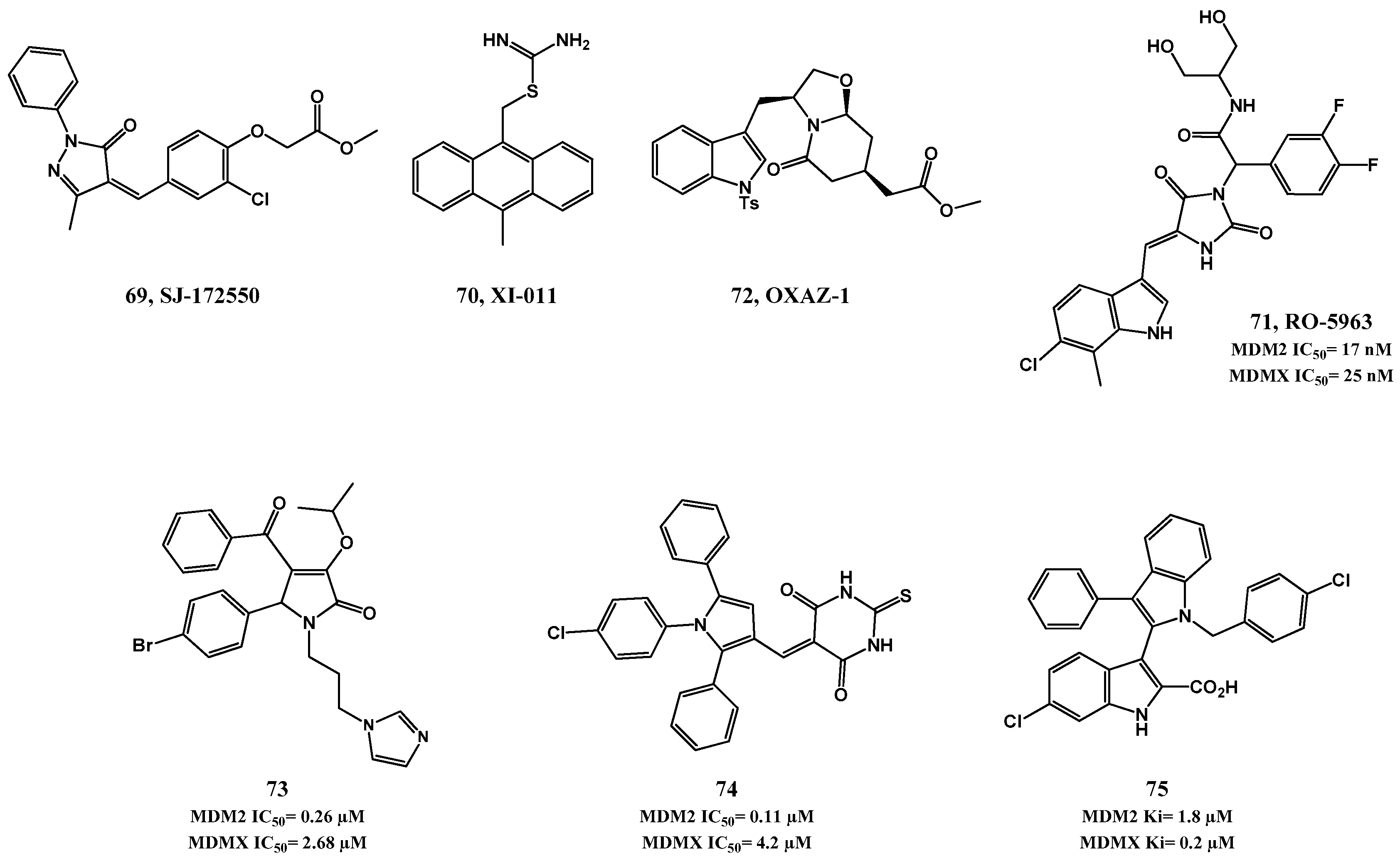
2.2. MDMX and Dual MDM2/MDMX Inhibitors
2.3. p53 Reactivators: wt p53 and Mut p53 Targeting Small Molecules
3. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. P53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Tumour suppression by p53: A role for the DNA damage response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B. Wild-type p53: Tumors can’t stand it. Cell 2007, 128, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Feldser, D.M.; Kostova, K.K.; Winslow, M.M.; Taylor, S.E.; Cashman, C.; Whittaker, C.A.; Sanchez-Rivera, F.J.; Resnick, R.; Bronson, R.; Hemann, M.T.; et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 2010, 468, 572–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christophorou, M.A.; Martin-Zanca, D.; Soucek, L.; Lawlor, E.R.; Brown-Swigart, L.; Verschuren, E.W.; Evan, G.I. Temporal dissection of p53 function in vitro and in vivo. Nat. Genet. 2005, 37, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Cheok, C.F.; Verma, C.S.; Baselga, J.; Lane, D.P. Translating p53 into the clinic. Nat. Rev. Clin. Oncol. 2011, 8, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Selivanova, G. Therapeutic targeting of p53 by small molecules. Semin. Cancer Biol. 2010, 20, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, Y. Targeting p53 for novel anticancer therapy. Transl. Oncol. 2010, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Elmets, C.A.; Kopelovich, L. Pharmacological activation of p53 in cancer cells. Curr. Pharm. Des. 2011, 17, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Dass, C.R. p53-targeted cancer pharmacotherapy: Move towards small molecule compounds. J. Pharm. Pharmacol. 2011, 63, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Essmann, F.; Schulze-Osthoff, K. Translational approaches targeting the p53 pathway for anti-cancer therapy. Br. J. Pharmacol. 2012, 165, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Stegh, A.H. Targeting the p53 signaling pathway in cancer therapy—The promises, challenges and perils. Expert Opin. Ther. Targets 2012, 16, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Cance, W.G. Targeting the p53 pathway. Surg. Oncol. Clin. N. Am. 2013, 22, 747–764. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; McGowan, P.M.; Crown, J.; O’Connor, D.; Gallagher, W.M. p53 as a target for the treatment of cancer. Cancer Treat. Rev. 2014, 40, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Hoe, K.K.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; van den Heuvel, A.P.J.; Prabhu, V.V.; Zhang, S.; El-Deiry, W.S. Targeting tumor suppressor p53 for cancer therapy: Strategies, challenges and opportunities. Curr. Drug Targets 2014, 15, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Selivanova, G. Wild type p53 reactivation: From lab bench to clinic. FEBS Lett. 2014, 588, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Narayanan, S.; Vazquez, A.; Carpizo, D.R. Small molecule compounds targeting the p53 pathway: Are we finally making progress? Apoptosis 2014, 19, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Zawacka-Pankau, J.; Selivanova, G. Pharmacological reactivation of p53 as a strategy to treat cancer. J. Intern. Med. 2015, 277, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Tazawa, H.; Kagawa, S.; Fujiwara, T. Advances in adenovirus-mediated p53 cancer gene therapy. Expert Opin. Biol. Ther. 2013, 13, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Chene, P. Inhibition of the p53-MDM2 interaction: Targeting a protein-protein interface. Mol. Cancer Res. 2004, 2, 20–28. [Google Scholar] [PubMed]
- Buolamwini, J.K.; Addo, J.; Kamath, S.; Patil, S.; Mason, D.; Ores, M. Small molecule antagonists of the MDM2 oncoprotein as anticancer agents. Curr. Cancer Drug Targets 2005, 5, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Fotouhi, N.; Graves, B. Small molecule inhibitors of p53/MDM2 interaction. Curr. Top. Med. Chem. 2005, 5, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.X.; Dayam, R.; Neamati, N. Patented small molecule inhibitors of p53-MDM2 interaction. Expert Opin. Ther. Patents 2006, 16, 165–188. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.M. Peptide, peptidomimetic, and small-molecule antagonists of the p53-HDM2 protein-protein interaction. Int. J. Pept. Res. Ther. 2006, 12, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Dudkina, A.S.; Lindsley, C.W. Small molecule protein-protein inhibitors for the p53-MDM2 interaction. Curr. Top. Med. Chem. 2007, 7, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, I.R. Inhibitors of the MDM2-p53 interaction as anticancer drugs. Drugs Future 2007, 32, 883–896. [Google Scholar] [CrossRef]
- Vassilev, L.T. MDM2 inhibitors for cancer therapy. Trends Mol. Med. 2007, 13, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-Q.; Hu, Y.-Z. Small molecule inhibitors of the p53-MDM2. Curr. Med. Chem. 2008, 15, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Player, M.R. Small-molecule inhibitors of the p53-HDM2 interaction for the treatment of cancer. Expert Opin. Investig. Drugs 2008, 17, 1865–1882. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Wang, S. Targeting the MDM2-p53 interaction for cancer therapy. Clin. Cancer Res. 2008, 14, 5318–5324. [Google Scholar] [CrossRef] [PubMed]
