
Autoimmunity and Immunodeficiency in Severe SARS-CoV-2 Infection and Prolonged COVID-19

 , and
, and 
Abstract
:1. Introduction
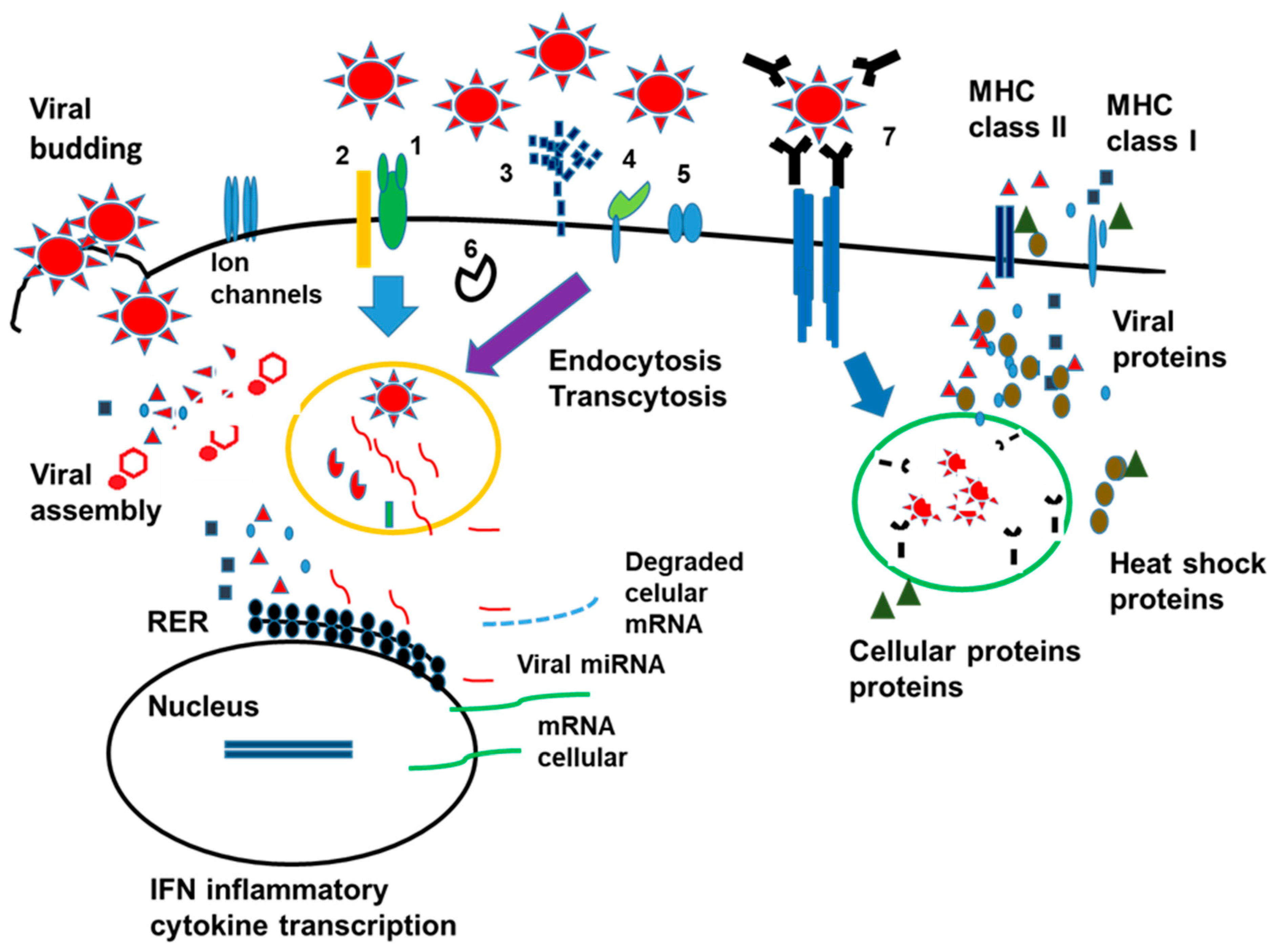
2. Brief Overview of the Immune Response against SARS-CoV-2
3. Autoimmunity and COVID-19
4. Primary Immunodeficiency and COVID-19
5. Conclusions
6. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chams, N.; Chams, S.; Badran, R.; Shams, A.; Araji, A.; Raad, M.; Mukhopadhyay, S.; Stroberg, E.; Duval, E.J.; Barton, L.M.; et al. COVID-19: A Multidisciplinary Review. Front. Public Health 2020, 8, 383. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [PubMed] [Green Version]
- Rahman, S.; Montero, M.T.V.; Rowe, K.; Kirton, R.; Kunik, F., Jr. Epidemiology, pathogenesis, clinical presentations, diagnosis and treatment of COVID-19: A review of current evidence. Expert Rev. Clin. Pharmacol. 2021, 14, 601–621. [Google Scholar] [CrossRef]
- Araf, Y.; Akter, F.; Tang, Y.D.; Fatemi, R.; Parvez, S.A.; Zhen, C.; Hossain, G. Omicron variant of SARS-CoV-2: Genomic, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Kared, H.; Wolf, A.S.; Alirezaylavasani, A.; Ravussin, A.; Solum, G.; Tran, T.T.; Lund-Johansen, F.; Vaage, J.T.; Nissen-Meyer, L.S.; Nygaard, U.C.; et al. Immune responses in Omicron SARS-CoV-2 breakthrough infection in vaccinated adults. Nat. Commun. 2022, 13, 4165. [Google Scholar] [CrossRef] [PubMed]
- Bansal, K.; Kumar, S. Mutational cascade of SARS-CoV-2 leading to evolution and emergence of omicron variant. Virus Res. 2022, 315, 198765. [Google Scholar] [CrossRef] [PubMed]
- Ochani, R.; Asad, A.; Yasmin, F.; Shaikh, S.; Khalid, H.; Batra, S.; Sohail, M.R.; Mahmood, S.F.; Ochani, R.; Hussham Arshad, M.; et al. COVID-19 pandemic: From origins to outcomes. A comprehensive review of viral pathogenesis, clinical manifestations, diagnostic evaluation, and management. Infez. Med. 2021, 29, 20–36. [Google Scholar]
- Riou, J.; Althaus, C.L. Pattern of early human-to-human transmission of Wuhan 2019 novel coronavirus [2019-nCoV), December 2019 to January 2020. Euro Surveill. 2020, 25, 2000058. [Google Scholar] [CrossRef] [Green Version]
- van Doremalen, N.; Bushmaker, T.; Morris, D.H.; Holbrook, M.G.; Gamble, A.; Williamson, B.N.; Tamin, A.; Harcourt, J.L.; Thornburg, N.J.; Gerber, S.I.; et al. Aerosol and Surface Stability of SARS-CoV-2 as Compared with SARS-CoV-1. N. Engl. J. Med. 2020, 382, 1564–1567. [Google Scholar] [CrossRef]
- Nikolaidis, M.; Papakyriakou, A.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G.; Amoutzias, G.D. Comparative Analysis of SARS-CoV-2 Variants of Concern, Including Omicron, Highlights Their Common and Distinctive Amino Acid Substitution Patterns, Especially at the Spike ORF. Viruses 2022, 14, 707. [Google Scholar] [CrossRef]
- Marik, P.E.; Iglesias, J.; Varon, J.; Kory, P. A scoping review of the pathophysiology of COVID-19. Int. J. Immunopathol. Pharmacol. 2021, 35. [Google Scholar] [CrossRef] [PubMed]
- Nalbadian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syn-drome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Alipoor, S.D.; Mirsaeidi, M. SARS-CoV-2 cell entry beyond the ACE2 receptor. Mol. Biol. Rep. 2022, 49, 10715–10727. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; De Diego, M.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe acute respiratory síndrome coronavirus envelope protein ion cannel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhuang, M.W.; Han, L.; Zhang, J.; Nan, M.L.; Zhan, P.; Kang, D.; Liu, X.; Gao, C.; Wang, P.H. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal. Transduct. Target Ther. 2020, 5, 299. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Wang, H.; Ji, Y.; Yang, J.; Xu, S.; Huang, X.; Wang, Z.; Qin, L.; Tien, P.; Zhou, X.; et al. The nucleocapsid protein of coronaviruses acts as viral suppressor of RNA silencing in mammalian cells. J. Virol. 2015, 89, 9029–9043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lee, L.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. Architecture of SARS-CoV-2 transcriptome. Cell 2020, 14, 914–921. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; Guo, F.; et al. activation and evasion of type I interferon responses by SARS.CoV.2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Lu, Y.; Michel, H.A.; Wang, P.H.; Smith, G.L. Manipulation of innate immune signaling pathways by SARS-CoV-2 non-structural proteins. Front. Microbiol. 2022, 13, 1027015. [Google Scholar] [CrossRef]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.J.; USFQ-COVID19 Consortium; Nakagawa, S.; et al. SARS-CoV-2 ORF3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: Open the door for novel therapies. Signal. Transduct. Target. Ther. 2022, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Rashid, F.; Xie, Z.; Suleman, M.; Shah, A.; Khan, S.; Luo, S. Roles and functions of SARS-CoV-2 proteins in host immune evasion. Front. Immunol. 2022, 13, 940756. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, J.B.; Garcia, A.; Garmendia, J.; Moreno, D.; Hajduch, M.; Radzioch, D. Importance of miRNA in SARS-CoV2 infection. Gac. Méd. Caracas 2020, 128 (Suppl. 1), S17–S22. [Google Scholar] [CrossRef]
- Roustai Geraylow, K.; Hemmati, R.; Kadkhoda, S.; Ghafouri-Fard, S. miRNA expression in COVID-19. Gene Rep. 2022, 28, 101641. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, Z.; Song, J.; Qian, W.; Gu, X.; Yang, C.; Shen, N.; Xue, F.; Tang, Y. SARS-CoV-2-Encoded MiRNAs Inhibit Host Type I Interferon Pathway and Mediate Allelic Differential Expression of Susceptible Gene. Front. Immunol. 2021, 12, 767726. [Google Scholar] [CrossRef]
- Zhang, J.J.; Dong, X.; Cao, Y.Y.; Yuan, Y.D.; Yang, Y.B.; Yan, Y.Q.; Akdis, C.A.; Gao, Y.D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020, 75, 1730–1741. [Google Scholar] [CrossRef]
- Lisco, G.; De Tullio, A.; Stragapede, A.; Solimando, A.G.; Albanese, F.; Capobianco, M.; Giagulli, V.A.; Guastamacchia, E.; De Pergola, G.; Vacca, A.; et al. COVID-19 and the Endocrine System: A Comprehensive Review on the Theme. J. Clin. Med. 2021, 10, 2920. [Google Scholar] [CrossRef]
- Smadja, D.M.; Mentzer, S.J.; Fontenay, M.; Laffan, M.A.; Ackermann, M.; Helms, J.; Jonigk, D.; Chocron, R.; Pier, G.B.; Gendron, N.; et al. COVID-19 is a systemic vascular hemopathy: Insight for mechanistic and clinical aspects. Angiogenesis 2021, 24, 755–788. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Hosseini, P.; Fallahi, M.S.; Erabi, G.; Pakdin, M.; Zarezadeh, S.M.; Faridzadeh, A.; Entezari, S.; Ansari, A.; Poudineh, M.; Deravi, N. Multisystem Inflammatory Syndrome and Autoimmune Diseases Following COVID-19: Molecular Mechanisms and Therapeutic Opportunities. Front. Mol. Biosci. 2022, 9, 804109. [Google Scholar] [CrossRef]
- Hoste, L.; Van Paemel, R.; Haerynck, F. Multisystem inflammatory syndrome in children related to COVID-19: A systematic review. Eur. J. Pediatr. 2021, 180, 2019–2034. [Google Scholar] [CrossRef] [PubMed]
- Kunal, S.; Ish, P.; Sakthivel, P.; Malhotra, N.; Gupta, K. The emerging threat of multisystem inflammatory syndrome in adults (MIS-A) in COVID-19: A systematic review. Heart Lung. 2022, 54, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.A.; Canna, S.W.; Friedman, K.G.; Gorelik, M.; Lapidus, S.K.; Bassiri, H.; Behrens, E.M.; Ferris, A.; Kernan, K.F.; Schulert, G.S.; et al. American College of Rheumatology Clinical Guidance for Multisystem Inflammatory Syndrome in Children Associated With SARS-CoV-2 and Hyperinflammation in Pediatric COVID-19: Version 2. Arthritis Rheumatol. 2021, 73, e13–e29. [Google Scholar] [CrossRef] [PubMed]
