
Identification of Age-Associated Transcriptomic Changes Linked to Immunotherapy Response in Primary Melanoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Cohorts
2.2. Data Selection and Processing
2.3. Differential Expression Analysis of Individual Genes
2.4. Functional and Pathway Enrichment Analysis
2.5. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Mapping Analysis
2.6. Principal Component Analysis
2.7. Statistical Analyses
3. Results
3.1. Demographic and Clinical Features of the Patients
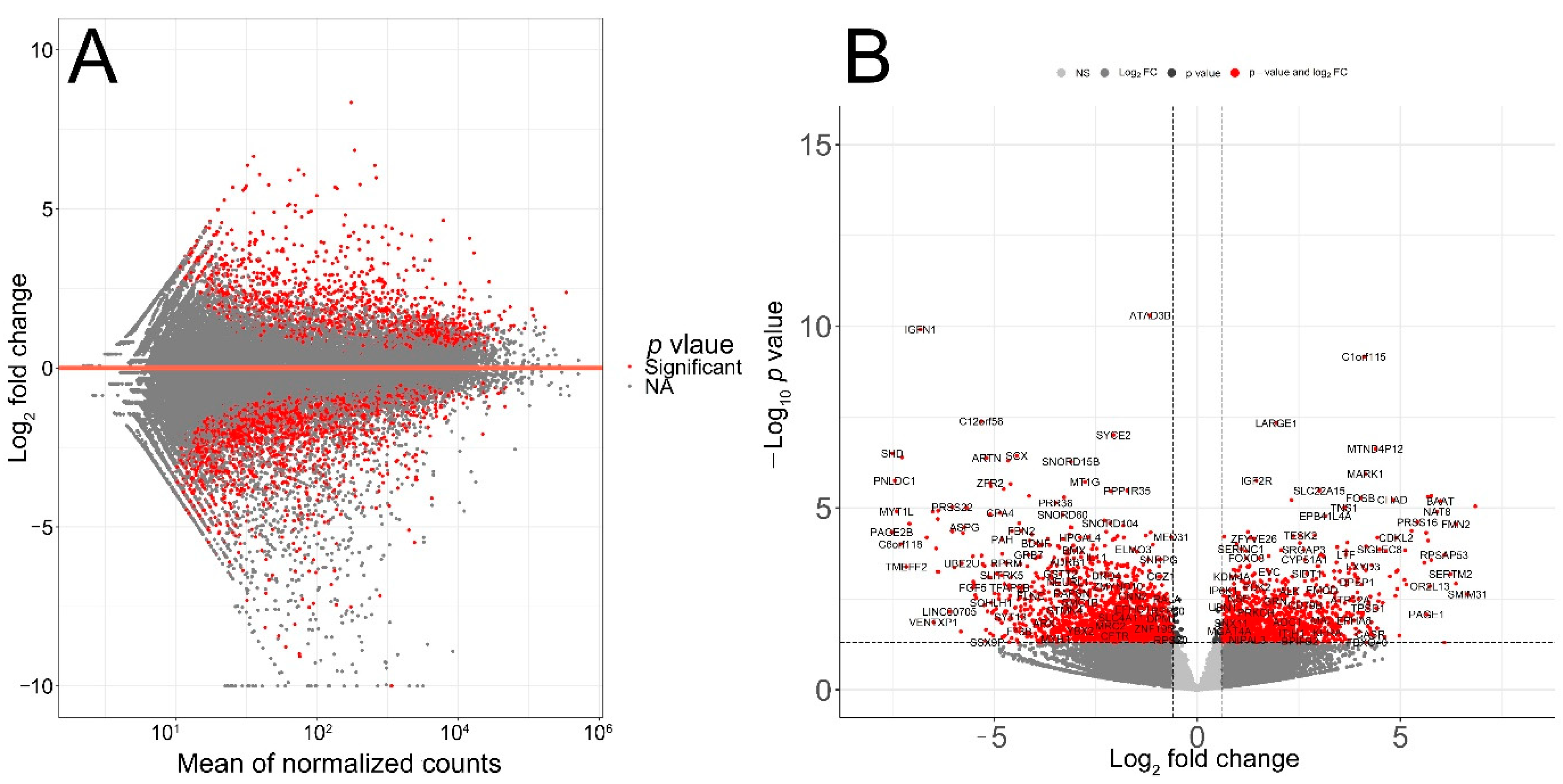
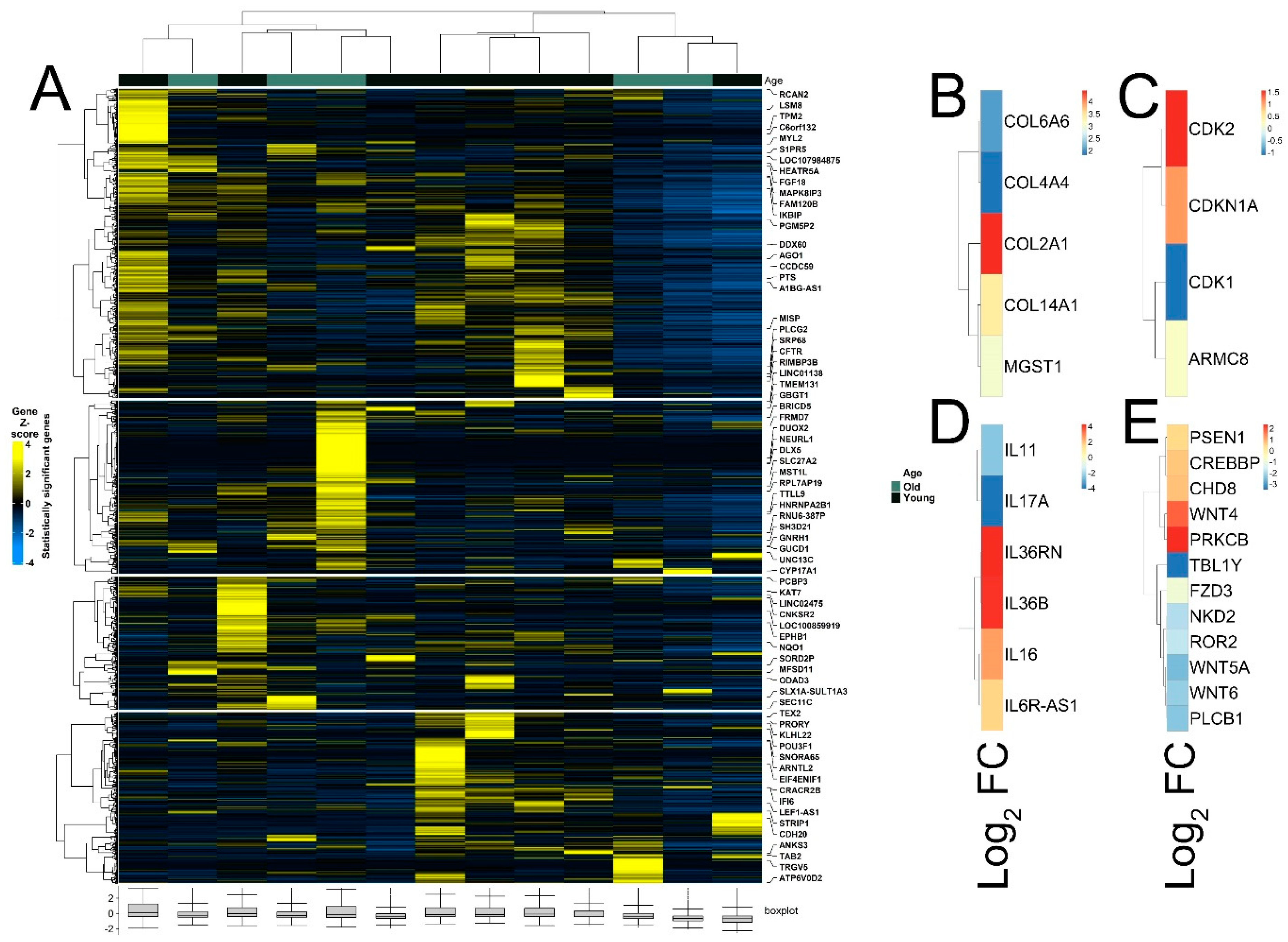
3.2. Identification of Age-Associated Differentially Expressed Genes in Melanoma
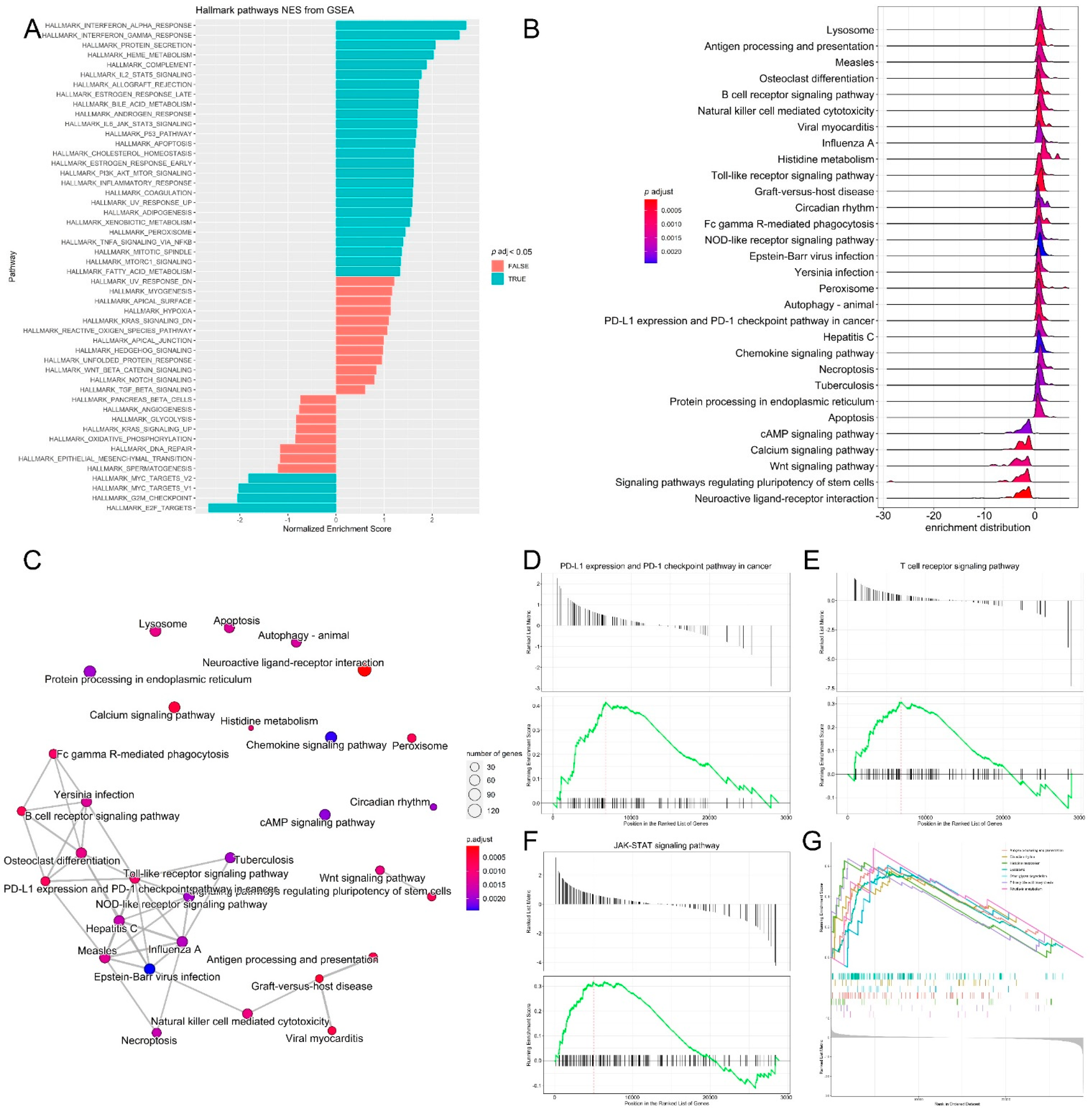
3.3. Pathway Analysis, Functional Annotation of Differentially Expressed Genes and Identification of Key Regulatory Genes of Response to Immunotherapy
3.4. Differential Expression of Treg Signature Genes and Metabolic Genes in Melanoma
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Noone, A.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review 1975–2015; National Cancer Institute: Bethesda, MD, USA, 2018. [Google Scholar]
- Centers for Disease Control and Prevention. Melanoma Incidence and Mortality, United States–2012–2016. U.S. Cancer Statistics Data Brief; No. 9; Centers for Disease Control and Prevention, US Department of Health and Human Services: Atlanta, GA, USA, 2019. [Google Scholar]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Moser, J.C.; Grossman, K.F. Adjuvant therapy for resected high-risk melanoma. Semin. Cutan. Med. Surg. 2018, 37, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Kakavand, H.; Rawson, R.V.; Pupo, G.M.; Yang, J.Y.; Menzies, A.M.; Carlino, M.S.; Kefford, R.F.; Howle, J.R.; Saw, R.P.; Thompson, J.F.; et al. PD-L1 expression and immune escape in melanoma resistance to MAPK inhibitors. Clin. Cancer Res. 2017, 23, 6054–6061. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Paulson, K.G.; Gupta, D.; Kim, T.S.; Veatch, J.R.; Byrd, D.R.; Bhatia, S.; Wojcik, K.; Chapuis, A.G.; Thompson, J.A.; Madeleine, M.M.; et al. Age-specific incidence of melanoma in the United States. JAMA Dermatol. 2020, 156, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M. Decreased survival rates of older-aged patients with melanoma: Biological differences or undertreatment? Ann. Surg. Oncol. 