
Disease Progression in CNGA3 and CNGB3 Retinopathy; Characteristics of Slovenian Cohort and Proposed OCT Staging Based on Pooled Data from 126 Patients from 7 Studies

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Genetic and Bioinformatic Analysis
2.3. Clinical Examination
2.3.1. Visual Function Tests
2.3.2. Imaging
2.3.3. Visual Electrodiagnostics
2.4. Analysis of Previously Published Data
3. Results
3.1. Genetic Findings
3.2. Clinical Presentation
3.2.1. Visual Function
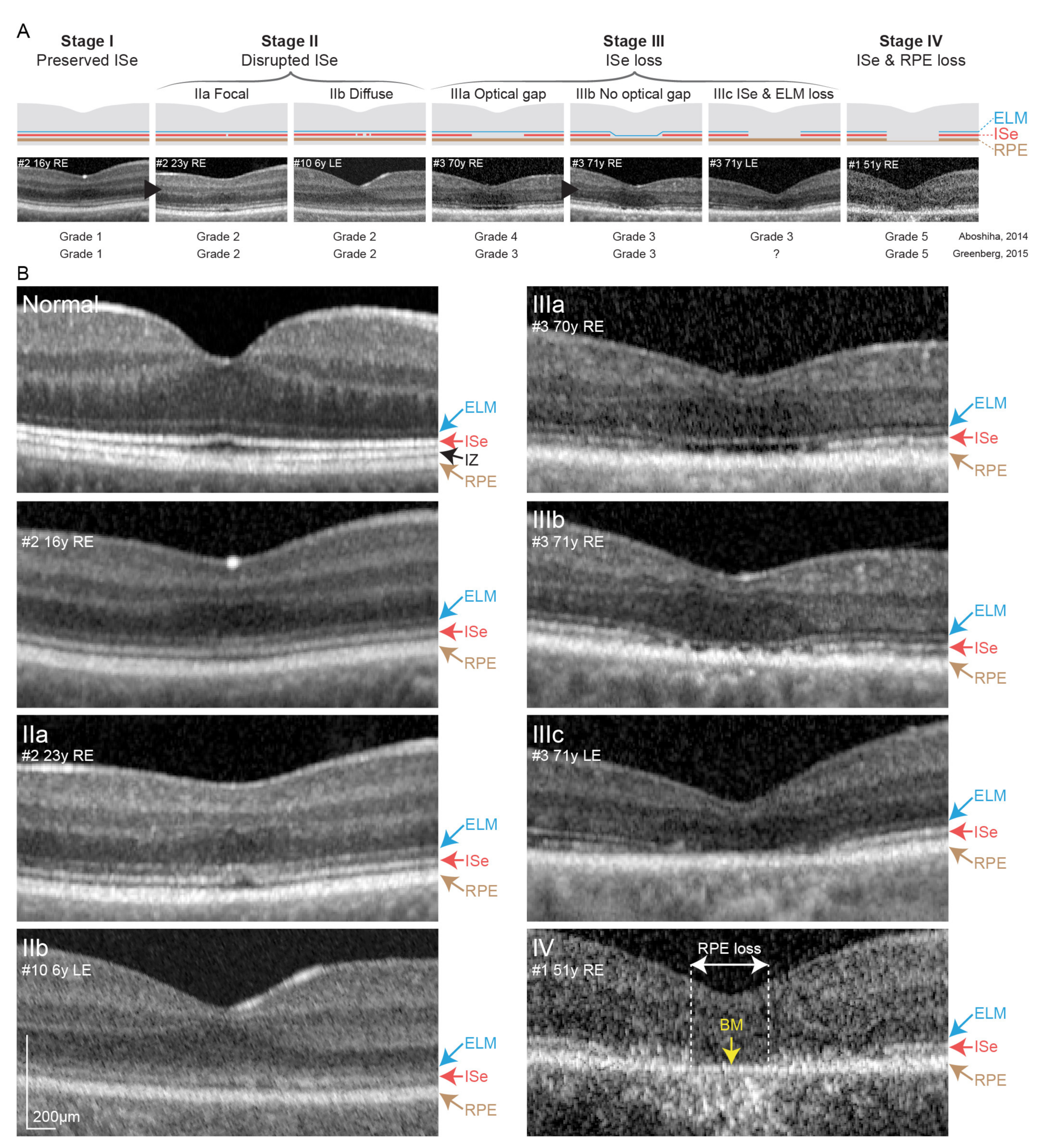
3.2.2. Optical Coherence Tomography and Fundus Autofluorescence Imaging
3.2.3. Electroretinography
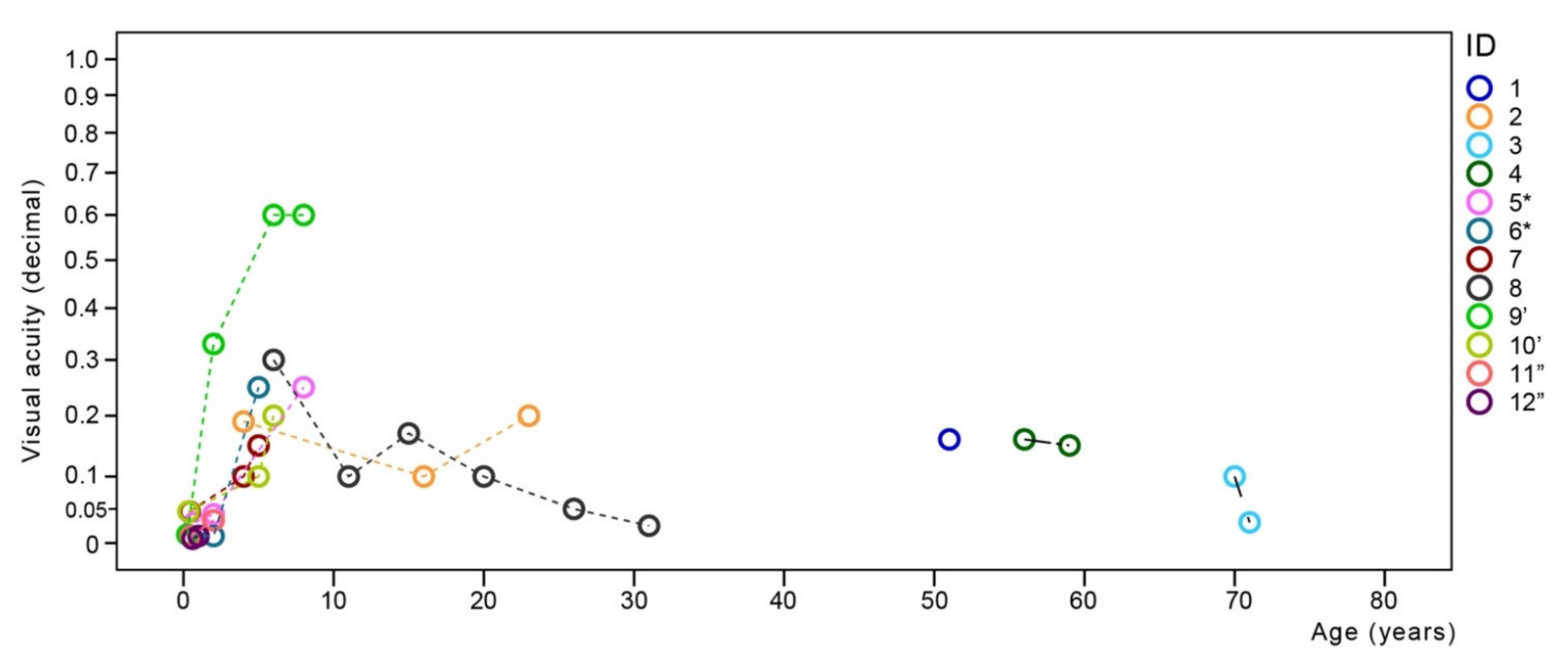
3.3. Correlation between Oct Stage and Age from Published Data
4. Discussion
4.1. Genetic Findings in the Slovenian Cohort
4.2. Visual Function
4.3. Electroretinography
4.4. CNGA3/CNGB3 Retinopathy Is Predominantly a Progressive Disease
4.5. Comparison between CNGA3 and CNGB3 Patients
4.6. Study Limitation and Strengths
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACHM | achromatopsia |
| BCVA | best corrected visual acuity |
| BE | both eyes |
| CNGA3 | Cyclic Nucleotide Gated Channel Subunit Alpha 3 |
| CNGB3 | Cyclic Nucleotide Gated Channel Subunit Beta 3 |
| CNV | copy number variation |
| DA | dark-adapted |
| ELM | external limiting membrane |
| ERG | electroretinography |
| FAF | fundus autofluorescence |
| GOSH | Great Ormond street hospital protocol |
| ISCE | International Society for Clinical Electrophysiology of vision protocol, |
| Ise band | inner segment ellipsoid band |
| LE | left eye |
| OCT | optical coherence tomography |
| RE | right eye |
| RGB | red green blue |
| RPE | retinal pigment epithelium |
| UMC LJ | University Medical Centre Ljubljana |
| VF | perimerty |
| VEP | visual evoked potentials |
References
- Michaelides, M.; Hunt, D.M.; Moore, A.T. The cone dysfunction syndromes. Br. J. Ophthalmol. 2004, 88, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Remmer, M.H.; Rastogi, N.; Ranka, M.P.; Ceisler, E.J. Achromatopsia: A review. Curr. Opin. Ophthalmol. 2015, 26, 333–340. [Google Scholar] [CrossRef]
- Hirji, N.; Aboshiha, J.; Georgiou, M.; Bainbridge, J.; Michaelides, M. Achromatopsia: Clinical features, molecular genetics, animal models and therapeutic options. Ophthalmic Genet. 2018, 39, 149–157. [Google Scholar] [CrossRef]
- Wissinger, B.; Jägle, H.; Kohl, S.; Broghammer, M.; Baumann, B.; Hanna, D.B.; Hedels, C.; Apfelstedt-Sylla, E.; Randazzo, G.; Jacobson, S.; et al. Human Rod Monochromacy: Linkage Analysis and Mapping of a Cone Photoreceptor Expressed Candidate Gene on Chromosome 2q11. Genome 1998, 51, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Sundin, O.H.; Yang, J.-M.; Li, Y.; Zhu, D.; Hurd, J.N.; Mitchell, T.N.; Silva, E.; Maumenee, I.H. Genetic basis of total colourblindness among the Pingelapese islanders. Nat. Genet. 