- Bizjak, M.; Emeršič, N.; Zajc Avramovič, M.; Barbone, F.; Ronchese, F.; Della Paolera, S.; Conversano, E.; Amoroso, S.; Vidoni, M.; Vesel Tajnšek, T.; et al. High incidence of multisystem inflammatory syndrome and other autoimmune diseases after SARS-CoV-2 infection compared to COVID-19 vaccination in children and adolescents in south central Europe. Clin. Exp. Rheumatol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, D.J.; Rasmussen, S.A. An update on COVID-19 and pregnancy. Am. J. Obstet. Gynecol. 2022, 226, 177–186. [Google Scholar] [CrossRef]
- SeyedAlinaghi, S.; Karimi, A.; Barzegary, A.; Mojdeganlou, H.; Vahedi, F.; Mirghaderi, S.P.; Shobeiri, P.; Ramezani, M.; Yousefi Konjdar, P.; Mirzapour, P.; et al. COVID-19 mortality in patients with immunodeficiency and its predictors: A systematic review. Eur. J. Med. Res. 2022, 27, 195. [Google Scholar] [CrossRef]
- Markarian, N.M.; Galli, G.; Patel, D.; Hemmings, M.; Nagpal, P.; Berghuis, A.M.; Abrahamyan, L.; Vidal, S.M. Identify-ing Markers of Emerging SARS-CoV-2 Variants in Patients with Secondary Immunodeficiency. Front. Microbiol. 2022, 13, 933983. [Google Scholar] [CrossRef]
- De Sanctis, J.B.; García, A.H.; Moreno, D.; Hajduch, M. Coronavirus infection: An immunologists’ perspective. Scand. J. Immunol. 2021, 93, e13043. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Norddahl, G.L.; Melsted, P.; Gunnarsdottir, K.; Holm, H.; Eythorsson, E.; Arnthorsson, A.O.; Helgason, D.; Bjarnadottir, K.; Ingvarsson, R.F.; et al. Humoral Immune Response to SARS-CoV-2 in Iceland. N. Engl. J. Med. 2020, 383, 1724–1734. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic , G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. [Google Scholar] [CrossRef]
- Vijay, R.; Perlman, S. Middle East respiratory syndrome and severe respiratory syndrome. Curr. Opin. Virol. 2016, 16, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyenholz, D.K.; Perlman, S. Dysregulated type I interferon and inflammatory monocytes-macrophages response cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Gao, D.; Ciancanelli, M.J.; Zhang, P.; Harschnitz, O.; Bondet, V.; Hasek, M.; Chen, J.; Mu, X.; Itan, Y.; Cobat, A.; et al. TLR3 controls constitutive IFN-β antiviral immunity in human fibroblasts and cortical neurons. J. Clin. Investig. 2021, 131, e134529. [Google Scholar] [CrossRef]
- Schultze, J.L.; Aschenbrenner, A.C. COVID-19 and the human innate immune system. Cell 2021, 184, 1671–1692. [Google Scholar] [CrossRef]
- Lee, J.S.; Park, S.; Jeong, H.W.; Ahn, J.Y.; Choi, S.J.; Lee, H.; Choi, B.; Nam, S.K.; Sa, M.; Kwon, J.S.; et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020, 5, eabd1554. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Miao, V.N.; Owings, A.H.; Navia, A.W.; Tang, Y.; Bromley, J.D.; Lotfy, P.; Sloan, M.; Laird, H.; Williams, H.B.; et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell 2021, 184, 4713–4733.e22. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The Trinity of COVID-19 immunity: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.A.; Canna, S.W.; Schulert, G.S.; Volpi, S.; Lee, P.Y.; Kernan, K.F.; Caricchio, R.; Mahmud, S.; Hazen, M.M.; Halyabar, O.; et al. On the Alert for Cytokine Storm: Immunopathology in COVID-19. Arthritis Rheumatol. 2020, 72, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thieme, C.J.; Anft, M.; Paniskaki, K.; Stervbo, U.; Roch, T.; Babel, N. Robust T cell response toward spike, membrane, and nucleocapsid SARS-CoV-2 proteins is not associated with recovery in critical COVID-19 patients. Cell Rep. Med. 2020, 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- RECOVERY Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalised Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar]