- Dickens, M.P.; Fitzgerald, R.; Fischer, P.M. Small-molecule inhibitors of MDM2 as new anticancer therapeutics. Semin. Cancer Biol. 2010, 20, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Weber, L. Patented inhibitors of p53-MDM2 interaction (2006–2008). Expert Opin. Ther. Patents 2010, 20, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Millard, M.; Pathania, D.; Grande, F.; Xu, S.; Neamati, N. Small-molecule inhibitors of p53-MDM2 interaction: The 2006–2010 update. Curr. Pharm. Des. 2011, 17, 536–559. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Maki, C.G. Pharmacologic activation of p53 by small-molecule MDM2 antagonists. Curr. Pharm. Des. 2011, 17, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Mohammed, A.A.; Shaik, T.B. p53-MDM2 inhibitors: Patent review (2009–2010). Expert Opin. Ther. Patents 2012, 22, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Khoury, K.; Doemling, A. p53 MDM2 inhibitors. Curr. Pharm. Des. 2012, 18, 4668–4678. [Google Scholar] [CrossRef] [PubMed]
- Carry, J.-C.; Garcia-Echeverria, C. Inhibitors of the p53/HDM2 protein-protein interaction-path to the clinic. Bioorg. Med. Chem. Lett. 2013, 23, 2480–2485. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Zhang, X.; Srivenugopal, K.S.; Wang, M.H.; Wang, W.; Zhang, R. Targeting MDM2-p53 interaction for cancer therapy: Are we there yet? Curr. Med. Chem. 2014, 21, 553–574. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.W.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53 MDM-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yu, H.; Hu, W. The regulation of MDM2 oncogene and its impact on human cancers. Acta Biochim. Biophys. Sin. 2014, 46, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: http://clinicaltrials.gov/ (accessed on 5 May 2016).
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Popowicz, G.M.; Doemling, A.; Holak, T.A. The structure-based design of MDM2/MDMX-p53 inhibitors gets serious. Angew. Chem. Int. Ed. 2011, 50, 2680–2688. [Google Scholar] [CrossRef] [PubMed]
- Stuhmer, T.; Chatterjee, M.; Hildebrandt, M.; Herrmann, P.; Gollasch, H.; Gerecke, C.; Theurich, S.; Cigliano, L.; Manz, R.A.; Daniel, P.T.; et al. Nongenotoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood 2005, 106, 3609–3617. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.L.; Vu, B.T.; Qing, W.G.; Packman, K.; et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Drakos, E.; Thomaides, A.; Medeiros, L.J.; Li, J.; Leventaki, V.; Konopleva, M.; Andreeff, M.; Rassidakis, G.Z. Inhibition of p53-murine double minute 2 interaction by nutlin-3a stabilizes p53 and induces cell cycle arrest and apoptosis in hodgkin lymphoma. Clin. Cancer Res. 2007, 13, 3380–3387. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhu, N.; Findley, H.W.; Zhou, M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2. Leukemia 2008, 22, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Wang, J.; Song, X.; Meng, X.; Pan, S.; Jiang, H.; Liu, L. Nutlin-3 cooperates with doxorubicin to induce apoptosis of human hepatocellular carcinoma cells through p53 or p73 signaling pathways. J. Cancer Res. Clin. Oncol. 2010, 136, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Ohnstad, H.O.; Paulsen, E.B.; Noordhuis, P.; Berg, M.; Lothe, R.A.; Vassilev, L.T.; Myklebost, O. MDM2 antagonist nutlin-3a potentiates antitumour activity of cytotoxic drugs in sarcoma cell lines. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.J.; Jiang, N.; Liu, J.J.; Zhao, C.L.; Glenn, K.; Wen, Y.; et al. Discovery of rg7112: A small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Blay, J.-Y.; Italiano, A.; le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the p53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Higgins, B.; Xia, M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, X.; Wang, W.; Zhang, L.; Tao, L.; Dong, X.; Sheng, R.; Yang, B.; Hu, Y. Design, synthesis, and biological evaluation of imidazoline derivatives as p53-MDM2 binding inhibitors. Bioorg. Med. Chem. 2011, 19, 5454–5461. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Dou, X.; Wu, Y.; Zhang, L.; Hua, Y. Design, synthesis and CoMFA studies of N1-amino acid substituted 2,4,5-triphenyl imidazoline derivatives as p53-MDM2 binding inhibitors. Bioorg. Med. Chem. 2012, 20, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hu, Y. Imidazoline derivatives: A patent review (2006–present). Expert Opin. Ther. Patents 2012, 22, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.; Pecak, A.; Rys, B.; Wladyka, B.; Doemling, A.; Weber, L.; Holak, T.A.; Dubin, G. MDM2 and MDMX inhibitors for the treatment of cancer: A patent review (2011–present). Expert Opin. Ther. Patents 2013, 23, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Kawato, H.; Naito, H.; Ikeda, M.; Miyazaki, M.; Kitagawa, M.; Seki, T.; Fukutake, S.; Aonuma, M.; Soga, T. Discovery of novel dihydroimidazothiazole derivatives as p53-MDM2 protein-protein interaction inhibitors: Synthesis, biological evaluation and structure-activity relationships. Bioorg. Med. Chem. Lett. 2012, 22, 6338–6342. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Uoto, K.; Sugimoto, Y.; Naito, H.; Yoshida, K.; Okayama, T.; Kawato, H.; Miyazaki, M.; Kitagawa, M.; Seki, T.; et al. Discovery of DS-5272 as a promising candidate: A potent and orally active p53-MDM2 interaction inhibitor. Bioorg. Med. Chem. 2015, 23, 2360–2367. [Google Scholar] [CrossRef] [PubMed]