2000, 25, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Aligianis, I.A.; Forshew, T.; Johnson, S.; Michaelides, M.; Johnson, C.A.; Trembath, R.C.; Hunt, D.M.; Moore, A.T.; Maher, E.R. Mapping of a novel locus for achromatopsia (ACHM4) to 1p and identification of a germline mutation in the α subunit of cone transducin (GNAT2). J. Med. Genet. 2002, 39, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.; Grau, T.; Dangel, S.; Hurd, R.; Jurklies, B.; Sener, E.C.; Andreasson, S.; Dollfus, H.; Baumann, B.; Bolz, S.; et al. A homologous genetic basis of the murine cpfl1 mutant and human achromatopsia linked to mutations in the PDE6C gene. Proc. Natl. Acad. Sci. USA 2009, 106, 19581–19586. [Google Scholar] [CrossRef] [Green Version]
- Kohl, S.; Coppieters, F.; Meire, F.; Schaich, S.; Roosing, S.; Brennenstuhl, C.; Bolz, S.; van Genderen, M.M.; Riemslag, F.C.; Lukowski, R.; et al. A Nonsense Mutation in PDE6H Causes Autosomal-Recessive Incomplete Achromatopsia. Am. J. Hum. Genet. 2012, 91, 527–532. [Google Scholar] [CrossRef] [Green Version]
- Kohl, S.; Zobor, D.; Chiang, W.-C.; Weisschuh, N.; Staller, J.; Menendez, I.G.; Chang, S.; Beck, S.C.; Garrido, M.G.; Sothilingam, V.; et al. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet. 2015, 47, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Kohl, S.; Marx, T.; Giddings, I.; Jägle, H.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Zrenner, E.; Sharpe, L.T.; WIssinger, B. Total colourblindness is caused by mutations in the gene encoding the α-subunit of the cone photoreceptor cGMP-gated cation channel. Nat. Genet. 1998, 19, 257–259. [Google Scholar] [CrossRef]
- Kohl, S.; Baumann, B.; Broghammer, M.; Jägle, H.; Sieving, P.; Kellner, U.; Spegal, R.; Anastasi, M.; Zrenner, E.; Sharpe, L.T.; et al. Mutations in the CNGB3 gene encoding the β-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q2. Hum. Mol. Genet. 2000, 9, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, V.; Wilde, C.; Aboshiha, J.; Cowing, J.; Han, C.; Langlo, C.S.; Chana, R.; Davidson, A.E.; Sergouniotis, P.I.; Bainbridge, J.; et al. Retinal Structure and Function in Achromatopsia. Ophthalmology 2014, 121, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Camps, I.; Barranco-Gonzalez, H.; Aviñó-Martínez, J.; Silva, E.; Harto-Castaño, M. Diagnosis and Treatment Options for Achromatopsia: A Review of the Literature. J. Pediatr. Ophthalmol. Strabismus 2018, 55, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Genead, M.A.; Fishman, G.A.; Rha, J.; Dubis, A.M.; Bonci, D.M.O.; Dubra, A.; Stone, E.M.; Neitz, M.; Carroll, J. Photoreceptor Structure and Function in Patients with Congenital Achromatopsia. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7298–7308. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Hamel, C. Clinical utility gene card for: Achromatopsia-update 2013. Eur. J. Hum. Genet. 