- Smadja, D.M.; Bonnet, G.; Gendron, N.; Weizman, O.; Khider, L.; Trimaille, A.; Mirault, T.; Fauvel, C.; Diehl, J.L.; Mika, D.; et al. Intermediate- vs. Standard-Dose Prophylactic Anticoagulation in Patients With COVID-19 Admitted in Medical Ward: A Propensity Score-Matched Cohort Study. Front. Med. 2021, 8, 747527. [Google Scholar] [CrossRef]
- Knyazev, E.; Nersisyan, S.; Tonevitsky, A. Endocytosis and transcytosis of SARS-CoV-2 across the intestinal epithelium and other tissue barriers. Front. Immunol. 2021, 12, 636966. [Google Scholar] [CrossRef]
- Nersisyan, S.A. Induction of Hypoxic Response in Caco-2 Cells Promote the Expression of Genes Involved in SARS-CoV-2 Endocytosis and Transcytosis. Dokl. Biochem. Biophys. 2022, 506, 206–209. [Google Scholar] [CrossRef]
- Wen, J.; Cheng, Y.; Ling, R.; Dai, Y.; Huang, B.; Huang, W.; Zhang, S.; Jiang, Y. Antibody-dependent enhancement of coronavirus. Int. J. Infect. Dis. 2020, 100, 483–489. [Google Scholar] [CrossRef]
- Binder, R.J. Functions of heat shock proteins in pathways of the innate and adaptive immune system. J. Immunol. 2014, 193, 5765–5771. [Google Scholar] [CrossRef] [Green Version]
- Kasperkiewicz, M. COVID-19, heat shock proteins, and autoimmune bullous diseases: A potential link deserving further attention. Cell Stress Chaperones 2021, 26, 1–2. [Google Scholar] [CrossRef]
- Vahabi, M.; Ghazanfari, T.; Sepehrnia, S. Molecular mimicry, hyperactive immune system, and SARS-COV-2 are three prerequisites of the autoimmune disease triangle following COVID-19 infection. Int. Immunopharmacol. 2022, 112, 109183. [Google Scholar] [CrossRef] [PubMed]
- Raghav, P.K.; Kalyanaraman, K.; Kumar, D. Human cell receptors: Potential drug targets to combat COVID-19. Amino Acids 2021, 53, 813–842. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.R.; Geng, R.; Li, Q.; Chen, Y.; Li, S.F.; Wang, Q.; Min, J.; Yang, Y.; Li, B.; Jiang, R.D.; et al. ACE2-independent infection of T lymphocytes by SARS-CoV-2. Signal. Transduct. Target Ther. 2022, 7, 83. [Google Scholar] [CrossRef]
- Huang, J.; Liu, X.; Wei, Y.; Li, X.; Gao, S.; Dong, L.; Rao, X.; Zhong, J. Emerging Role of Dipeptidyl Peptidase-4 in Autoimmune Disease. Front. Immunol. 2022, 13, 830863. [Google Scholar] [CrossRef] [PubMed]
- Sebastián-Martín, A.; Sánchez, B.G.; Mora-Rodríguez, J.M.; Bort, A.; Díaz-Laviada, I. Role of Dipeptidyl Peptidase-4 (DPP4) on COVID-19 Physiopathology. Biomedicines 2022, 10, 2026. [Google Scholar] [CrossRef]
- Luan, J.; Zhang, K.; Yang, P.; Zhang, Y.; Feng, F.; Zhu, Y.M.; Zhu, P.; Chen, Z.N. The combination of FK506 and an anti-CD147 mAb exerts potential therapeutic effects on a mouse model of collagen-induced arthritis. Mol. Immunol. 2018, 101, 1–9. [Google Scholar] [CrossRef]
- Cordero, O.J.; Viéitez, I.; Altabás, I.; Nuño-Nuño, L.; Villalba, A.; Novella-Navarro, M.; Peiteado, D.; Miranda-Carús, M.E.; Balsa, A.; Varela-Calviño, R.; et al. Study of Plasma Anti-CD26 Autoantibody Levels in a Cohort of Treatment-Naïve Early Arthritis Patients. Arch. Immunol. Exp. 2022, 70, 12. [Google Scholar] [CrossRef]
- Urata, R.; Ikeda, K.; Yamazaki, E.; Ueno, D.; Katayama, A.; Shin-Ya, M.; Ohgitani, E.; Mazda, O.; Matoba, S. Senescent endothelial cells are predisposed to SARS-CoV-2 infection and subsequent endothelial dysfunction. Sci. Rep. 2022, 12, 11855. [Google Scholar] [CrossRef]