- Grasberger, B.L.; Lu, T.B.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.J.; LaFrance, L.V.; Calvo, R.R.; Milkiewicz, K.L.; Gupta, V.; Lattanze, J.; Ramachandren, K.; Carver, T.E.; Petrella, E.C.; Cummings, M.D.; et al. 1,4-benzodiazepine-2,5-diones as small molecule antagonists of the HDM2-p53 interaction: Discovery and sar. Bioorg. Med. Chem. Lett. 2005, 15, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Cummings, M.D.; Schubert, C.; Parks, D.J.; Calvo, R.R.; LaFrance, L.V.; Lattanze, J.; Milkiewicz, K.L.; Lu, T.B. Substituted 1,4-benzodiazepine-2,5-diones as alpha-helix mimetic antagonists of the HDM2-p53 protein-protein interaction. Chem. Biol. Drug Des. 2006, 67, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Raboisson, P.; Marugan, J.J.; Schubert, C.; Koblish, H.K.; Lu, T.B.; Zhao, S.Y.; Player, M.R.; Maroney, A.C.; Reed, R.L.; Huebert, N.D.; et al. Structure-based design, synthesis, and biological evaluation of novel 1,4-diazepines as HDM2 antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 1857–1861. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.J.; LaFrance, L.V.; Calvo, R.R.; Milkiewicz, K.L.; Marugan, J.J.; Raboisson, P.; Schubert, C.; Koblish, H.K.; Zhao, S.Y.; Franks, C.F.; et al. Enhanced pharmacokinetic properties of 1,4-benzodiazepine-2,5-dione antagonists of the HDM2-p53 protein-protein interaction through structure-based drug design. Bioorg. Med. Chem. Lett. 2006, 16, 3310–3314. [Google Scholar] [CrossRef] [PubMed]
- Leonard, K.; Marugan, J.J.; Raboisson, P.; Calvo, R.; Gushue, J.M.; Koblish, H.K.; Lattanze, J.; Zhao, S.Y.; Cummings, M.D.; Player, M.R.; et al. Novel 1,4-benzodiazepine-2,5-diones as HDM2 antagonists with improved cellular activity. Bioorg. Med. Chem. Lett. 2006, 16, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Marugan, J.J.; Leonard, K.; Raboisson, P.; Gushue, J.M.; Calvo, R.; Koblish, H.K.; Lattanze, J.; Zhao, S.Y.; Cummings, M.D.; Player, M.R.; et al. Enantiomerically pure 1,4-benzodiazepine-2,5-diones as HDM2 antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 3115–3120. [Google Scholar] [CrossRef] [PubMed]
- Koblish, H.K.; Zhao, S.Y.; Franks, C.F.; Donatelli, R.R.; Tominovich, R.M.; LaFrance, L.V.; Leonard, K.A.; Gushue, J.M.; Parks, D.J.; Calvo, R.R.; et al. Benzodiazepinedione inhibitors of the HDM2: P53 complex suppress human tumor cell proliferation in vitro and sensitize tumors to doxorubicin in vivo. Mol. Cancer Ther. 2006, 5, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wolf, S.; Bista, M.; Meireles, L.; Camacho, C.; Holak, T.A.; Doemling, A. 1,4-thienodiazepine-2,5-diones via MCR (I): Synthesis, virtual space and p53-MDM2 activity. Chem. Biol. Drug Des. 2010, 76, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Miao, Z.; Zhu, L.; Zhang, Y.; Guo, Z.; Yao, J.; Dong, G.; Wang, S.; Liu, Y.; Chen, H.; et al. Synthesis and biological evaluation of thio-benzodiazepines as novel small molecule inhibitors of the p53-MDM2 protein-protein interaction. Eur. J. Med. Chem. 2011, 46, 5654–5661. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhuang, C.; Zhu, L.; Zhang, Y.; Yao, J.; Dong, G.; Wang, S.; Liu, Y.; Chen, H.; Sheng, C.; et al. Structure-activity relationship and antitumor activity of thio-benzodiazepines as p53-MDM2 protein-protein interaction inhibitors. Eur. J. Med. Chem. 2012, 56, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhuang, C.; Wu, Y.; Guo, Z.; Li, J.; Dong, G.; Yao, J.; Sheng, C.; Miao, Z.; Zhang, W. Design, synthesis and biological evaluation of sulfamide and triazole benzodiazepines as novel p53-MDM2 inhibitors. Int. J. Mol. Sci. 2014, 15, 15741–15753. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, I.R.; Ahmed, S.U.; Atkins, H.; Calvert, A.H.; Curtin, N.J.; Farnie, G.; Golding, B.T.; Griffin, R.J.; Guyenne, S.; Hutton, C.; et al. Isoindolinone-based inhibitors of the MDM2-p53 protein-protein interaction. Bioorg. Med. Chem. Lett. 2005, 15, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, I.R.; Ahmed, S.U.; Atkins, H.; Farnie, G.; Golding, B.T.; Griffin, R.J.; Guyenne, S.; Hutton, C.; Kallblad, P.; Kemp, S.J.; et al. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction based on an isoindolinone scaffold. J. Med. Chem. 2006, 49, 6209–6221. [Google Scholar] [CrossRef] [PubMed]
- Riedinger, C.; Endicott, J.A.; Kemp, S.J.; Smyth, L.A.; Watson, A.; Valeur, E.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Noble, M.E.; et al. Analysis of chemical shift changes reveals the binding modes of isoindolinone inhibitors of the MDM2-p53 interaction. J. Am. Chem. Soc. 2008, 130, 16038–16044. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, I.R.; Liu, J.; Valeur, E.; Watson, A.; Ahmed, S.U.; Blackburn, T.J.; Bennaceur, K.; Clegg, W.; Drummond, C.; Endicott, J.A.; et al. Isoindolinone inhibitors of the murine double minute 2 (MDM2)-p53 protein-protein interaction: Structure-activity studies leading to improved potency. J. Med. Chem. 2011, 54, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Riedinger, C.; Noble, M.E.; Wright, D.J.; Mulks, F.; Hardcastle, I.R.; Endicott, J.A.; McDonnell, J.M. Understanding small-molecule binding to MDM2: Insights into structural effects of isoindolinone inhibitors from NMR spectroscopy. Chem. Biol. Drug Des. 2011, 77, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.F.; Liu, J.; Bennaceur, K.; Drummond, C.J.; Endicott, J.A.; Golding, B.T.; Griffin, R.J.; Haggerty, K.; Lu, X.; McDonnell, J.M.; et al. MDM2-p53 protein-protein interaction inhibitors: A-ring substituted isoindolinones. Bioorg. Med. Chem. Lett. 2011, 21, 5916–5919. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Pereira, N.A.L.; Monteiro, A.; Leao, M.; Bessa, C.; dos Santos, D.; Rairnundo, L.; Queiroz, G.; Bisio, A.; Inga, A.; et al. Oxazoloisoindolinones with in vitro antitumor activity selectively activate a p53-pathway through potential inhibition of the p53-MDM2 interaction. Eur. J. Pharm. Sci. 2015, 66, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.G.; Bourbeau, M.P.; Wohlhieter, G.E.; Bartberger, M.D.; Michelsen, K.; Hungate, R.; Gadwood, R.C.; Gaston, R.D.; Evans, B.; Mann, L.W.; et al. Discovery and optimization of chromenotriazolopyrimidines as potent inhibitors of the mouse double minute 2-tumor protein 53 protein-protein interaction. J. Med. Chem. 2009, 52, 7044–7053. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.P.; DeGraffenreid, M.; Fox, B.; Allen, J.G.; Rew, Y.; Schneider, S.; Saiki, A.Y.; Yu, D.Y.; Oliner, J.D.; Salyers, K.; et al. Improvement of the synthesis and pharmacokinetic properties of chromenotriazolopyrimidine MDM2-p53 protein-protein inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2752–2755. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.S.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P.P.; Tomita, Y.; et al. Structure-based design of potent non-peptide MDM2 inhibitors. J. Am. Chem. Soc. 2005, 127, 10130–10131. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2006, 49, 3432–3435. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Wang, G.P.; Deschamps, J.R.; Parrish, D.A.; Wang, S.M. Synthesis of spirooxindoles via asymmetric 1,3-dipolar cycloaddition. Tetrahedron Lett. 2005, 46, 5949–5951. [Google Scholar] [CrossRef]
- Yu, S.; Qin, D.; Shangary, S.; Chen, J.; Wang, G.; Ding, K.; McEachern, D.; Qiu, S.; Nikolovska-Coleska, Z.; Miller, R.; et al. Potent and orally active small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2009, 52, 7970–7973. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yu, S.; Sun, W.; Liu, L.; Lu, J.; McEachern, D.; Shargary, S.; Bernard, D.; Li, X.; Zhao, T.; et al. A potent small-molecule inhibitor of the MDM2-p53 interaction (MI-888) achieved complete and durable tumor regression in mice. J. Med. Chem. 2013, 56, 5553–5561. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barriere, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, L.; Sun, W.; Lu, J.; McEachern, D.; Li, X.; Yu, S.; Bernard, D.; Ochsenbein, P.; Ferey, V.; et al. Diastereomeric spirooxindoles as highly potent and efficacious MDM2 inhibitors. J. Am. Chem. Soc. 2013, 135, 7223–7234. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, A.; Sun, W.; Liu, L.; Lu, J.; McEachern, D.; Bernard, D.; Deschamps, J.R.; Wang, S. Design of chemically stable, potent, and efficacious MDM2 inhibitors that exploit the retro-mannich ring-opening-cyclization reaction mechanism in spiro-oxindoles. J. Med. Chem. 2014, 57, 10486–10498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ding, Q.; Liu, J.-J.; Zhang, J.; Jiang, N.; Chu, X.-J.; Bartkovitz, D.; Luk, K.-C.; Janson, C.; Tovar, C.; et al. Discovery of potent and selective spiroindolinone MDM2 inhibitor, RO8994, for cancer therapy. Bioorg. Med. Chem. 2014, 22, 4001–4009. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.H.; Li, Z.Z.; Gu, C.; Fishlock, D. Synthesis of a spiroindolinone pyrrolidinecarboxamide MDM2 antagonist. Org. Process Res. Dev. 2013, 17, 247–256. [Google Scholar] [CrossRef]
- Zhang, Z.; Chu, X.-J.; Liu, J.-J.; Ding, Q.; Zhang, J.; Bartkovitz, D.; Jiang, N.; Karnachi, P.; So, S.-S.; Tovar, C.; et al. Discovery of potent and orally active p53-MDM2 inhibitors RO5353 and RO2468 for potential clinical development. ACS Med. Chem. Lett. 2014, 5, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Monterrey, I.; Bertamino, A.; Porta, A.; Carotenuto, A.; Musella, S.; Aquino, C.; Granata, I.; Sala, M.; Brancaccio, D.; Picone, D.; et al. Identification of the spiro(oxindole-3,3′-thiazolidine)-based derivatives as potential p53 activity modulators. J. Med. Chem. 2010, 53, 8319–8329. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Bendinelli, S.; Gabelloni, P.; Da Pozzo, E.; Daniele, S.; Scatena, F.; Vanacore, R.; Campiglia, P.; Bertamino, A.; Gomez-Monterrey, I.; et al. Human glioblastoma multiforme: p53 reactivation by a novel MDM2 inhibitor. PLoS ONE 2013, 8, e72281. [Google Scholar] [CrossRef] [PubMed]
