
Assessment of T Cell Receptor Complex Expression Kinetics in Natural Killer Cells

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Plasmids
2.2. Lentiviral Production and Transduction of NK Cells
2.3. Flow Cytometry
2.4. Treatment of TCR-Transduced NK92 Cells with Inhibitors
2.5. Peptides and Target Cell Pulsing
2.6. In Vitro NK Cell Activation
2.7. Labeling of Genetically Modified NK92 Cells
2.8. Flow Cytometry-Based and Live-Cell Imaging of NK92 Cell Cytotoxicity
2.9. Statistical Analysis
3. Results
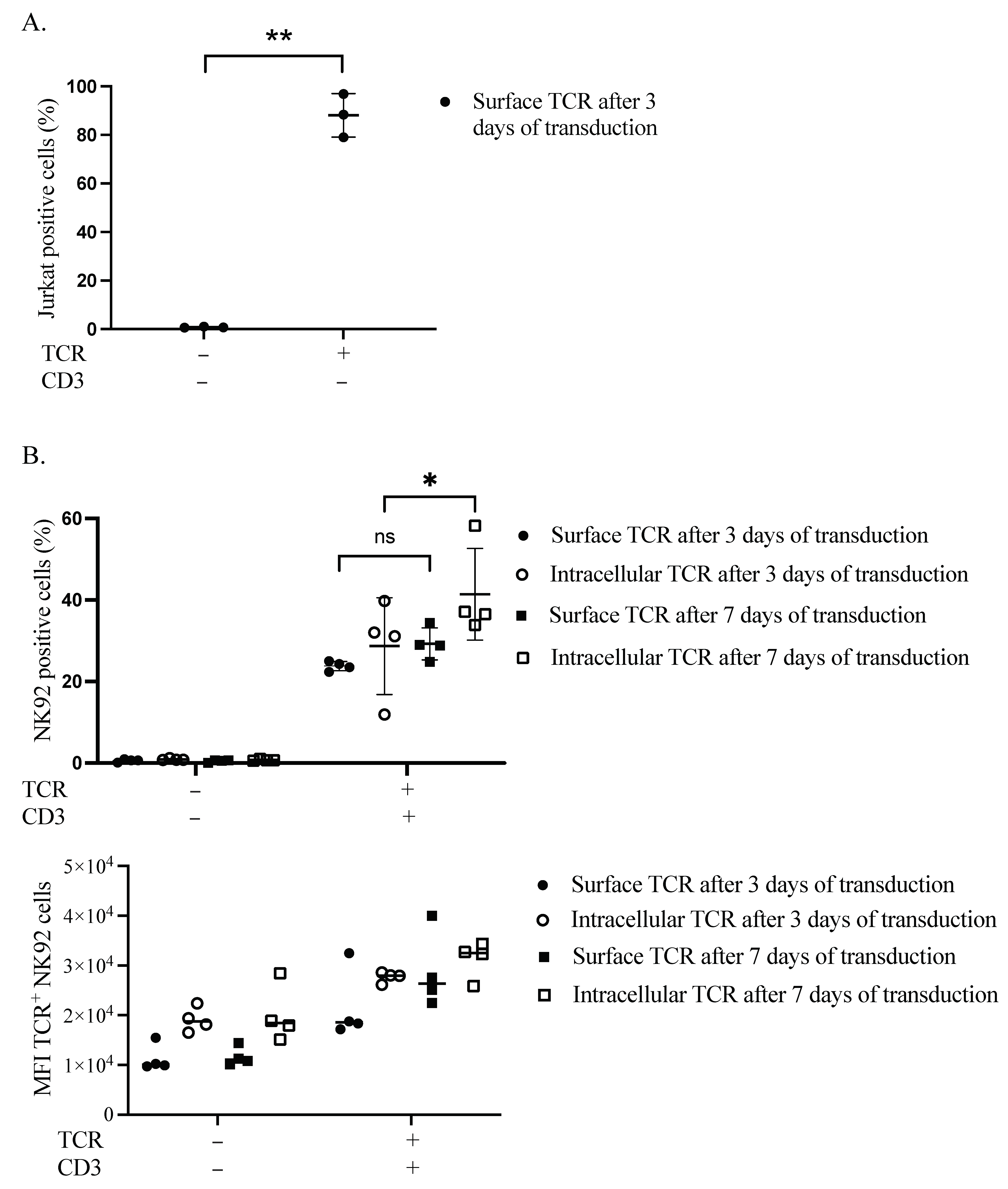
3.1. Influence of Time on TCR Complex Expression
3.2. TCR Complex Expression Requirements in the Plasma Membrane of NK92 Cells
3.3. Influence of Inhibitors on TCR Complex Expression
3.4. Functional Evaluation of Genetically Modified NK92 Cells
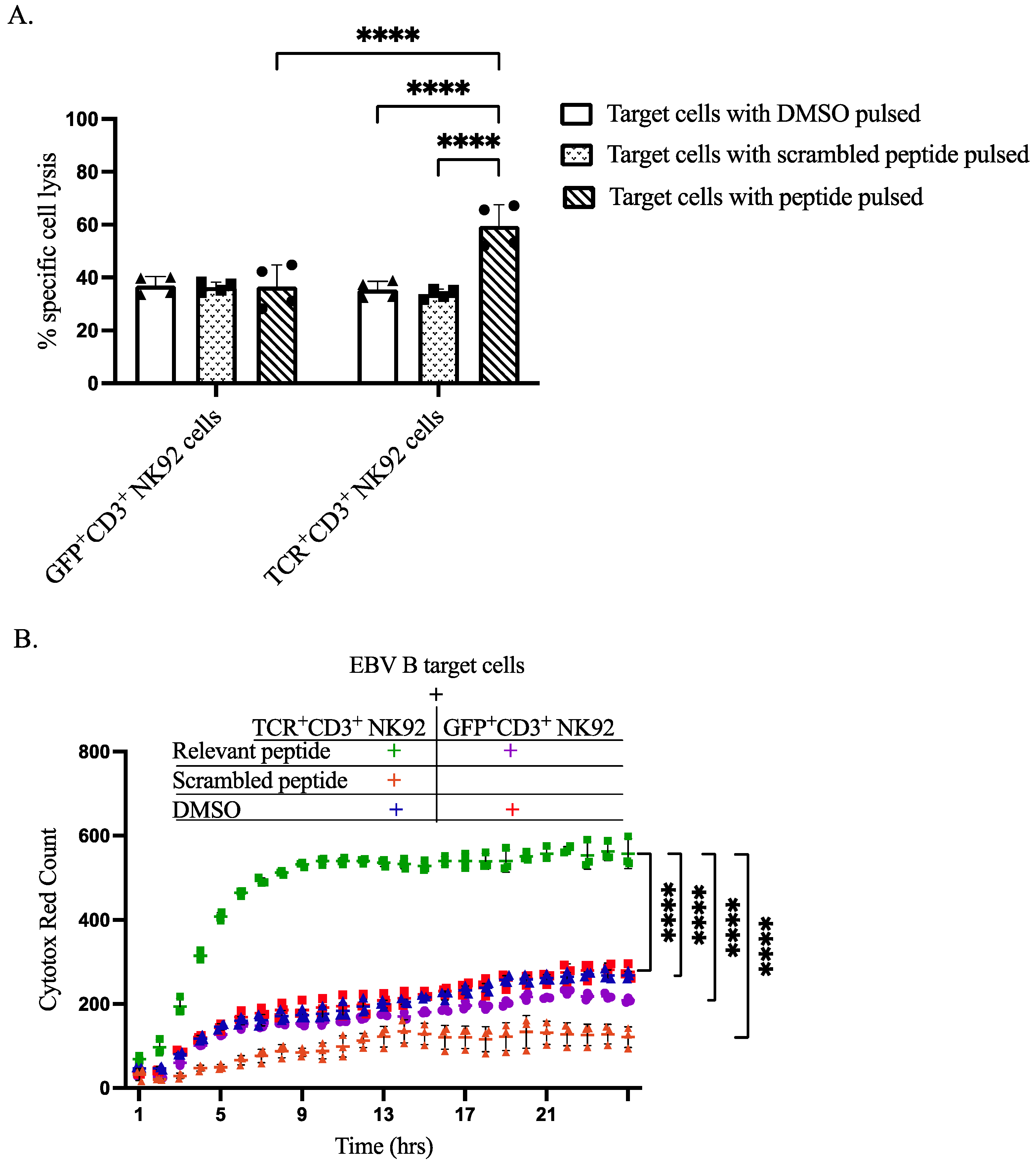
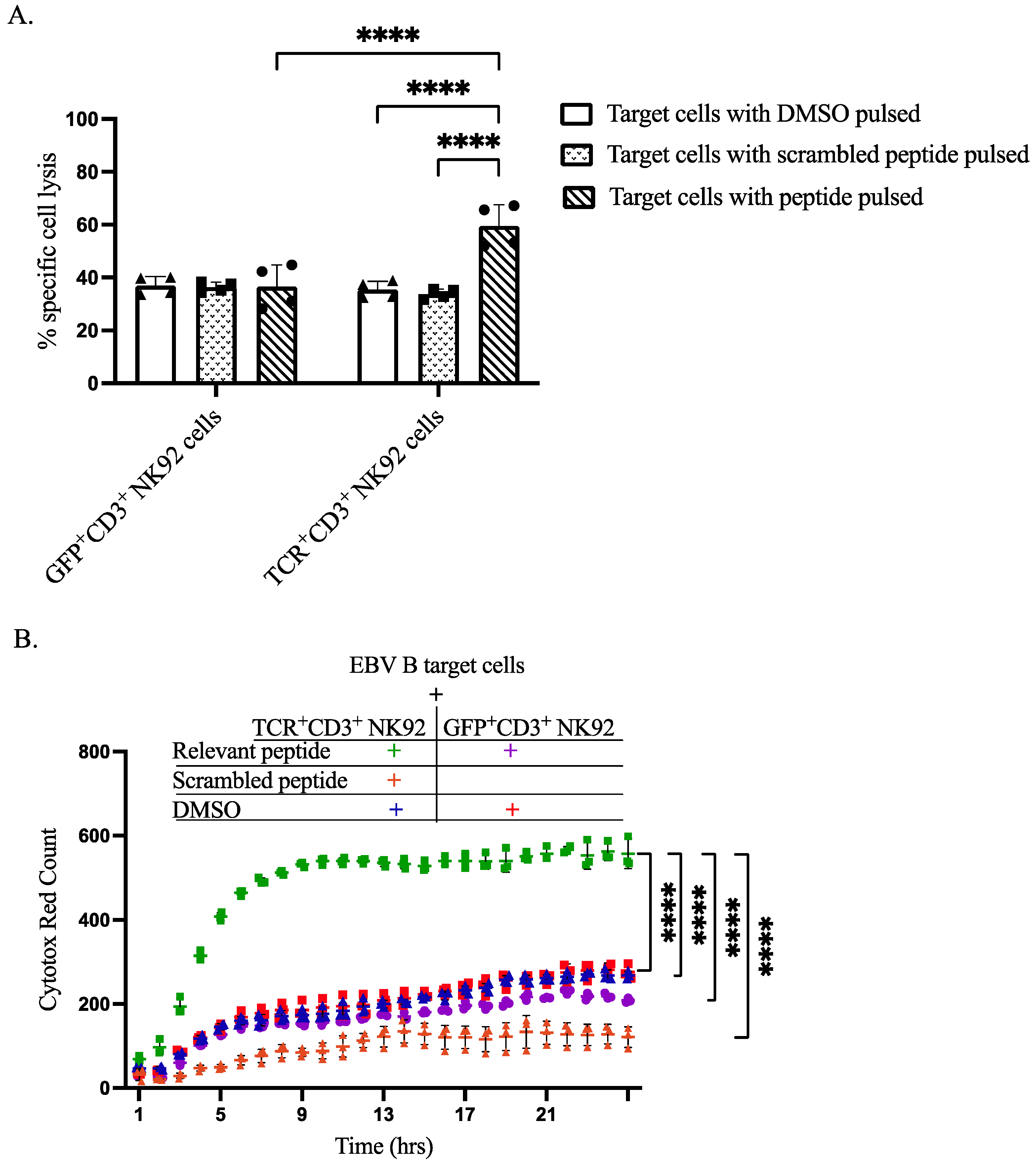
3.5. Specific Cytotoxicity of TCR+ NK92 Cells against Selective Target Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med. J. 2015, 6, e0004. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, M.H.M.; Hoogeboom, M.; De Paus, R.A.; Kester, M.G.D.; Van Der Hoorn, M.A.W.G.; Goulmy, E.A.J.M.; Willemze, R.; Falkenburg, J.H.F. Redirection of antileukemic reactivity of peripheral T lymphocytes using gene transfer of minor histocompatibility antigen HA-2-specific T-cell receptor complexes expressing a conserved alpha joining region. Blood 2003, 102, 3530–3540. [Google Scholar] [CrossRef] [PubMed]
- Calogero, A.; Hospers, G.A.; Krüse, K.M.; Schrier, P.I.; Mulder, N.H.; Hooijberg, E.; de Leij, L.F. Retargeting of a T cell line by anti MAGE-3/HLA-A2 alpha beta TCR gene transfer. Anticancer. Res. 2000, 20, 1793–1799. [Google Scholar]
- Alcover, A.; Alarcón, B.; Di Bartolo, V. Cell Biology of T Cell Receptor Expression and Regulation. Annu. Rev. Immunol. 2018, 36, 103–125. [Google Scholar] [CrossRef]