2013, 21, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskowitz, A.; Hansen, R.M.; Akula, J.D.; Eklund, S.E.; Fulton, A.B. Rod and Rod-Driven Function in Achromatopsia and Blue Cone Monochromatism. Invest. Ophthalmol. Vis. Sci. 2009, 50, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Zobor, D.; Werner, A.; Stanzial, F.; Benedicenti, F.; Rudolph, G.; Kellner, U.; Hamel, C.; Andréasson, S.; Strasser, T.; Wissinger, B.; et al. The Clinical Phenotype of CNGA3 -Related Achromatopsia: Pretreatment Characterization in Preparation of a Gene Replacement Therapy Trial. Investig. Ophthalmol. Vis. Sci. 2017, 58, 821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiadens, A.A.H.J.; Somervuo, V.; Born, L.I.V.D.; Roosing, S.; van Schooneveld, M.J.; Kuijpers, R.W.A.M.; van Moll-Ramirez, N.; Cremers, F.P.M.; Hoyng, C.B.; Klaver, C.C.W. Progressive Loss of Cones in Achromatopsia: An Imaging Study Using Spectral-Domain Optical Coherence Tomography. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5952–5957. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, J.P.; Sherman, J.; Zweifel, S.A.; Chen, R.W.S.; Duncker, T.; Kohl, S.; Baumann, B.; Wissinger, B.; Yannuzzi, L.A.; Tsang, S.H. Spectral-Domain Optical Coherence Tomography Staging and Autofluorescence Imaging in Achromatopsia. JAMA Ophthalmol. 2014, 132, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, R.; Karali, M.; Melillo, P.; Di Iorio, V.; De Benedictis, A.; Iaccarino, G.; Testa, F.; Banfi, S.; Simonelli, F. Clinical and Molecular Characterization of Achromatopsia Patients: A Longitudinal Study. Int. J. Mol. Sci. 2021, 22, 1681. [Google Scholar] [CrossRef]
- Fahim, A.; Khan, N.W.; Zahid, S.; Schachar, I.H.; Branham, K.; Kohl, S.; Wissinger, B.; Elner, V.M.; Heckenlively, J.R.; Jayasundera, T. Diagnostic Fundus Autofluorescence Patterns in Achromatopsia. Am. J. Ophthalmol. 2013, 156, 1211–1219.e2. [Google Scholar] [CrossRef]
- Aboshiha, J.; Dubis, A.M.; A Cowing, J.; Fahy, R.T.A.; Sundaram, V.; Bainbridge, J.; Ali, R.; Dubra, A.; Nardini, M.; Webster, A.R.; et al. A Prospective Longitudinal Study of Retinal Structure and Function in Achromatopsia. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5733–5743. [Google Scholar] [CrossRef] [Green Version]
- Georgiou, M.; Fujinami, K.; Michaelides, M. Retinal imaging in inherited retinal diseases. Ann. Eye Sci. 2020, 5, 25. [Google Scholar] [CrossRef]
- Simunovic, M.P. Acquired color vision deficiency. Surv. Ophthalmol. 2016, 61, 132–155. [Google Scholar] [CrossRef]
- Hirji, N.; Georgiou, M.; Kalitzeos, A.; Bainbridge, J.W.; Kumaran, N.; Aboshiha, J.; Carroll, J.; Michaelides, M. Longitudinal Assessment of Retinal Structure in Achromatopsia Patients With Long-Term Follow-up. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5735–5744. [Google Scholar] [CrossRef] [Green Version]