- Bertamino, A.; Soprano, M.; Musella, S.; Rusciano, M.R.; Sala, M.; Vernieri, E.; di Sarno, V.; Limatola, A.; Carotenuto, A.; Cosconati, S.; et al. Synthesis, in vitro, and in cell studies of a new series of indoline-3,2′-thiazolidine-based p53 modulators. J. Med. Chem. 2013, 56, 5407–5421. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; del Giudice, C.; Bertamino, A.; Ciccarelli, M.; Gomez-Monterrey, I.; Campiglia, P.; Novellino, E.; Illario, M.; Trimarco, B.; de Luca, N.; et al. New small molecules, ISA27 and SM13, inhibit tumour growth inducing mitochondrial effects of p53. Br. J. Cancer 2015, 112, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gupta, G.; Bishnoi, A.K.; Saxena, R.; Saini, K.S.; Konwar, R.; Kumar, S.; Dwivedi, A. Design and synthesis of new bioisosteres of spirooxindoles (MI-63/219) as anti-breast cancer agents. Bioorg. Med. Chem. 2015, 23, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Ivanenkov, Y.A.; Vasilevski, S.V.; Beloglazkina, E.K.; Kukushkin, M.E.; Machulkin, A.E.; Veselov, M.S.; Chufarova, N.V.; Chernyaginab, E.S.; Vanzcool, A.S.; Zyk, N.V.; et al. Design, synthesis and biological evaluation of novel potent MDM2/p53 small-molecule inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.J.A.; Kumar, S.P.; Moreira, R.; Santos, M.M.M. Efficient synthesis of spiroisoxazoline oxindoles. Tetrahedron Lett. 2012, 53, 281–284. [Google Scholar] [CrossRef]
- Ribeiro, C.J.A.; Amaral, J.D.; Rodrigues, C.M.P.; Moreira, R.; Santos, M.M.M. Synthesis and evaluation of spiroisoxazoline oxindoles as anticancer agents. Bioorg. Med. Chem. 2014, 22, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.J.A.; Amaral, J.D.; Rodrigues, C.M.P.; Moreira, R.; Santos, M.M.M. Spirooxadiazoline oxindoles with promising in vitro antitumor activities. Med. Chem. Comm. 2016, 7, 420–425. [Google Scholar] [CrossRef]
- Monteiro, A.; Goncalves, L.M.; Santos, M.M.M. Synthesis of novel spiropyrazoline oxindoles and evaluation of cytotoxicity in cancer cell lines. Eur. J. Med. Chem. 2014, 79, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.M.M. Recent advances in the synthesis of biologically active spirooxindoles. Tetrahedron 2014, 70, 9735–9757. [Google Scholar] [CrossRef]
- Czarna, A.; Beck, B.; Srivastava, S.; Popowicz, G.M.; Wolf, S.; Huang, Y.; Bista, M.; Holak, T.A.; Doemling, A. Robust generation of lead compounds for protein-protein interactions by computational and mcr chemistry: P53/HDM2 antagonists. Angew. Chem. Int. Ed. 2010, 49, 5352–5356. [Google Scholar] [CrossRef] [PubMed]
- Popowicz, G.M.; Czarna, A.; Wolf, S.; Wang, K.; Wang, W.; Domling, A.; Holak, T.A. Structures of low molecular weight inhibitors bound to MDMX and MDM2 reveal new approaches for p53-MDMX/MDM2 antagonist drug discovery. Cell Cycle 2010, 9, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Wolf, S.; Koes, D.; Popowicz, G.M.; Camacho, C.J.; Holak, T.A.; Domling, A. Exhaustive fluorine scanning toward potent p53-MDM2 antagonists. ChemMedChem 2012, 7, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.-J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.-J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Higgins, B.; Glenn, K.; Walz, A.; Tovar, C.; Filipovic, Z.; Hussain, S.; Lee, E.; Kolinsky, K.; Tannu, S.; Adames, V.; et al. Preclinical optimization of MDM2 antagonist scheduling for cancer treatment by using a model-based approach. Clin. Cancer Res. 2014, 20, 3742–3752. [Google Scholar] [CrossRef] [PubMed]
- De Turiso, F.G.-L.; Sun, D.; Rew, Y.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Correll, T.L.; Huang, X.; et al. Rational design and binding mode duality of MDM2-p53 inhibitors. J. Med. Chem. 2013, 56, 4053–4070. [Google Scholar] [CrossRef] [PubMed]
- Rew, Y.; Sun, D.; de Turiso, F.G.-L.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Deignan, J.; Fox, B.M.; et al. Structure-based design of novel inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2012, 55, 4936–4954. [Google Scholar] [CrossRef] [PubMed]
- Michelsen, K.; Jordan, J.B.; Lewis, J.; Long, A.M.; Yang, E.; Rew, Y.; Zhou, J.; Yakowec, P.; Schnier, P.D.; Huang, X.; et al. Ordering of the N-terminus of human MDM2 by small molecule inhibitors. J. Am. Chem. Soc. 2012, 134, 17059–17067. [Google Scholar] [CrossRef] [PubMed]
- Rew, Y.; Sun, D. Discovery of a small molecule MDM2 inhibitor (AMG 232) for treating cancer. J. Med. Chem. 2014, 57, 6332–6341. [Google Scholar] [CrossRef] [PubMed]
- Lucas, B.S.; Fisher, B.; McGee, L.R.; Olson, S.H.; Medina, J.C.; Cheung, E. An expeditious synthesis of the MDM2-p53 inhibitor AM-8553. J. Am. Chem. Soc. 2012, 134, 12855–12860. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Zhao, Y.; Wang, S. AM-8553: A novel MDM2 inhibitor with a promising outlook for potential clinical development. J. Med. Chem. 2012, 55, 4934–4935. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, J.; Liu, J.; Chen, X.; Mihalic, J.; Deignan, J.; Yu, M.; Sun, D.; Kayser, F.; McGee, L.R.; et al. Optimization beyond AMG 232: Discovery and SAR of sulfonamides on a piperidinone scaffold as potent inhibitors of the MDM2-p53 protein-protein interaction. Bioorg. Med. Chem. Lett. 2014, 24, 3782–3785. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, Y.; Zhu, J.; Bartberger, M.D.; Canon, J.; Chen, A.; Chow, D.; Eksterowicz, J.; Fox, B.; Fu, J.; et al. Discovery of potent and simplified piperidinone-based inhibitors of the MDM2-p53 interaction. ACS Med. Chem. Lett. 2014, 5, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Osgood, T.; Olson, S.H.; Saiki, A.Y.; Robertson, R.; Yu, D.; Eksterowicz, J.; Ye, Q.; Jin, L.; Chen, A.; et al. The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol. Cancer Ther. 2015, 14, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.Z.; Li, Z.H.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Duquette, J.; Eksterowicz, J.; Fox, B.M.; Fu, J.S.; et al. Novel inhibitors of the MDM2-p53 interaction featuring hydrogen bond acceptors as carboxylic acid isosteres. J. Med. Chem. 2014, 57, 2963–2988. [Google Scholar] [CrossRef] [PubMed]