- Ramos-Martínez, I.E.; Ramos-Martínez, E.; Segura-Velázquez, R.Á.; Saavedra-Montañez, M.; Cervantes-Torres, J.B.; Cerbón, M.; Papy-Garcia, D.; Zenteno, E.; Sánchez-Betancourt, J.I. Heparan Sulfate and Sialic Acid in Viral Attachment: Two Sides of the Same Coin? Int. J. Mol. Sci. 2022, 23, 9842. [Google Scholar] [CrossRef]
- López-Muñoz, A.D.; Kosik, I.; Holly, J.; Yewdell, J.W. Cell surface SARS-CoV-2 nucleocapsid protein modulates innate and adaptive immunity. Sci. Adv. 2022, 8, eabp9770. [Google Scholar] [CrossRef]
- Chen, L.; Guan, W.-J.; Qiu, Z.-E.; Xu, J.-B.; Bai, X.; Hou, X.-C.; Sun, J.; Qu, S.; Huang, Z.X.; Lei, T.L.; et al. SARS-CoV-2 nucleocapsid protein triggers hyperinflammation via protein-protein interaction-mediated intracellular Cl− accumulation in respiratory epithelium. Signal. Transduct. Target. Ther. 2022, 7, 255. [Google Scholar] [CrossRef]
- ACTIV-3/TICO Study Group. The Association of Baseline Plasma SARS-CoV-2 Nucleocapsid Antigen Level and Out-comes in Patients Hospitalised with COVID-19. Ann. Intern. Med. 2022, 175, 1401–1410. [Google Scholar]
- Schoeman, D.; Fielding, B.C. Is There a Link Between the Pathogenic Human Coronavirus Envelope Protein and Im-munopathology? A Review of the Literature. Front. Microbiol. 2020, 11, 2086. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Cloete, R.; Fielding, B.C. The Flexible, Extended Coil of the PDZ-Binding Motif of the Three Deadly Hu-man Coronavirus E Proteins Plays a Role in Pathogenicity. Viruses 2022, 14, 1707. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Law, R.; Korosec, C.S.; Zhou, C.; Koh, W.H.; Ghaemi, M.S.; Samaan, P.; Ooi, H.K.; Matveev, V.; Yue, F.; et al. Longitudinal Assessment of SARS-CoV-2-Specific T Cell Cytokine-Producing Responses for 1 Year Reveals Persistence of Multicytokine Proliferative Responses, with Greater Immunity Associated with Disease Severity. J. Virol. 2022, 96, e0050922. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, Z.; Li, S.; Xu, W.; Zhang, Q.; Silva, I.T.; Li, C.; Wu, Y.; Jiang, Q.; Liu, Z.; et al. Enhancement versus neutralisation by SARS-CoV-2 antibodies from convalescent donor associates with distinct epitopes on the RBD. Cell Rep. 2021, 34, 108699. [Google Scholar] [CrossRef]
- Negro, F. Is antibody-dependent enhancement playing a role in COVID-19 pathogenesis? Swiss Med. Wkly. 2020, 150, w20249. [Google Scholar] [CrossRef]
- De Sanctis, J.B.; Garmendia, J.V.; Hajdúch, M. Overview of Memory NK Cells in Viral Infections: Possible Role in SARS-CoV-2 Infection. Immunology 2022, 2, 52–67. [Google Scholar] [CrossRef]
- Chouaki Benmansour, N.; Carvelli, J.; Vivier, É. Complement cascade in severe forms of COVID-19: Recent advances in therapy. Eur. J. Immunol. 2021, 51, 1652–1659. [Google Scholar] [CrossRef]
- Ameratunga, R. Assessing Disease Severity in Common Variable Immunodeficiency Disorders (CVID) and CVID-Like Disorders. Front. Immunol. 2018, 9, 2130. [Google Scholar] [CrossRef] [Green Version]
- Ameratunga, R.; Longhurst, H.; Steele, R.; Lehnert, K.; Leung, E.; Brooks, A.E.S.; Woon, S.T. Common Variable Immunodeficiency Disorders, T-Cell Responses to SARS-CoV-2 Vaccines, and the Risk of Chronic COVID-19. J. Allergy Clin. Immunol. Pr. 2021, 9, 3575–3583. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Barrett, B.S.; Morrison, J.H.; Mickens, K.L.; Vladar, E.K.; Hasenkrug, K.J.; Poeschla, E.M.; Santiago, M.L. Interferon resistance of emerging SARS-CoV-2 variants. Proc. Natl. Acad. Sci. USA 2022, 119, e2203760119. [Google Scholar] [CrossRef] [PubMed]