- Georgiou, M.; Singh, N.; Kane, T.; Zaman, S.; Hirji, N.; Aboshiha, J.; Kumaran, N.; Kalitzeos, A.; Carroll, J.; Weleber, R.G.; et al. Long-Term Investigation of Retinal Function in Patients with Achromatopsia. Investig. Ophthalmol. Vis. Sci. 2020, 61, 38. [Google Scholar] [CrossRef] [PubMed]
- Langlo, C.S.; Erker, L.R.; Parker, M.; Patterson, E.J.; Higgins, B.P.; Summerfelt, P.; Razeen, M.M.; Collison, F.T.; Fishman, G.A.; Kay, C.N.; et al. Repetability and longitudinal assessment of foveal cone structure in CNGB3-associated achromatopsia. Retina 2017, 37, 1956–1966. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; McLean, R.J.; Kohl, S.; Sheth, V.; Gottlob, I. Early signs of longitudinal progressive cone photoreceptor degeneration in achromatopsia. Br. J. Ophthalmol. 2012, 96, 1232–1236. [Google Scholar] [CrossRef]
- Georgiou, M.; Robson, A.G.; Singh, N.; Pontikos, N.; Kane, T.; Hirji, N.; Ripamonti, C.; Rotsos, T.; Dubra, A.; Kalitzeos, A.; et al. Deep Phenotyping ofPDE6C-Associated Achromatopsia. Investig. Ophthalmol. Vis. Sci. 2019, 60, 5112–5123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Thomas, M.; Kumar, A.; Mohammad, S.; Proudlock, F.A.; Engle, E.C.; Andrews, C.; Chan, W.-M.; Thomas, S.; Gottlob, I. Structural Grading of Foveal Hypoplasia Using Spectral-Domain Optical Coherence Tomography: A Predictor of Visual Acuity? Ophthalmology 2011, 118, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Kriss, A.; Jeffrey, B.; Taylor, D. The Electroretinogram in Infants and Young Children. J. Clin. Neurophysiol. 1992, 9, 373–393. [Google Scholar] [CrossRef]
- Robson, A.G.; Nilsson, J.; Li, S.; Jalali, S.; Fulton, A.B.; Tormene, A.P.; Holder, G.E.; Brodie, S. ISCEV guide to visual electrodiagnostic procedures. Doc. Ophthalmol. 2018, 136, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawlina, M.; Konec, B. New noncorneal HK-loop electrode for clinical electroretinography. Doc. Ophthalmol. 1992, 81, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Marmoy, O.R.; Moinuddin, M.; Thompson, D.A. An alternative electroretinography protocol for children: A study of diagnostic agreement and accuracy relative to ISCEV standard electroretinograms. Acta Ophthalmol. 2021. [Google Scholar] [CrossRef]
- Yang, P.; Michaels, K.V.; Courtney, R.J.; Wen, Y.; Greninger, D.A.; Reznick, L.; Karr, D.J.; Wilson, L.B.; Weleber, R.G.; Pennesi, M.E. Retinal Morphology of Patients With Achromatopsia During Early Childhood: Implications for Gene Therapy. JAMA Ophthalmol. 2014, 132, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Perlman, I.; Kondo, M.; Chelva, E.; Robson, A.G.; Holder, G.E. ISCEV extended protocol for the S-cone ERG. Doc. Ophthalmol. 2019, 140, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiadens, A.A.H.J.; Slingerland, N.W.R.; Roosing, S.; van Schooneveld, M.J.; van Lith-Verhoeven, J.J.C.; van Moll-Ramirez, N.; van der Born, L.I.; Hoyng, C.B.; Cremers, F.P.M.; Claver, C.C.W. Genetic Etiology and Clinical Consequences of Complete and Incomplete Achromatopsia. Ophthalmology 2009, 116, 1984–1989.e1. [Google Scholar] [CrossRef] [PubMed]