2015, 22, 2101–2103. [Google Scholar] [CrossRef]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H.; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 2016, 532, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Kugel, C.H.; Douglass, S.M.; Webster, M.R.; Kaur, A.; Liu, Q.; Yin, X.; Weiss, S.A.; Darvishian, F.; Al-Rohil, R.N.; Ndoye, A.; et al. Age correlates with response to anti-PD1, reflecting age-related differences in intratumoral effector and regulatory T-cell populations. Clin. Cancer Res. 2018, 24, 5347–5356. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68. [Google Scholar] [CrossRef]
- Love, M.; Anders, S.; Huber, M. Differential gene expression analysis based on the negative binomial distribution. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. Enhanced Volcano: Publication-Ready Volcano Plots with 667 Enhanced Colouring and Labeling; R Package Version 1.6. 0. 668; EnhancedVolcano, 2020; Available online: https://github.com/kevinblighe (accessed on 13 July 2022).
- Yuan, J.; Jiang, L.; Guo, C. The micro RNA hsa-miR-377-3p inhibits tumor growth in malignant melanoma. RSC Adv. 2019, 9, 19057–19064. [Google Scholar] [CrossRef]
- Fan, C.; Zhao, Y.; Mao, X.; Miao, Y.; Lin, X.; Jiang, G.; Zhang, X.; Han, Q.; Luan, L.; Wang, E. Armc8 expression was elevated during atypia-to-carcinoma progression and associated with cancer development of breast carcinoma. Tumor Biol. 2014, 35, 11337–11343. [Google Scholar] [CrossRef]
- Zhao, Y.; Peng, S.; Jia, C.; Xu, F.; Xu, Y.; Dai, C. Armc8 regulates the invasive ability of hepatocellular carcinoma through E-cadherin/catenin complex. Tumor Biol. 2016, 37, 11219–11224. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Jiang, G.; Fan, C.; Zhang, X.; Zhang, Y.; Miao, Y.; Lin, X.; Wu, J.; Wang, L.; Liu, Y.; et al. ARMC8α promotes proliferation and invasion of non-small cell lung cancer cells by activating the canonical Wnt signaling pathway. Tumor Biol. 2014, 35, 8903–8911. [Google Scholar] [CrossRef]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. IL-17 can promote tumor growth through an IL-6–Stat3 signaling pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Fujio, K.; Shoda, H.; Okamoto, A.; Tsuno, N.H.; Takahashi, K.; Yamamoto, K. IL-17B and IL-17C are associated with TNF-α production and contribute to the exacerbation of inflammatory arthritis. J. Immunol. 2007, 179, 7128–7136. [Google Scholar] [CrossRef] [PubMed]