- Rubinstein, M.P.; Kadima, A.N.; Salem, M.; Nguyen, C.L.; Gillanders, W.E.; Nishimura, M.I.; Cole, D.J. Transfer of TCR genes into mature T cells is accompanied by the maintenance of parental T cell avidity. J. Immunol. 2003, 170, 1209–1217. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- Ikeda, H. T-cell adoptive immunotherapy using tumor-infiltrating T cells and genetically engineered TCR-T cells. Int. Immunol. 2016, 28, 349–353. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef]
- Stärck, L.; Popp, K.; Pircher, H.; Uckert, W. Immunotherapy with TCR-redirected T cells: Comparison of TCR-transduced and TCR-engineered hematopoietic stem cell–derived T cells. J. Immunol. 2014, 192, 206–213. [Google Scholar] [CrossRef]
- Schumacher, T.N.M. T-cell-receptor gene therapy. Nat. Rev. Immunol. 2002, 2, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Orbelyan, G.A.; Tang, F.; Sally, B.; Solus, J.; Meresse, B.; Ciszewski, C.; Grenier, J.-C.; Barreiro, L.B.; Lanier, L.L.; Jabri, B. Human NKG2E is expressed and forms an intracytoplasmic complex with CD94 and DAP12. J. Immunol. 2014, 193, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Kruschinski, A.; Moosmann, A.; Poschke, I.; Norell, H.; Chmielewski, M.; Seliger, B.; Kiessling, R.; Blankenstein, T.; Abken, H.; Charo, J. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc. Natl. Acad. Sci. USA 2008, 105, 17481–17486. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, R.; Zhu, X.; Wang, L.; Ma, J.; Han, H.; Wang, X.; Zhang, G.; He, W.; Wang, W.; et al. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol. Cell Biol. 2013, 91, 615–624. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Mensali, N.; Dillard, P.; Hebeisen, M.; Lorenz, S.; Theodossiou, T.; Myhre, M.R.; Fåne, A.; Gaudernack, G.; Kvalheim, G.; Myklebust, J.H.; et al. NK cells specifically TCR-dressed to kill cancer cells. EBioMedicine 2019, 40, 106–117. [Google Scholar] [CrossRef]