- Rocco, J.M.; Laghetti, P.; Di Stefano, M.; Sereti, I.; Ortega-Villa, A.; Wang, J.; Rupert, A.; Chironna, M.; Ye, L.; Liu, X.; et al. Impact of Innate Immunity, Endothelial Damage, and Metabolic Biomarkers on COVID-19 Severity and Mortality. Open Forum Infect. Dis. 2020, 9, ofac427. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, M.T.; Di Natale, M.; García-Martínez, E.; Navarro, J.; Muñoz-Blanco, J.L.; Demelo-Rodríguez, P.; Sánchez-Mateos, P. Immunoserologic Detection and Diagnostic Relevance of Cross-Reactive Autoantibodies in Corona-virus Disease 2019 Patients. J. Infect. Dis. 2020, 222, 1439–1443. [Google Scholar] [CrossRef]
- Tandel, D.; Sah, V.; Singh, N.K.; Potharaju, P.S.; Gupta, D.; Shrivastava, S.; Sowpati, D.T.; Harshan, K.H. SARS-CoV-2 Variant Delta Potently Suppresses Innate Immune Response and Evades Interferon-Activated Antiviral Responses in Human Colon Epithelial Cells. Microbiol. Spectr. 2022, 10, e0160422. [Google Scholar] [CrossRef]
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; Wong, P.; et al. Diverse functional autoantibodies in patients with COVID-19. Nature 2021, 595, 283–288. [Google Scholar] [CrossRef]
- Trahtemberg, U.; Rottapel, R.; Dos Santos, C.C.; Slutsky, A.S.; Baker, A.; Fritzler, M.J. Anticardiolipin and other antiphospholipid antibodies in critically ill COVID-19 positive and negative patients. Ann. Rheum. Dis. 2021, 80, 1236–1240. [Google Scholar] [CrossRef]
- Pascolini, S.; Vannini, A.; Deleonardi, G.; Ciordinik, M.; Sensoli, A.; Carletti, I.; Veronesi, L.; Ricci, C.; Pronesti, A.; Mazzanti, L.; et al. COVID-19 and Immunological Dysregulation: Can Autoantibodies be Useful?). Clin. Transl. Sci. 2021, 14, 502–508. [Google Scholar] [CrossRef]
- Lee, L.E.; Jeong, W.; Park, Y.B.; Jeong, S.J.; Lee, S.W. Clinical Significance of Antineutrophil Cytoplasmic Antibody Positivity in Patients Infected with SARS-CoV-2. J. Clin. Med. 2022, 11, 4152. [Google Scholar] [CrossRef]
- Christodoulou, M.; Iatridi, F.; Chalkidis, G.; Lioulios, G.; Nikolaidou, C.; Badis, K.; Fylaktou, A.; Papagianni, A.; Stangou, M. ANCA-Associated Vasculitis May Result as a Complication to Both SARS-CoV-2 Infection and Vaccination. Life 2022, 12, 1072. [Google Scholar] [CrossRef]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockman, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic autoantibodies in serum from patients hospitalised with COVID-19. Sci. Transl. Med. 2020, 12, eabd3876. [Google Scholar] [CrossRef] [PubMed]
- Pascolini, S.; Granito, A.; Muratori, L.; Lenzi, M.; Muratori, P. Coronavirus disease associated immune thrombocytopenia: Causation or correlation? J. Microbiol. Immunol. Infect. 2021, 54, 531–533. [Google Scholar] [CrossRef] [PubMed]
- de Laat, B.; Stragier, H.; de Laat-Kremers, R.; Ninivaggi, M.; Mesotten, D.; Thiessen, S.; Van Pelt, K.; Roest, M.; Penders, J.; Vanelderen, P.; et al. Population-wide persistent hemostatic changes after vaccination with ChAdOx1-S. Front. Cardiovasc. Med. 2022, 9, 966028. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Quan, Y.; Cassady, K.; Zou, Z.; Gao, Y.; Zhang, X. Clinical characteristics in immune thrombocytopenia patients after COVID-19 vaccination. Hum. Vaccines Immunother. 2022, 18, 2119043. [Google Scholar] [CrossRef] [PubMed]
- See, I.; Su, J.R.; Lale, A.; Woo, E.J.; Guh, A.Y.; Shimabukuro, T.T.; Streiff, M.B.; Rao, A.K.; Wheeler, A.P.; Beavers, S.F.; et al. US Case Reports of Cerebral Venous Sinus Thrombosis With Thrombocytopenia After Ad26.COV2.S Vaccination, March 2 to April 21, 2021. JAMA 2021, 325, 2448–2456. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, Y.; Rojas, M.; Beltrán, S.; Polo, F.; Camacho-Domínguez, L.; Morales, S.D.; Gershwin, M.E.; Anaya, J.M. Autoimmune and autoinflammatory conditions after COVID-19 vaccination. New case reports and updated literature review. J. Autoimmun. 2022, 132, 102898. [Google Scholar] [CrossRef]