- Sirikanjanapong, S.; Lanson, B.; Amin, M.; Martiniuk, F.; Kamino, H.; Wang, B.Y. Collision tumor of primary laryngeal mucosal melanoma and invasive squamous cell carcinoma with IL-17A and CD70 gene over-expression. Head Neck Pathol. 2010, 4, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Wróblewska, J.P.; Lach, M.S.; Kulcenty, K.; Galus, Ł.; Suchorska, W.M.; Rösel, D.; Brábek, J.; Marszałek, A. The Analysis of Inflammation-Related Proteins in a Cargo of Exosomes Derived from the Serum of Uveal Melanoma Patients Reveals Potential Biomarkers of Disease Progression. Cancers 2021, 13, 3334. [Google Scholar] [CrossRef] [PubMed]
- Nishina, T.; Deguchi, Y.; Ohshima, D.; Takeda, W.; Ohtsuka, M.; Shichino, S.; Ueha, S.; Yamazaki, S.; Kawauchi, M.; Nakamura, E.; et al. Interleukin-11-expressing fibroblasts have a unique gene signature correlated with poor prognosis of colorectal cancer. Nat. Commun. 2021, 12, 2281. [Google Scholar] [CrossRef]
- Johnstone, C.N.; Chand, A.; Putoczki, T.L.; Ernst, M. Emerging roles for IL-11 signaling in cancer development and progression: Focus on breast cancer. Cytokine Growth Factor Rev. 2015, 26, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Putoczki, T.L.; Ernst, M. IL-11 signaling as a therapeutic target for cancer. Immunotherapy 2015, 7, 441–453. [Google Scholar] [CrossRef]
- Krishnamurthy, K.; Urioste, S.N.; Cusnir, M.; Schwartz, M.; Alghamdi, S.; Sriganeshan, V.; Poppiti, R. Analysis of Genetic Alterations in Cutaneous Malignant Melanomas Unveils Unique Loco-Regional Variations and Novel Predictors of Metastatic Potential. Am. J. Dermatopathol. 2021, 43, e185–e189. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Targeting invasive properties of melanoma cells. FEBS J. 2017, 284, 2148–2162. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.R.; Fane, M.E.; Alicea, G.M.; Basu, S.; Kossenkov, A.V.; Marino, G.E.; Douglass, S.M.; Kaur, A.; Ecker, B.L.; Gnanapradeepan, K.; et al. Paradoxical role for wild-type p53 in driving therapy resistance in melanoma. Mol. Cell 2020, 77, 633–644. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q.; et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov. 2013, 3, 1378–1393. [Google Scholar] [CrossRef]