- Wiszniewski, W.; Lewis, R.A.; Lupski, J.R. Achromatopsia: The CNGB3 p.T383fsX mutation results from a founder effect and is responsible for the visual phenotype in the original report of uniparental disomy 14. Qual. Life Res. 2007, 121, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.L.; Maurer, D. Multiple sensitive periods in human visual development: Evidence from visually deprived children. Dev. Psychobiol. 2005, 46, 163–183. [Google Scholar] [CrossRef]
- Kurent, A.; Stirn-Kranjc, B.; Brecelj, J. Electroretinographic Characteristics in Children with Infantile Nystagmus Syndrome and Early-Onset Retinal Dystrophies. Eur. J. Ophthalmol. 2015, 25, 33–42. [Google Scholar] [CrossRef]
- Zelinger, L.; Cideciyan, A.V.; Kohl, S.; Schwartz, S.B.; Rosenmann, A.; Eli, D.; Sumaroka, A.; Roman, A.J.; Luo, X.; Brown, C.; et al. Genetics and Disease Expression in the CNGA3 Form of Achromatopsia. Ophthalmology 2015, 122, 997–1007. [Google Scholar] [CrossRef]
- Thomas, M.; Kumar, A.; Kohl, S.; Proudlock, F.A.; Gottlob, I. High-Resolution In Vivo Imaging in Achromatopsia. Ophthalmology 2011, 118, 882–887. [Google Scholar] [CrossRef]
- Michalakis, S.; Geiger, H.; Haverkamp, S.; Hofmann, F.; Gerstner, A.; Biel, M. Impaired Opsin Targeting and Cone Photoreceptor Migration in the Retina of Mice Lacking the Cyclic Nucleotide-Gated Channel CNGA3. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Morris, L.; Fliesler, S.J.; Sherry, D.M.; Ding, X.-Q. Early-Onset, Slow Progression of Cone Photoreceptor Dysfunction and Degeneration in CNG Channel Subunit CNGB3 Deficiency. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3557–3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komáromy, A.M.; Alexander, J.J.; Rowlan, J.S.; Garcia, M.M.; Chiodo, V.A.; Kaya, A.; Tanaka, J.C.; Acland, G.M.; Hauswirth, W.W.; Aguirre, D.E. Gene therapy rescues cone function in congenital achromatopsia. Hum. Mol. Genet. 2010, 19, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.; Xu, J.; Pearson, R.; Smith, A.J.; Bainbridge, J.; Morris, L.M.; Fliesler, S.J.; Ding, X.-Q.; Ali, R.R. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum. Mol. Genet. 2011, 20, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Kobal, N.; Krašovec, T.; Šuštar, M.; Volk, M.; Peterlin, B.; Hawlina, M.; Fakin, A. Stationary and Progressive Phenotypes Caused by the p.G90D Mutation in Rhodopsin Gene. Int. J. Mol. Sci. 2021, 22, 2133. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | ClinVar ID and Intrepretation | LOVD | GnomAD Allele Frequency | Reference PMID | Functional Study PMID |