- Seeßle, J.; Waterboer, T.; Hippchen, T.; Simon, J.; Kirchner, M.; Lim, A.; Müller, B.; Merle, U. Persistent Symptoms in Adult Patients 1 Year After Coronavirus Disease 2019 (COVID-19): A Prospective Cohort Study. Clin. Infect. Dis. 2022, 74, 1191–1198. [Google Scholar] [CrossRef]
- Bastard, P.; Gervais, A.; Le Voyer, T.; Rosain, J.; Philippot, Q.; Manry, J. Autoantibodies neutralising type I IFNs are present in ~ 4% of uninfected individuals over 70 years old and account for ~ 20% of COVID-19 deaths. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Bastard, P.; Orlova, E.; Sozaeva, L.; Lévy, R.; James, A.; Schmitt, M.M.; Ochoa, S.; Kareva, M.; Rodina, Y.; Gervais, A.; et al. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J. Exp. Med. 2021, 218, e20210554. [Google Scholar] [CrossRef]
- Bastard, P.; Vazquez, S.; Liu, J.; Laurie, M.T.; Wang, C.Y.; Gervais, A.; Le Voyer, T.; Bizien, L.; Zamecnik, C.; Philippot, Q.; et al. Vaccine breakthrough hypoxemic COVID-19 pneumonia in patients with auto-Abs neutralising type I IFNs. Sci. Immunol. 2022, 14, eabp8966. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Mommert, M.; Mouton, W.; Pizzorno, A.; Brengel-Pesce, K.; Mezidi, M.; Villard, M.; Lina, B.; Richard, J.C.; Fassier, J.B.; et al. Early nasal type I IFN immunity against SARS-CoV-2 is compromised in patients with autoantibodies against type I IFNs. J. Exp. Med. 2021, 218, e20211211. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Yang, C.D.; Cheng, X.B. Anti-Interferon Autoantibodies in Adult-Onset Immunodeficiency Syndrome and Severe COVID-19 Infection. Front. Immunol. 2021, 12, 788368. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Bolze, A.; Jouanguy, E.; Zhang, S.Y.; COVID Human Genetic Effort; Cobat, A.; Notarangelo, L.D.; Su, H.C.; Casanova, J.-L.; et al. Life-Threatening COVID-19: Defective Interferons Unleash Excessive Inflammation. Medicine 2020, 1, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Matuozzo, D.; Le Pen, J.; Lee, D.; Moens, L.; Asano, T.; Bohlen, J.; Liu, Z.; Moncada-Velez, M.; Kendir-Demirkol, Y.; et al. Recessive inborn errors of type I IFN immunity in children with COVID-19 pneumonia. J. Exp. Med. 2022, 219, e20220131. [Google Scholar] [CrossRef] [PubMed]
- Steels, S.; Van Elslande, J.; Leuven COVID-Study Group; De Munter, P.; Bossuyt, X. Transient Increase of Pre-existing Anti-IFN-α2 Antibodies Induced by SARS-CoV-2 Infection. J. Clin. Immunol. 2022, 42, 742–745. [Google Scholar] [CrossRef] [PubMed]
- Kreye, J.; Reincke, S.M.; Prüss, H. Do cross-reactive antibodies cause neuropathology in COVID-19? Nat. Rev. Immunol. 2020, 20, 645–646. [Google Scholar] [CrossRef]
- Franke, C.; Ferse, C.; Kreye, J.; Reincke, S.M.; Sanchez-Sendin, E.; Rocco, A.; Steinbrenner, M.; Angermair, S.; Treskatsch, S.; Zickler, D.; et al. High frequency of cerebrospinal fluid autoantibodies in COVID-19 patients with neurological symptoms. Brain. Behav. Immun. 2021, 93, 415–419. [Google Scholar] [CrossRef]
- Wang, W.; Shen, M.; Tao, Y.; Fairley, C.K.; Zhong, Q.; Li, Z.; Chen, H.; Ong, J.J.; Zhang, D.; Zhang, K.; et al. Elevated glucose level leads to rapid COVID-19 progression and high fatality. BMC Pulm. Med. 2021, 21, 64. [Google Scholar] [CrossRef]
- Marchand, L.; Pecquet, M.; Luyton, C. Type 1 diabetes onset triggered by COVID-19. Acta Diabetol. 2020, 57, 1265–1266. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.B.; Javed, N.; Sheikh, A.A.E.; Upadhyay, S.; Shekhar, R. Diabetes insipidus and concomitant myocarditis: A late sequelae of COVID-19 infection. J. Investig. Med. High Impact Case Rep. 2021, 9, 2324709621999954. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.B.; Javaid, M.A.; Sheikh, A.A.E.; Shekhar, R. Central adrenal insufficiency and diabetes insipidus as potential endocrine manifestations of COVID-19 infection: A case report. Pan. Afr. Med. J. 2021, 38, 222. [Google Scholar] [PubMed]