- O’Connell, M.P.; Fiori, J.L.; Xu, M.; Carter, A.D.; Frank, B.P.; Camilli, T.C.; French, A.D.; Dissanayake, S.K.; Indig, F.E.; Bernier, M.; et al. The orphan tyrosine kinase receptor, ROR2, mediates Wnt5A signaling in metastatic melanoma. Oncogene 2010, 29, 34–44. [Google Scholar] [CrossRef]
- Weeraratna, A.T.; Jiang, Y.; Hostetter, G.; Rosenblatt, K.; Duray, P.; Bittner, M.; Trent, J.M. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 2002, 1, 279–288. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Biological hallmarks of cancer. In Holland-Frei Cancer Medicine; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 1–10. [Google Scholar]
- Johnson, D.B.; Bordeaux, J.; Kim, J.Y.; Vaupel, C.; Rimm, D.L.; Ho, T.H.; Joseph, R.W.; Daud, A.I.; Conry, R.M.; Gaughan, E.M.; et al. Quantitative spatial profiling of PD-1/PD-L1 interaction and HLA-DR/IDO-1 predicts improved outcomes of anti–PD-1 therapies in metastatic melanoma. Clin. Cancer Res. 2018, 24, 5250–5260. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [Green Version]
- Neitzke, D.J.; Bowers, J.S.; Andrijauskaite, K.; O’Connell, N.S.; Garrett-Mayer, E.; Wrangle, J.; Li, Z.; Paulos, C.M.; Cole, D.J.; Rubinstein, M.P. Murine Th17 cells utilize IL-2 receptor gamma chain cytokines but are resistant to cytokine withdrawal-induced apoptosis. Cancer Immunol. Immunother. 2017, 66, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Gorlov, I.; Orlow, I.; Ringelberg, C.; Hernando, E.; Ernstoff, M.S.; Cheng, C.; Her, S.; Parker, J.S.; Thompson, C.L.; Gerstenblith, M.R.; et al. Identification of gene expression levels in primary melanoma associated with clinically meaningful characteristics. Melanoma Res. 2018, 28, 380. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, J.D.; Nishino, M.; Severgnini, M.; Manos, M.; Mak, R.H.; Hodi, F.S. Pneumonitis resulting from radiation and immune checkpoint blockade illustrates characteristic clinical, radiologic and circulating biomarker features. J. Immunother. Cancer 2019, 7, 112. [Google Scholar] [CrossRef]
- Zhou, G.; Levitsky, H.I. Natural regulatory T cells and de novo-induced regulatory T cells contribute independently to tumor-specific tolerance. J. Immunol. 2007, 178, 2155–2162. [Google Scholar] [CrossRef]