|---|---|---|---|---|---|
| NM_001298.2(CNGA3):c.829C>T | 9481 | CNGA3_000033 | 0,0000955 | 11536077 | 20238023 |
| (p.Arg277Cys) | Pathogenic/Likely pathogenic | (no homozygotes) | |||
| NM_001298.2(CNGA3):c.847C>T | 9474 | CNGA3_000034 | 0,0000995 | 9662398, 26992781, 24504161, 11536077, 25637600, 9662398, 25637600 | 17693388, 20238023 |
| (Arg283Trp) | Pathogenic/Likely pathogenic | (no homozygotes) | |||
| NM_001298.2(CNGA3):c.1279C>T | 497256 | CNGA3_000038 | 0,000395 | 11536077, 23972307, 28559085, 18445228 | 18445228 |
| (Arg427Cys) | Pathogenic | (1 homozygote) | |||
| NM_001298.2(CNGA3):c.1641C>A | 9478 | CNGA3_000044 | 0,000151 | 14757870, 23972307, 30682209, 11536077, 31456290, 9662398 | 17693388 |
| (p.Phe547Leu) | Pathogenic/Likely pathogenic | (1 homozygote) | |||
| NM_019098.4(CNGB3):c.1578+1G>A | 189031 | CNGB3_000034 | 0,0000199 | 10958649, 12187429 | n/a |
| Pathogenic/Likely pathogenic | (no homozygotes) | ||||
| NM_019098.4(CNGB3):c.1148delC (p.Thr383IlefsTer13) | 5225 | CNGB3_000001 | 0,00172 | 25770143, 17265047 | 12815043, 23805033 |
| Conflicting interpretations of pathogenicity | (2 homozygotes) | ||||
| NM_019098.4(CNGB3):c.2104-2A>G | Novel variant (PVS1, PM2); Likely pathogenic | n/a | n/a | 15657609 | n/a |
| NM_019098.4(CNGB3):c.819_826delCAGACTCC (p.Arg274ValfsTer13) | 374027 | CNGB3_000044 | 0,0000517 | 10888875, 20079539, 29769798 | n/a |
| Pathogenic | (no homozygotes) | ||||
| CNGB3:dup ex7 | Pathogenic/Likely pathogenic | n/a | n/a | 28795510 | n/a |
| (NC_000008.11:g.86,652,314_86,662,912dup) |
| ID | Sex | Gene | Variants | Nysta-Gmus | Foveal Hypo-Plasia | Age at Exam | Color Vision (Ishihara) | BCVA | BCVA Better Eye Decimal (logMAR) | OCT Machine | CRT OD (um) | CRT OS (um) | OCT Grade | Foveal FAF (BE) | ERG Rod Response | ERG Cone Response | Measuring Standard |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | CNGA3 | p.Arg277Cys (hmz) | no | 1a | 51 | n/a | RE 0.16 LE 0.16 | 0.16 (0.80) | Spectralis | 211 | 233 | BE 5 | decreased with hyperAF ring | normal | undetectable | ISCEV |
| 2 | F | CNGA3 | p.Arg283Trp (hmz) | yes | 1b | 4 | n/a | RE 3/16 LE 2/16 | 0.19 (0.73) | n/a | n/a | n/a | n/a | n/a | normal | degraded | GOSH |
| 16 | RE 0/15 LE 0/15 | RE 0.1 LE 0.1 | 0.1 (1.00) | Spectralis | 268 | 271 | BE 1 | decreased | normal | undetectable | ISCEV | ||||||
| 23 | RE 1/15 LE 1/15 | RE 0.2 LE 0.2 | 0.2 (0.70) | Spectralis | 267 | 269 | RE 2 LE 1 | n/a | n/a | n/a | n/a | ||||||
| 3 | F | CNGA3 | p.Arg283Trp (hmz) | yes | 1a | 70 | n/a | RE CF 0.5 m LE 0.1 | 0.1 (1.00) | Spectralis | 180 | 224 | RE 3 LE 4 | decreased with hyperAF ring | reduced | undetectable | ISCEV extended |
| 71 | RE 0/15 LE 1/15 | RE CF 0.5 m LE CF 2 m | 0.03 (1.52) | Spectralis | 177 | 219 | RE 3 LE 3 | decreased with hyperAF ring | n/a | n/a | n/a | ||||||