- Rew, Y.; Sun, D.Q.; Yan, X.L.; Beck, H.P.; Canon, J.; Chen, A.; Duquette, J.; Eksterowicz, J.; Fox, B.M.; Fu, J.S.; et al. Discovery of AM-7209, a potent and selective 4-amidobenzoic acid inhibitor of the MDM2-p53 interaction. J. Med. Chem. 2014, 57, 10499–10511. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.Z.; Eksterowicz, J.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Duquette, J.; Fox, B.M.; Fu, J.; et al. Selective and potent morpholinone inhibitors of the MDM2-p53 protein-protein interaction. J. Med. Chem. 2014, 57, 2472–2488. [Google Scholar] [CrossRef] [PubMed]
- Gessier, F.; Kallen, J.; Jacoby, E.; Chène, P.; Stachyra-Valat, T.; Ruetz, S.; Jeay, S.; Holzer, P.; Masuya, K.; Furet, P. Discovery of dihydroisoquinolinone derivatives as novel inhibitors of the p53-MDM2 interaction with a distinct binding mode. Bioorg. Med. Chem. Lett. 2015, 17, 3621–3625. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Yin, H.; Farooqi, B.; Sebti, S.; Hamilton, A.D.; Chen, J.D. P53 alpha-helix mimetics antagonize p53/MDM2 interaction and activate p53. Mol. Cancer Ther. 2005, 4, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Lee, G.I.; Park, H.S.; Payne, G.A.; Rodriguez, J.M.; Sebti, S.M.; Hamilton, A.D. Terphenyl-based helical mimetics that disrupt the p53/HDM2 interaction. Angew. Chem. Int. Ed. 2005, 44, 2704–2707. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Miao, Z.; Zhu, L.; Dong, G.; Guo, Z.; Wang, S.; Zhang, Y.; Wu, Y.; Yao, J.; Sheng, C.; et al. Discovery, synthesis, and biological evaluation of orally active pyrrolidone derivatives as novel inhibitors of p53-MDM2 protein-protein interaction. J. Med. Chem. 2012, 55, 9630–9642. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, Y.; Guo, Z.; Zhuang, C.; Yao, J.; Dong, G.; Yu, Z.; Min, X.; Wang, S.; Liu, Y.; et al. Discovery of 1-arylpyrrolidone derivatives as potent p53-MDM2 inhibitors based on molecule fusing strategy. Bioorg. Med. Chem. Lett. 2014, 24, 2648–2650. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Miao, Z.; Wu, Y.; Guo, Z.; Li, J.; Yao, J.; Xing, C.; Sheng, C.; Zhang, W. Double-edged swords as cancer therapeutics: Novel, orally active, small molecules simultaneously inhibit p53-MDM2 interaction and the NF-kappaB pathway. J. Med. Chem. 2014, 57, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Sheng, C.; Shin, W.S.; Wu, Y.; Li, J.; Yao, J.; Dong, G.; Zhang, W.; Sham, Y.Y.; Miao, Z.; et al. A novel drug discovery strategy: Mechanistic investigation of an enantiomeric antitumor agent targeting dual p53 and NF-κB pathways. Oncotarget 2014, 5, 10830–10839. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lahue, B.R.; Shipps, G.W., Jr.; Brookes, J.; Wang, Y. Substituted piperidines as HDM2 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Lahue, B.R.; Ma, Y.; Nair, L.G.; Shipps, G.W., Jr.; Wang, Y.; Doll, R.; Bogen, S.L. Core modification of substituted piperidines as novel inhibitors of HDM2-p53 protein-protein interaction. Bioorg. Med. Chem. Lett. 2014, 24, 1983–1986. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lahue, B.R.; Gibeau, C.R.; Shipps, G.W.; Bogen, S.L.; Wang, Y.L.; Guo, Z.Y.; Guzi, T.J. Pivotal role of an aliphatic side chain in the development of an HDM2 inhibitor. ACS Med. Chem. Lett. 2014, 5, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.-H.; Shen, J.-J.; Zhan, Y.-C.; Yi, H.; Xue, S.-T.; Wang, Z.; Ji, X.-Y.; Li, Z.-R. Design, synthesis and in vitro and in vivo antitumour activity of 3-benzylideneindolin-2-one derivatives, a novel class of small-molecule inhibitors of the MDM2-p53 interaction. Eur. J. Med. Chem. 2014, 81, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.P.; Nikolovska-Coleska, Z.; Fang, X.L.; Gao, W.; Shangary, S.; Qiu, S.; Qin, D.G.; Wang, S.M. Discovery of a nanomolar inhibitor of the human murine double minute 2 (MDM2)-p53 interaction through an integrated, virtual database screening strategy. J. Med. Chem. 2006, 49, 3759–3762. [Google Scholar] [CrossRef] [PubMed]
- Bowman, A.L.; Nikolovska-Coleska, Z.; Zhong, H.; Wang, S.; Carlson, H.A. Small molecule inhibitors of the MDM2-p53 interaction discovered by ensemble-based receptor models. J. Am. Chem. Soc. 2007, 129, 12809–12814. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, A.; Bold, G.; de Pover, A.; Stachyra-Valat, T.; Hergovich-Lisztwan, J.; Kallen, J.; Masuya, K.; Furet, P. Tetra-substituted imidazoles as a new class of inhibitors of the p53-MDM2 interaction. Bioorg. Med. Chem. Lett. 2014, 24, 2110–2114. [Google Scholar] [CrossRef] [PubMed]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.A.; van Oordt, W.V.; Hateboer, G.; van der Eb, A.J.; et al. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [PubMed]
- Marine, J.C.; Jochemsen, A.G. MDMX as an essential regulator of p53 activity. Biochem. Biophys. Res. Commun. 2005, 331, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Warr, M.R.; Martins, C.P.; Swigart, L.B.; Passegue, E.; Evan, G.I. Validation of MDMX as a therapeutic target for reactivating p53 in tumors. Genes Dev. 2011, 25, 1746–1757. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.; Shen, Y.; Shelat, A.A.; Arnold, L.A.; Ferreira, A.M.; Zhu, F.; Mills, N.; Smithson, D.C.; Regni, C.A.; Bashford, D.; et al. Identification and characterization of the first small molecule inhibitor of MDMX. J. Biol. Chem. 2010, 285, 10786–10796. [Google Scholar] [CrossRef] [PubMed]