- Mourmouras, V.; Fimiani, M.; Rubegni, P.; Epistolato, M.C.; Malagnino, V.; Cardone, C.; Cosci, E.; Nisi, M.D.; Miracco, C. Evaluation of tumour-infiltrating CD4 + CD25 + FOXP3 + regulatory T cells in human cutaneous benign and atypical naevi, melanomas and melanoma metastases. Br. J. Dermatol. 2007, 157, 531–539. [Google Scholar] [CrossRef]
- Ladányi, A.; Mohos, A.; Somlai, B.; Liszkay, G.; Gilde, K.; Fejős, Z.; Gaudi, I.; Tímár, J. FOXP3+ cell density in primary tumor has no prognostic impact in patients with cutaneous malignant melanoma. Pathol. Oncol. Res. 2010, 16, 303–309. [Google Scholar] [CrossRef]
- Vence, L.; Palucka, A.K.; Fay, J.W.; Ito, T.; Liu, Y.J.; Banchereau, J.; Ueno, H. Circulating tumor antigen-specific regulatory T cells in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2007, 104, 20884–20889. [Google Scholar] [CrossRef]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef]
- Kumar, P.; Bhattacharya, P.; Prabhakar, B.S. A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J. Autoimmun. 2018, 95, 77–99. [Google Scholar] [CrossRef]
- Nizar, S.; Meyer, B.; Galustian, C.; Kumar, D.; Dalgleish, A. T regulatory cells, the evolution of targeted immunotherapy. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2010, 1806, 7–17. [Google Scholar] [CrossRef]
- Jennings, E.; Elliot, T.A.; Thawait, N.; Kanabar, S.; Yam-Puc, J.C.; Ono, M.; Toellner, K.M.; Wraith, D.C.; Anderson, G.; Bending, D. Nr4a1 and Nr4a3 reporter mice are differentially sensitive to T cell receptor signal strength and duration. Cell Rep. 2020, 33, 108328. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Lu, H.; Li, J.; Yan, X.; Xiao, M.; Hao, J.; Alekseev, A.; Khong, H.; Chen, T.; et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 2019, 567, 525–529. [Google Scholar] [CrossRef]
- Sekiya, T.; Kashiwagi, I.; Inoue, N.; Morita, R.; Hori, S.; Waldmann, H.; Rudensky, A.Y.; Ichinose, H.; Metzger, D.; Chambon, P.; et al. The nuclear orphan receptor Nr4a2 induces Foxp3 and regulates differentiation of CD4+ T cells. Nat. Commun. 2011, 2, 269. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, N.; Sakaguchi, S. Transcriptional and epigenetic basis of Treg cell development and function: Its genetic anomalies or variations in autoimmune diseases. Cell Res. 2020, 30, 465–474. [Google Scholar] [CrossRef]