| 4 | F | CNGA3 | p.Phe547Leu (hmz) | no | 1a | 56 | n/a | RE 0.1 LE 0.16 | 0.16 (0.80) | Spectralis | 247 | 242 | BE 4 | decreased with hyperAF ring | normal | undetectable | ISCEV |
| 59 | RE 1/15 LE 1/15 | RE 0.1 LE 0.15 | 0.15 (0.82) | Spectralis | 241 | 235 | BE 4 | decreased with hyperAF ring | n/a | n/a | n/a | ||||||
| 5 * | M | CNGA3 | p. Arg283Trp; p.Phe547Leu | yes | no | 9 m | n/a | binocular ≥ 6/190 | 0.03 (1.52) | n/a | n/a | n/a | n/a | n/a | normal | undetectable | GOSH |
| 14 m | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | normal | undetectable | ISCEV with skin electrodes | ||||||
| 2 | n/a | binocular 6/12 at 0.5 m | 0.042 (1.38) | Triton | 216 | 207 | BE 1 | n/a | n/a | n/a | n/a | ||||||
| 8 | n/a | binocular 6/24 | 0.25 (0.60) | Triton | 198 | 200 | BE 1 | n/a | n/a | n/a | n/a | ||||||
| 6 * | F | CNGA3 | p. Arg283Trp; p.Phe547Leu | yes | no | 6 m | n/a | binocular ≥ 6/620 | 0.01 (2.00) | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a |
| 9 m | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | normal | undetectable | GOSH | ||||||
| 2 | n/a | binocular ≥ 6/620 | 0.01 (2.00) | Triton | 172 | 179 | BE 1 | n/a | n/a | n/a | n/a | ||||||
| 5 | n/a | binocular 6/24 | 0.25 (0.60) | Triton | 192 | 182 | BE 1 | increased | n/a | n/a | n/a | ||||||
| 7 | M | CNGA3 | Arg265Trp; p.Arg409Cys | yes | no | 5 m | n/a | binocular 6/130 | 0.046 (1.34) | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a |
| 7 m | n/a | n/a | n/a | n/a | n/a | n/a | n/a | reduced | undetectable | GOSH | |||||||
| 4 | n/a | binocular 0.1 | 0.1 (1.00) | Triton | 251 | 195 | RE 2 LE 1 | n/a | n/a | n/a | n/a | ||||||
| 5 | n/a | RE 0.15 LE 0.15 | 0.15 (0.82) | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||||||
| 8 | F | CNGB3 | p.Thr383IlefsTer13 (pat,mat), c.2104-2A>G (pat) | yes | 1b | 5 m | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a |
| 6 | n/a | RE 0.3 LE 0.2 | 0.3 (0.52) | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||||||
| 11 | 1/12 | RE 0.1 LE 0.1 | 0.1 (1.00) | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||||||
| 15 | 3/12 | BE 4/24 (E signs) | 0.17 (0.78) | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||||||
| 20 | n/a | RE 0.1 LE 0.1 | 0.1 (1.00) | n/a | n/a | n/a | n/a | n/a | reduced | undetectable | ISCEV | ||||||
| 21 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | reduced | undetectable | ISCEV extended | ||||||
| 22 | n/a | n/a | n/a | Triton | 214 | 205 | BE 4 | n/a | n/a | n/a | n/a | ||||||
| 26 | n/a | RE 0.05 LE 0.05 | 0.05 (1.30) | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||||||
| 31 | RE 3/15 LE 2/15 | RE CF 1.5 m LE CF 1.5 m | 0.025 (1.60) | Triton | 216 | 205 | BE 4 | decreased with hyperAF ring | n/a | n/a | n/a | ||||||
| 9 ’ | M | CNGB3 | p.Arg274ValfsTer13; dup ex7 | yes | 1a | 3 m | n/a | RE 6/500 LE 6/500 | 0.012 (1.92) | n/a | n/a | n/a | n/a | n/a | normal | undetectable | GOSH |