- Bista, M.; Smithson, D.; Pecak, A.; Salinas, G.; Pustelny, K.; Min, J.; Pirog, A.; Finch, K.; Zdzalik, M.; Waddell, B.; et al. On the mechanism of action of SJ-172550 in inhibiting the interaction of MDM4 and p53. PLoS ONE 2012, 7, e37518. [Google Scholar] [CrossRef] [PubMed]
- Berkson, R.G.; Hollick, J.J.; Westwood, N.J.; Woods, J.A.; Lane, D.P.; Lain, S. Pilot screening programme for small molecule activators of p53. Int. J. Cancer 2005, 115, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, C. A small-molecule p53 activator induces apoptosis through inhibiting MDMX expression in breast cancer cells. Neoplasia 2011, 13, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.-L.; Park, J.Y.; Kim, E.H. XI-011 enhances cisplatin-induced apoptosis by functional restoration of p53 in head and neck cancer. Apoptosis 2014, 19, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Graves, B.; Thompson, T.; Xia, M.; Janson, C.; Lukacs, C.; Deo, D.; di Lello, P.; Fry, D.; Garvie, C.; Huang, K.-S.; et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc. Natl. Acad. Sci. USA 2012, 109, 11788–11793. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Raimundo, L.; Pereira, N.A.L.; dos Santos, D.J.V.A.; Perez, M.; Queiroz, G.; Leao, M.; Santos, M.M.M.; Saraiva, L. A tryptophanol-derived oxazolopiperidone lactam is cytotoxic against tumors via inhibition of p53 interaction with murine double minute proteins. Pharmacol. Res. 2015, 95–96, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, T.J.; Ahmed, S.; Coxon, C.R.; Liu, J.; Lu, X.; Golding, B.T.; Griffin, R.J.; Hutton, C.; Newell, D.R.; Ojo, S.; et al. Diaryl- and triaryl-pyrrole derivatives: Inhibitors of the MDM2-p53 and MDMX-p53 protein-protein interactions. Med. Chem. Commun. 2013, 4, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Neochoritis, C.G.; Wang, K.; Estrada-Ortiz, N.; Herdtweck, E.; Kubica, K.; Twarda, A.; Zak, K.M.; Holak, T.A.; Domling, A. 2,3′-bis(1′H-indole) heterocycles: New p53/MDM2/MDMX antagonists. Bioorg. Med. Chem. Lett. 2015, 25, 5661–5666. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Bou-Hanna, C.; Jarry, A.; Lode, L.; Schmitz, I.; Schulze-Osthoff, K.; Kury, S.; Bezieau, S.; Mosnier, J.-F.; Laboisse, C.L. Acute cytotoxicity of MIRA-1/NSC19630, a mutant p53-reactivating small molecule, against human normal and cancer cells via a caspase-9-dependent apoptosis. Cancer Lett. 2015, 359, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, M.; Ozdowy, P.; D’Silva, L.; Rothweiler, U.; Holak, T.A. NMR indicates that the small molecule RITA does not block p53-MDM2 binding in vitro. Nat. Med. 2005, 11, 1135–1136. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Y.; Grinkevich, V.V.; Nikulenkov, F.; Bao, W.; Selivanova, G. Rescue of the apoptotic-inducing function of mutant p53 by small molecule RITA. Cell Cycle 2010, 9, 1847–1855. [Google Scholar] [CrossRef] [PubMed]
- Burmakin, M.; Shi, Y.; Hedstrom, E.; Kogner, P.; Selivanova, G. Dual targeting of wild-type and mutant p53 by small molecule RITA results in the inhibition of N-Myc and key survival oncogenes and kills neuroblastoma cells in vivo and in vitro. Clin. Cancer Res. 2013, 19, 5092–5103. [Google Scholar] [CrossRef] [PubMed]
- Spinnler, C.; Hedstrom, E.; Li, H.; de Lange, J.; Nikulenkov, F.; Teunisse, A.F.A.S.; Verlaan-de Vries, M.; Grinkevich, V.; Jochemsen, A.G.; Selivanova, G. Abrogation of Wip1 expression by RITA-activated p53 potentiates apoptosis induction via activation of ATM and inhibition of HDMX. Cell Death Differ. 2011, 18, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Jin, X.; Bu, Y.; Cao, D.; Zhang, N.; Li, S.; Sun, Q.; Tan, C.; Gao, C.; Jiang, Y. Efficient synthesis of RITA and its analogues: Derivation of analogues with improved antiproliferative activity via modulation of p53/MIR-34a pathway. Org. Biomol. Chem. 2012, 10, 9734–9746. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ding, C.; Li, L.; Gao, C.; Jiang, Y.; Tan, C.; Hua, R. Synthesis and antiproliferative activity of RITA and its analogs. Tetrahedron Lett. 2014, 55, 6635–6638. [Google Scholar] [CrossRef]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, R.; Liu, X.; Zimmermann, M.O.; Rutherford, T.J.; Fersht, A.R.; Joerger, A.C.; Boeckler, F.M. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J. Am. Chem. Soc. 2012, 134, 6810–6818. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Zhang, Z.; Liang, G.; Yao, Z.; Wu, H.; Wang, B.; Zhang, J.; Tariq, M.; Ying, M.; Yang, B. SAHA triggered MET activation contributes to SAHA tolerance in solid cancer cells. Cancer Lett. 2015, 356, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Wassman, C.D.; Baronio, R.; Demir, O.; Wallentine, B.D.; Chen, C.K.; Hall, L.V.; Salehi, F.; Lin, D.W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, R.; Wang, W.G.; Dicker, D.T.; Rastinejad, F.; Lyssikatos, J.; El-Deiry, W.S. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells and can stabilize wild-type p53 protein. Cancer Biol. Ther. 2002, 1, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.G.; Takimoto, R.; Rastinejad, F.; El-Deiry, W.S. Stabilization of p53 by CP-31398 inhibits ubiquitination without altering phosphorylation at serine 15 or 20 or MDM2 binding. Mol. Cell Biol. 2003, 23, 2171–2181. [Google Scholar] [CrossRef] [PubMed]