- Baine, I.; Basu, S.; Ames, R.; Sellers, R.S.; Macian, F. Helios induces epigenetic silencing of IL2 gene expression in regulatory T cells. J. Immunol. 2013, 190, 1008–1016. [Google Scholar] [CrossRef]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.; van den Braber, M.; Rozeman, E.A.; Haanen, J.B.; Blank, C.U.; et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 2020, 181, 747. [Google Scholar] [CrossRef]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef]
- Yingjuan, W.; Li, Z.; Wei, C.; Xiaoyuan, W. Identification of prognostic genes and construction of a novel gene signature in the skin melanoma based on the tumor microenvironment. Medicine 2021, 100, e26017. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’S Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef]
- Jaiswal, A.R.; Liu, A.J.; Pudakalakatti, S.; Dutta, P.; Jayaprakash, P.; Bartkowiak, T.; Ager, C.R.; Wang, Z.Q.; Reuben, A.; Cooper, Z.A.; et al. Melanoma evolves complete immunotherapy resistance through the acquisition of a hypermetabolic phenotype. Cancer Immunol. Res. 2020, 8, 1365. [Google Scholar] [CrossRef]
- Filipp, F.V.; Ratnikov, B.; De Ingeniis, J.; Smith, J.W.; Osterman, A.L.; Scott, D.A. Glutamine-fueled mitochondrial metabolism is decoupled from glycolysis in melanoma. Pigment Cell Melanoma Res. 2012, 25, 732–739. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.E.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef]
- De Ingeniis, J.; Ratnikov, B.; Richardson, A.D.; Scott, D.A.; Aza-Blanc, P.; De, S.K.; Kazanov, M.; Pellecchia, M.; Ronai, Z.E.; Osterman, A.L.; et al. Functional specialization in proline biosynthesis of melanoma. PLoS ONE 2012, 7, e45190. [Google Scholar] [CrossRef]
- Qin, J.-Z.; Xin, H.; Nickoloff, B. Targeting glutamine metabolism sensitizes melanoma cells to TRAIL-induced death. Biochem. Biophys. Res. Commun. 2010, 398, 146–152. [Google Scholar] [CrossRef]
- Jessen, C.; Kreß, J.K.; Baluapuri, A.; Hufnagel, A.; Schmitz, W.; Kneitz, S.; Roth, S.; Marquardt, A.; Appenzeller, S.; Ade, C.P.; et al. The transcription factor NRF2 enhances melanoma malignancy by blocking differentiation and inducing COX2 expression. Oncogene 2020, 39, 6841–6855. [Google Scholar] [CrossRef]