- Parlar, A.; Sayitoglu, E.C.; Ozkazanc, D.; Georgoudaki, A.M.; Pamukcu, C.; Aras, M.; Josey, B.J.; Chrobok, M.; Branecki, S.; Zahedimaram, P.; et al. Engineering antigen-specific NK cell lines against the melanoma-associated antigen tyrosinase via TCR gene transfer. Eur. J. Immunol. 2019, 49, 1278–1290. [Google Scholar] [CrossRef]
- Morton, L.T.; Wachsmann, T.L.A.; Meeuwsen, M.H.; Wouters, A.K.; Remst, D.F.G.; van Loenen, M.M.; Falkenburg, J.H.F.; Heemskerk, M.H.M. T cell receptor engineering of primary NK cells to therapeutically target tumors and tumor immune evasion. J. ImmunoTherapy Cancer 2022, 10, e003715. [Google Scholar] [CrossRef]
- Klausner, R.D.; Lippincott-Schwartz, J.; Bonifacino, J.S. The T cell antigen receptor: Insights into organelle biology. Annu. Rev. Cell Biol. 1990, 6, 403–431. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.; Kastrup, J.; Lauritsen, J.P.; Menné, C.; von Bülow, F.; Geisler, C. TCRzeta is transported to and retained in the Golgi apparatus independently of other TCR chains: Implications for TCR assembly. Eur. J. Immunol. 1999, 29, 1719–1728. [Google Scholar] [CrossRef]
- Restifo, N.P.; Marincola, F.M.; Kawakami, Y.; Taubenberger, J.; Yannelli, J.R.; Rosenberg, S.A. Loss of functional beta2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. JNCI J. Natl. Cancer Inst. 1996, 88, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Christopher, M.J.; Petti, A.A.; Rettig, M.P.; Miller, C.A.; Chendamarai, E.; Duncavage, E.J.; Klco, J.M.; Helton, N.M.; O’Laughlin, M.; Fronick, C.C.; et al. Immune escape of relapsed AML cells after allogeneic transplantation. N. Engl. J. Med. 2018, 379, 2330–2341. [Google Scholar] [CrossRef] [PubMed]