- Zache, N.; Lambert, J.M.R.; Rokaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykou, V.N. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2008, 2, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [PubMed]
- Demma, M.; Maxwell, E.; Ramos, R.; Liang, L.; Li, C.; Hesk, D.; Rossman, R.; Mallams, A.; Doll, R.; Liu, M.; et al. SCH529074, a small molecule activator of mutant p53, which binds p53 DNA binding domain (DBD), restores growth-suppressive function to mutant p53 and interrupts HDM2-mediated ubiquitination of wild type p53. J. Biol. Chem. 2010, 285, 10198–10212. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Blanden, A.R.; Narayanan, S.; Jayakumar, L.; Lubin, D.; Augeri, D.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism. Oncotarget 2014, 5, 8879–8892. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Hwang, S.-Y.; Cao, S.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small-molecule reactivation of mutant p53 to wild-type-like p53 through the p53-HSP40 regulatory axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Raimundo, L.; Pereira, N.A.L.; Monteiro, A.; Gomes, S.; Bessa, C.; Pereira, C.; Queiroz, G.; Bisio, A.; Fernandes, J.; et al. Reactivation of wild-type and mutant p53 by tryptophanol-derived oxazoloisoindolinone SLMP53-1, a novel anticancer small-molecule. Oncotarget 2016, 7, 4326–4343. [Google Scholar] [PubMed]
- Li, D.; Marchenko, N.D.; Moll, U.M. Saha shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-HSP90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule retra suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wang, W.; El-Deiry, W.S. Non-genotoxic anti-neoplastic effects of ellipticine derivative NSC176327 in p53-deficient human colon carcinoma cells involve stimulation of p73. Cancer Biol. Ther. 2008, 7, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Rao, B.; Lain, S.; Thompson, A.M. P53-based cyclotherapy: Exploiting the “guardian of the genome” to protect normal cells from cytotoxic therapy. Br. J. Cancer 2013, 109, 2954–2958. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
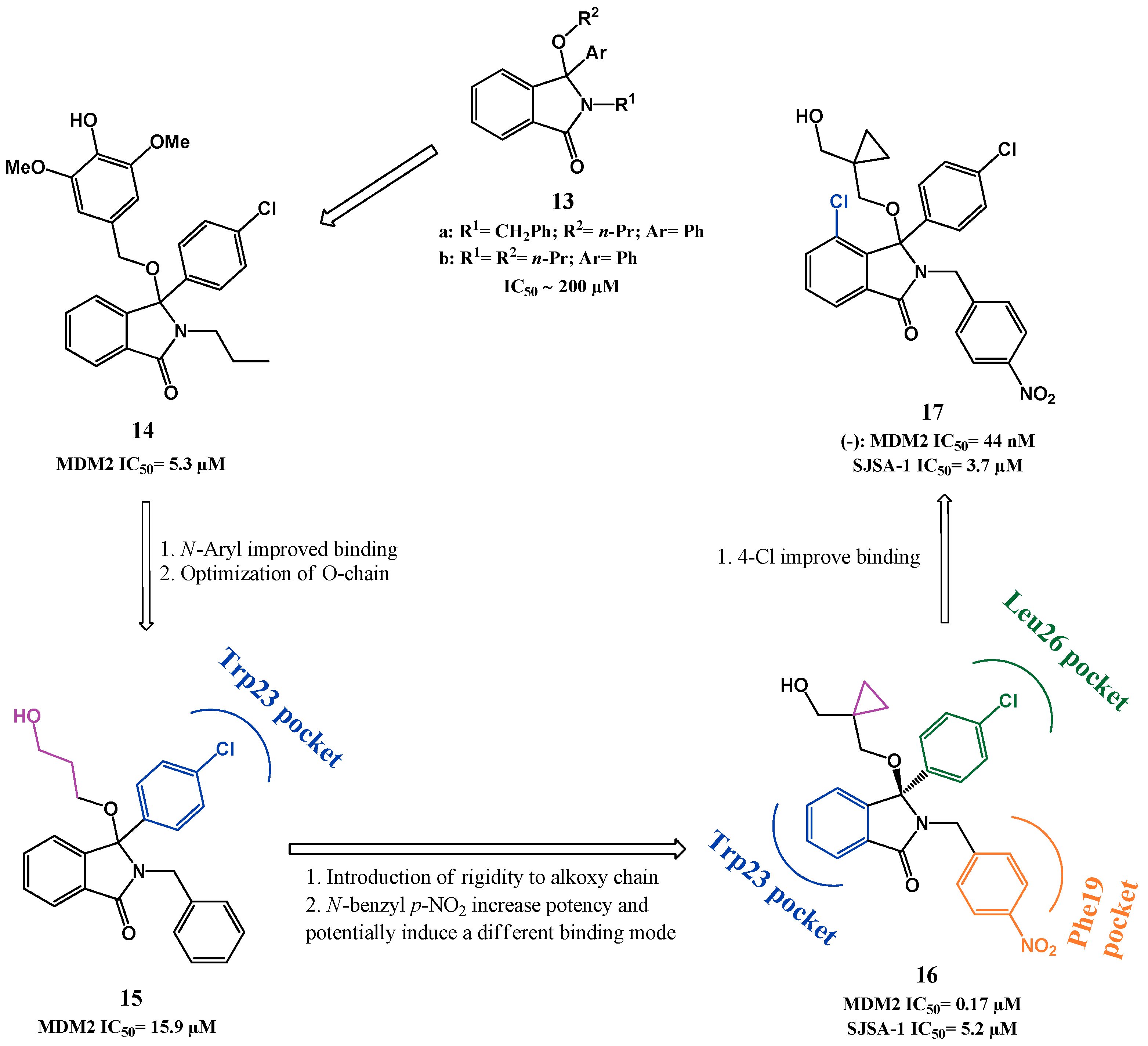{kind=link}
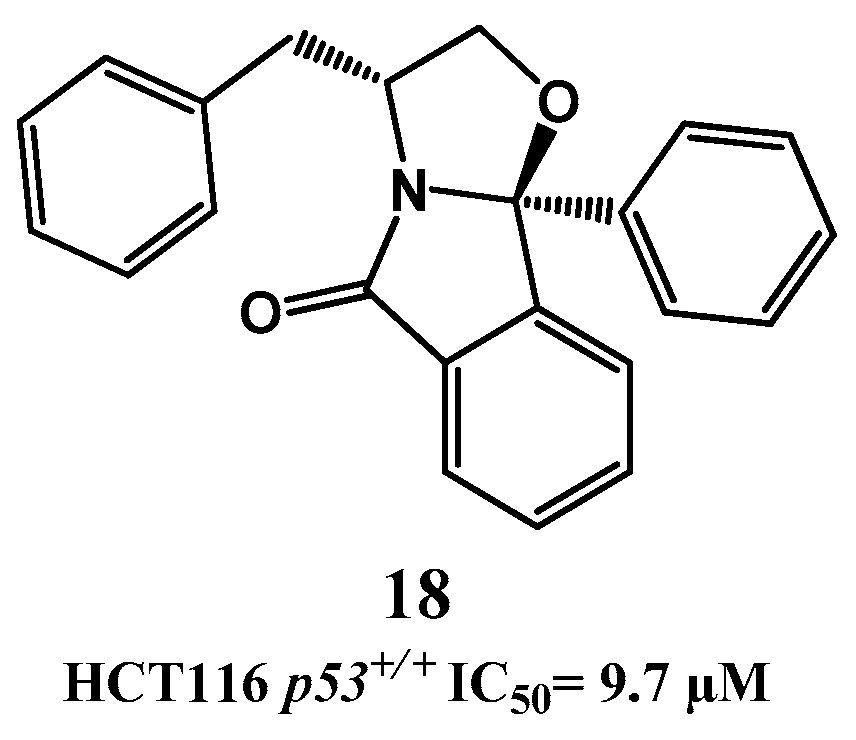{kind=link}
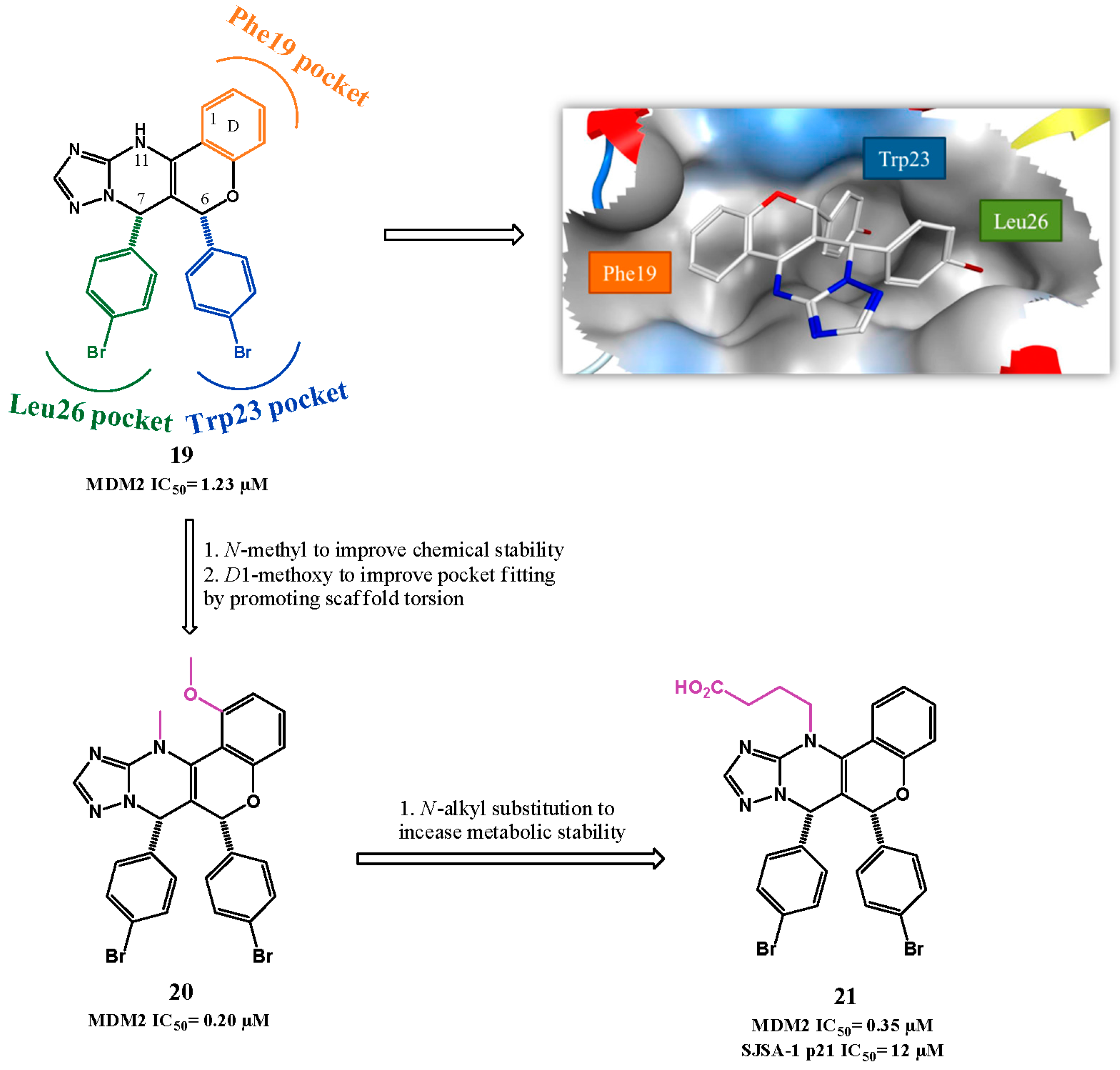{kind=link}
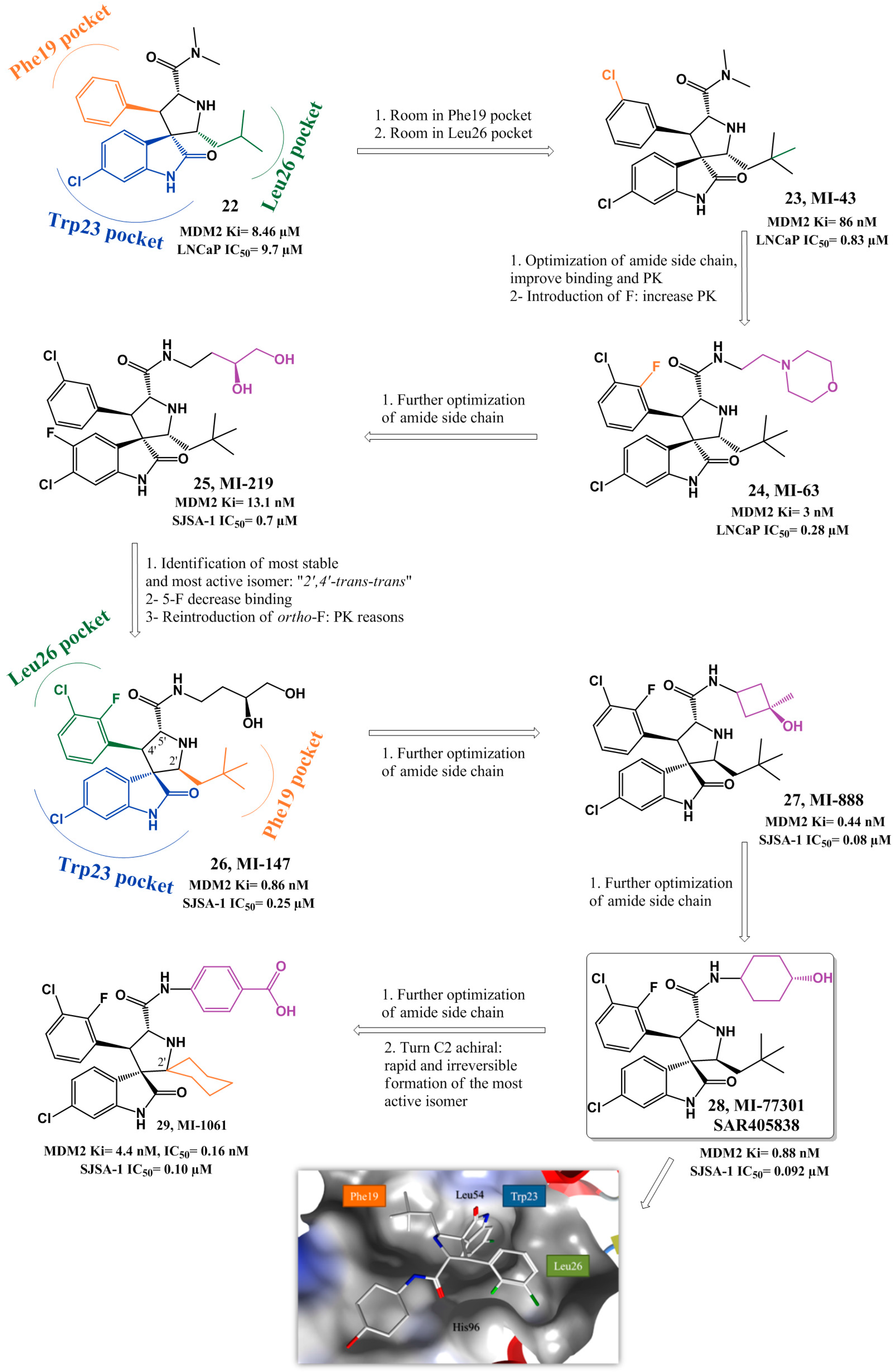{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell-Free Binding Assays | |
| SPR | Surface plasmon resonance |
| HTRF | Homogeneous time resolved fluorescence |
| FP | Fluorescence polarization |
| NMR-AIDA | NMR-based antagonist induced dissociation assay |
| ThermoFluor | Thermal denaturation screening assay |
| TR-FRET | Time-resolved fluorescence energy transfer |
| ELISA | Enzyme-linked immunosorbent assay |
| Cell-Based Assays | |
| BrdU | Bromo-2′-deoxyuridine |
| EdU | 5-Ethynyl-2′-deoxyuridine |
| LCVA | Luminescent cell viability assay |
| MTT | Tetrazolium salt |
| SRB | Sulforhodamine B |
| WST-8 | Water soluble tetrazolium salt |
| Cell Lines | |
| A549 | Human lung carcinoma—wild-type p53 |
| Fro | Human anaplastic thyroid carcinoma—null p53 |
| HCT116 p53(+/+) | Human colorectal cancer—wild-type p53 |
| JAR | Human choriocarcinoma—wild-type p53 |
| Kat-4 | Human thyroid tumor—mutant p53 |
| LNCaP | Human prostatic adenocarcinoma—wild-type p53 |
| MCF-7 | Human breast adenocarcinoma—wild-type p53 |
| MDA-MB-231 | Human breast adenocarcinoma—mutant p53 |
| MHM | Human osteosarcoma—wild-type p53 |
| SJSA-1 | Human osteosarcoma—wild-type p53 |
| U-2OS | Human osteosarcoma—wild-type p53 |
| U937 | Human lung lymphoblast—wild-type p53 |
| Chemical Family | Compound | MDM2 (IC50 or Ki) | Cell-Based Assay (IC50) |
|---|---|---|---|
| Dihydroisoquinolinones [131] | 60 | IC50 = 2.3 nM a | 1.2 μM (SJSA-1) |
| Terphenyl derivatives [132,133] | 61 | Ki = 180 nM b | ND f |
| Pyrrolidones [134,135] | 62 | IC50 = 90 nM c | ND f |
| Pyrrolo[3,4-c]pyrazoles [136,137] | 63 | IC50 = 83 nM c | 5.8 µM (A549) d |
| Piperidines [138,139,140] | 64 | IC50 = 41 nM c | 1.0 µM (SJSA-1) d |
| 3-benzylideneindolin-2-ones [141] | 65 | Ki = 93 nM c | 13.4 µM (HCT116 p53(+/+)) d |
| 8-hydroxyquinoline [142] | NSC66811 (66) | Ki = 120 nM c | ND f |
| Pyridine derivative [143] | 67 | Ki = 110 nM c | ND f |
| Imidazole [144] | 68 | IC50 = 2 nM a | 0.5 µM (SJSA-1) e |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, C.J.A.; Rodrigues, C.M.P.; Moreira, R.; Santos, M.M.M. Chemical Variations on the p53 Reactivation Theme. Pharmaceuticals 2016, 9, 25. https://doi.org/10.3390/ph9020025
Ribeiro CJA, Rodrigues CMP, Moreira R, Santos MMM. Chemical Variations on the p53 Reactivation Theme. Pharmaceuticals. 2016; 9(2):25. https://doi.org/10.3390/ph9020025
Chicago/Turabian StyleRibeiro, Carlos J. A., Cecília M. P. Rodrigues, Rui Moreira, and Maria M. M. Santos. 2016. "Chemical Variations on the p53 Reactivation Theme" Pharmaceuticals 9, no. 2: 25. https://doi.org/10.3390/ph9020025
APA StyleRibeiro, C. J. A., Rodrigues, C. M. P., Moreira, R., & Santos, M. M. M. (2016). Chemical Variations on the p53 Reactivation Theme. Pharmaceuticals, 9(2), 25. https://doi.org/10.3390/ph9020025