- León-Letelier, R.A.; Bonifaz, L.C.; Fuentes-Pananá, E.M. OMIC signatures to understand cancer immunosurveillance and immunoediting: Melanoma and immune cells interplay in immunotherapy. J. Leukoc. Biol. 2019, 105, 915–933. [Google Scholar] [CrossRef]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Zakka, L.R.; Mihm, M.C., Jr.; Schatton, T. Tumour-infiltrating lymphocytes in melanoma prognosis and cancer immunotherapy. Pathology 2016, 48, 177–187. [Google Scholar] [CrossRef]
- Morgenstern, R.; Zhang, J.; Johansson, K. Microsomal glutathione transferase 1: Mechanism and functional roles. Drug Metab. Rev. 2011, 43, 300–306. [Google Scholar] [CrossRef]
- Zeng, F.; Su, J.; Peng, C.; Liao, M.; Zhao, S.; Guo, Y.; Chen, X.; Deng, G. Prognostic implications of metabolism related gene signature in cutaneous melanoma. Front. Oncol. 2020, 10, 1710. [Google Scholar] [CrossRef] [PubMed]
- Prall, W.C.; Czibere, A.; Jäger, M.; Spentzos, D.; Libermann, T.A.; Gattermann, N.; Haas, R.; Aivado, M. Age-related transcription levels of KU70, MGST1 and BIK in CD34+ hematopoietic stem and progenitor cells. Mech. Ageing Dev. 2007, 128, 503–510. [Google Scholar] [CrossRef]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell MotilityHAPLN1 Loss in Aged Skin Promotes Melanoma Metastasis. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Miskolczi, Z.; Smith, M.P.; Rowling, E.J.; Ferguson, J.; Barriuso, J.; Wellbrock, C. Collagen abundance controls melanoma phenotypes through lineage-specific microenvironment sensing. Oncogene 2018, 37, 3166–3182. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Kim, Y.J.; Robert, L.; Tsoi, J.; Comin-Anduix, B.; Berent-Maoz, B.; Cochran, A.J.; Economou, J.S.; Tumeh, P.C.; Puig-Saus, C.; et al. Immunotherapy resistance by inflammation-induced dedifferentiation. Cancer Discov. 2018, 8, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, S.K.; Olkhanud, P.B.; O’Connell, M.P.; Carter, A.; French, A.D.; Camilli, T.C.; Emeche, C.D.; Hewitt, K.J.; Rosenthal, D.T.; Leotlela, P.D.; et al. Wnt5A regulates expression of tumor-associated antigens in melanoma via changes in signal transducers and activators of transcription 3 phosphorylation. Cancer Res. 2008, 68, 10205–10214. [Google Scholar] [CrossRef]
- Behera, R.; Marchbank, K.; Kaur, A.; Dang, V.; Webster, M.; O’Connell, M.; Xu, X.; Weeraratna, A. Abstract A11: Crosstalk between klotho and wnt5A drives age-related melanoma progression. Cancer Res. 2015, 75, A11. [Google Scholar] [CrossRef]