| 7 m | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | normal | undetectable | GOSH | ||||||
| 2 | n/a | binocular 6/18 | 0.33 (0.48) | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||||||
| 6 | n/a | RE 0.2 LE 0.4 | 0.4 (0.40) | Spectralis | 274 | 271 | BE 2 | decreased | n/a | n/a | n/a | ||||||
| 7 | n/a | n/a | n/a | n/a | n/a | n/a | BE 2 | decreased | normal | undetectable | ISCEV with skin electrodes | ||||||
| 8 | n/a | RE 0.25 LE 0.6 | 0.6 (0.22) | Spectralis | 270 | 271 | BE 2 | n/a | n/a | n/a | n/a | ||||||
| 10 ’ | F | CNGB3 | p.Arg274ValfsTer13; dup ex7 | yes | 1a | 4 m | n/a | binocular 6/130 | 0.046 (1.34) | Triton | n/a | n/a | RE 1 LE n/a | n/a | normal | undetectable | GOSH |
| 5 | n/a | RE 0.1 LE 0.1 | 0.1 (1.00) | Spectralis | 237 | 245 | BE 1 | n/a | normal | undetectable | ISCEV with skin electrodes | ||||||
| 6 | n/a | RE 0.2 LE 0.2 | 0.2 (0.70) | Triton | 219 | 206 | BE 2 | n/a | n/a | n/a | n/a | ||||||
| 11 ” | F | CNGB3 | c.1578+1G>A; p.Thr383IlefsTer13 | yes | n/a | 6 m | n/a | binocular 6/620 | 0.01 (2.00) | n/a | n/a | n/a | n/a | n/a | normal | undetectable | GOSH |
| 14 m | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | normal | undetectable | GOSH | ||||||
| 2 | n/a | binocular 0.1 at 2 m | 0.033 (1.48) | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||||||
| 12 ” | M | CNGB3 | c.1578+1G>A; p.Thr383IlefsTer13 | yes | n/a | 7 m | n/a | binocular 6/1000 | 0.006 (2.22) | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a |
| 1 | n/a | binocular 6/620 | 0.01 (2.00) | n/a | n/a | n/a | n/a | n/a | normal | undetectable | GOSH |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tekavčič Pompe, M.; Vrabič, N.; Volk, M.; Meglič, A.; Jarc-Vidmar, M.; Peterlin, B.; Hawlina, M.; Fakin, A. Disease Progression in CNGA3 and CNGB3 Retinopathy; Characteristics of Slovenian Cohort and Proposed OCT Staging Based on Pooled Data from 126 Patients from 7 Studies. Curr. Issues Mol. Biol. 2021, 43, 941-957. https://doi.org/10.3390/cimb43020067
Tekavčič Pompe M, Vrabič N, Volk M, Meglič A, Jarc-Vidmar M, Peterlin B, Hawlina M, Fakin A. Disease Progression in CNGA3 and CNGB3 Retinopathy; Characteristics of Slovenian Cohort and Proposed OCT Staging Based on Pooled Data from 126 Patients from 7 Studies. Current Issues in Molecular Biology. 2021; 43(2):941-957. https://doi.org/10.3390/cimb43020067
Chicago/Turabian StyleTekavčič Pompe, Manca, Nika Vrabič, Marija Volk, Andrej Meglič, Martina Jarc-Vidmar, Borut Peterlin, Marko Hawlina, and Ana Fakin. 2021. "Disease Progression in CNGA3 and CNGB3 Retinopathy; Characteristics of Slovenian Cohort and Proposed OCT Staging Based on Pooled Data from 126 Patients from 7 Studies" Current Issues in Molecular Biology 43, no. 2: 941-957. https://doi.org/10.3390/cimb43020067