- Burr, M.; Sparbier, C.E.; Chan, K.L.; Chan, Y.-C.; Kersbergen, A.; Lam, E.Y.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O.; et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 2019, 36, 385–401.e8. [Google Scholar] [CrossRef]
- Villalobos, I.B.; Takahashi, Y.; Akatsuka, Y.; Muramatsu, H.; Nishio, N.; Hama, A.; Yagasaki, H.; Saji, H.; Kato, M.; Ogawa, S.; et al. Relapse of leukemia with loss of mismatched HLA resulting from uniparental disomy after haploidentical hematopoietic stem cell transplantation. Blood 2010, 115, 3158–3161. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Wileman, T.; Kane, L.P.; Young, J.; Carson, G.R.; Terhorst, C. Associations between subunit ectodomains promote T cell antigen receptor assembly and protect against degradation in the ER. J. Cell Biol. 1993, 122, 67–78. [Google Scholar] [CrossRef]
- Yu, H.; Kaung, G.; Kobayashi, S.; Kopito, R.R. Cytosolic degradation of T-cell receptor alpha chains by the proteasome. J. Biol. Chem. 1997, 272, 20800–20804. [Google Scholar] [CrossRef]
- Valitutti, S.; Müller, S.; Salio, M.; Lanzavecchia, A. Degradation of T cell receptor (TCR)-CD3-zeta complexes after antigenic stimulation. J. Exp. Med. 1997, 185, 1859–1864. [Google Scholar] [CrossRef]
- Iliopoulou, E.G.; Kountourakis, P.; Karamouzis, M.V.; Doufexis, D.; Ardavanis, A.; Baxevanis, C.N.; Rigatos, G.; Papamichail, M.; Perez, S.A. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Li, L.; Mccarty, J.; Kaur, I.; Yvon, E.; Shaim, H.; Muftuoglu, M.; Liu, E.; Orlowski, R.; Cooper, L.; et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br. J. Haematol. 2017, 177, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Sarukhan, A.; Garcia, C.; Lanoue, A.; von Boehmer, H. Allelic inclusion of T cell receptor α genes poses an autoimmune hazard due to low-level expression of autospecific receptors. Immunity 1998, 8, 563–570. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Neudorfer, J.; Weinhold, M.; Leisegang, M.; Engels, B.; Noessner, E.; Heemskerk, M.H.; Charo, J.; Schendel, D.J.; Blankenstein, T.; et al. Designer T cells by T cell receptor replacement. Eur. J. Immunol. 2006, 36, 3052–3059. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H.-G.; Wong, E.; Maki, G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 1996, 2, 68–75. [Google Scholar]
- Yan, Y.; Steinherz, P.; Klingemann, H.G.; Dennig, D.; Childs, B.H.; McGuirk, J.; O’Reilly, R.J. Antileukemia activity of a natural killer cell line against human leukemias. Clin. Cancer Res. 1998, 4, 2859–2868. [Google Scholar]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasul, K.H.; Hussain, A.; Reilly, H.; Karvouni, M.; Dahlberg, C.I.M.; Al-Attar, M.S.; Wagner, A.K.; Alici, E.; Mohammad, D.K. Assessment of T Cell Receptor Complex Expression Kinetics in Natural Killer Cells. Curr. Issues Mol. Biol. 2022, 44, 3859-3871. https://doi.org/10.3390/cimb44090265
Rasul KH, Hussain A, Reilly H, Karvouni M, Dahlberg CIM, Al-Attar MS, Wagner AK, Alici E, Mohammad DK. Assessment of T Cell Receptor Complex Expression Kinetics in Natural Killer Cells. Current Issues in Molecular Biology. 2022; 44(9):3859-3871. https://doi.org/10.3390/cimb44090265
Chicago/Turabian StyleRasul, Khder H., Alamdar Hussain, Hazel Reilly, Maria Karvouni, Carin I. M. Dahlberg, Mustafa S. Al-Attar, Arnika K. Wagner, Evren Alici, and Dara K. Mohammad. 2022. "Assessment of T Cell Receptor Complex Expression Kinetics in Natural Killer Cells" Current Issues in Molecular Biology 44, no. 9: 3859-3871. https://doi.org/10.3390/cimb44090265
APA StyleRasul, K. H., Hussain, A., Reilly, H., Karvouni, M., Dahlberg, C. I. M., Al-Attar, M. S., Wagner, A. K., Alici, E., & Mohammad, D. K. (2022). Assessment of T Cell Receptor Complex Expression Kinetics in Natural Killer Cells. Current Issues in Molecular Biology, 44(9), 3859-3871. https://doi.org/10.3390/cimb44090265