- Behera, R.; Kaur, A.; Webster, M.R.; Kim, S.; Ndoye, A.; Kugel, C.H.; Alicea, G.M.; Wang, J.; Ghosh, K.; Cheng, P.; et al. Inhibition of Age-Related Therapy Resistance in Melanoma by Rosiglitazone-Mediated Induction of KlothoIncreasing Klotho in Aged Mice Reduces Tumor Burden. Clin. Cancer Res. 2017, 23, 3181–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, D.; Verburg, F.; Renner, K.; Peeper, D.S.; Lacroix, R.; Blank, C.U. Metabolic profiles of regulatory T cells in the tumour microenvironment. Cancer Immunol. Immunother. 2021, 70, 2417–2427. [Google Scholar] [CrossRef]
- Jacobs, J.F.; Nierkens, S.; Figdor, C.G.; de Vries, I.J.M.; Adema, G.J. Regulatory T cells in melanoma: The final hurdle towards effective immunotherapy? Lancet Oncol. 2012, 13, e32–e42. [Google Scholar] [CrossRef]
- Wu, Y.; Wei, J.; Chen, X.; Qin, Y.; Mao, R.; Song, J.; Fan, Y. Comprehensive transcriptome profiling in elderly cancer patients reveals aging-altered immune cells and immune checkpoints. Int. J. Cancer 2019, 144, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.M.; Adamoski, D.; Dos Reis, L.M.; Ascenção, C.F.; de Oliveira, K.R.; Mafra, A.C.P.; da Silva Bastos, A.C.; Quintero, M.; de G Cassago, C.; Ferreira, I.M.; et al. GLS2 is protumorigenic in breast cancers. Oncogene 2020, 39, 690–702. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Helbawy, N.F.; El Zowalaty, A.E. Identification of Age-Associated Transcriptomic Changes Linked to Immunotherapy Response in Primary Melanoma. Curr. Issues Mol. Biol. 2022, 44, 4118-4131. https://doi.org/10.3390/cimb44090282
El-Helbawy NF, El Zowalaty AE. Identification of Age-Associated Transcriptomic Changes Linked to Immunotherapy Response in Primary Melanoma. Current Issues in Molecular Biology. 2022; 44(9):4118-4131. https://doi.org/10.3390/cimb44090282
Chicago/Turabian StyleEl-Helbawy, Nehal Farid, and Ahmed Ezat El Zowalaty. 2022. "Identification of Age-Associated Transcriptomic Changes Linked to Immunotherapy Response in Primary Melanoma" Current Issues in Molecular Biology 44, no. 9: 4118-4131. https://doi.org/10.3390/cimb44090282
APA StyleEl-Helbawy, N. F., & El Zowalaty, A. E. (2022). Identification of Age-Associated Transcriptomic Changes Linked to Immunotherapy Response in Primary Melanoma. Current Issues in Molecular Biology, 44(9), 4118-4131. https://doi.org/10.3390/cimb44090282