- Wheatland, R. Molecular mimicry of ACTH in SARS—Implications for corticosteroid treatment and prophylaxis. Med. Hypotheses 2004, 63, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Mirza, S.A.; Sheikh, A.A.E.; Barbera, M.; Ijaz, Z.; Javaid, M.A.; Shekhar, R.; Pal, S.; Sheikh, A.B. COVID-19 and the Endocrine System: A Review of the Current Information and Misinformation. Infect. Dis. Rep. 2022, 14, 184–197. [Google Scholar] [CrossRef]
- Pérez-Torres, D.; Díaz-Rodríguez, C.; Armentia-Medina, A. Anti-ACTH antibodies in critically ill COVID-19 patients: A potential immune evasion mechanism of SARS-CoV-2. Med. Intensiv. 2022, 46, 472–474. [Google Scholar] [CrossRef]
- Jensterle, M.; Herman, R.; Janež, A.; Mahmeed, W.A.; Al-Rasadi, K.; Al-Alawi, K.; Banach, M.; Banerjee, Y.; Ceriello, A.; Cesur, M.; et al. The Relationship between COVID-19 and Hypothalamic-Pituitary-Adrenal Axis: A Large Spectrum from Glucocorticoid Insufficiency to Excess-The CAPISCO International Expert Panel. Int. J. Mol. Sci. 2022, 23, 7326. [Google Scholar] [CrossRef]
- Vakhshoori, M.; Heidarpour, M.; Bondariyan, N.; Sadeghpour, N.; Mousavi, Z. Adrenal Insufficiency in Coronavirus Disease 2019 (COVID-19)-Infected Patients without Preexisting Adrenal Diseases: A Systematic Literature Review. Int. J. Endocrinol. 2021, 2021, 2271514. [Google Scholar] [CrossRef]
- Davies, T.F. Infection and autoimmune thyroid disease. J. Clin. Endocrinol. Metab. 2008, 93, 674–676. [Google Scholar] [CrossRef] [Green Version]
- Mateu-Salat, M.; Urgell, E.; Chico, A. SARS-COV-2 as a trigger for autoimmune disease: Report of two cases of Graves’ disease after COVID-19. J. Endocrinol. Investig. 2020, 43, 1527–1528. [Google Scholar] [CrossRef]
- Tee, L.Y.; Harjanto, S.; Rosario, B.H. COVID-19 complicated by Hashimoto’s thyroiditis. Singap. Med. J. 2021, 62, 265. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, M.C.; Ramonell, R.P.; Haddad, N.S.; Anam, F.A.; Rudolph, M.E.; Walker, T.A.; Truong, A.D.; Dixit, A.N.; Han, J.E.; Cabrera-Mora, M.; et al. Dysregulated naïve B cells and de novo autoreactivity in severe COVID-19. Nature 2022, 611, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Castleman, M.J.; Stumpf, M.M.; Therrien, N.R.; Smith, M.J.; Lesteberg, K.E.; Palmer, B.E.; Maloney, J.P.; Janssen, W.J.; Mould, K.J.; Beckham, J.D.; et al. Autoantibodies elicited with SARS-CoV-2 infection are linked to alterations in double negative B cells. Front. Immunol. 2022, 13, 988125. [Google Scholar] [CrossRef] [PubMed]
- Bomhof, G.; Mutsaers, P.G.N.J.; Leebeek, F.W.G.; Te Boekhorst, P.A.W.; Hofland, J.; Croles, F.N.; Jansen, A.J.G. COVID-19-associated immune thrombocytopenia. Br. J. Haematol. 2020, 190, e61–e64. [Google Scholar] [CrossRef]
- Bonometti, R.; Sacchi, M.C.; Stobbione, P.; Lauritano, E.C.; Tamiazzo, S.; Marchegiani, A.; Novara, E.; Molinaro, E.; Benedetti, I.; Massone, L.; et al. The first case of systemic lupus erythematosus (SLE) triggered by COVID-19 infection. Eur. Rev. Med. Pharm. Sci. 2020, 24, 9695–9697. [Google Scholar]
- Satheesh, N.J.; Salloum-Asfar, S.; Abdulla, S.A. The Potential Role of COVID-19 in the Pathogenesis of Multiple Sclerosis-A Preliminary Report. Viruses 2021, 13, 2091. [Google Scholar] [CrossRef]
- Palao, M.; Fernández-Díaz, E.; Gracia-Gil, J.; Romero-Sánchez, C.M.; Díaz-Maroto, I.; Segura, T. Multiple sclerosis fol-lowing SARS-CoV-2 infection. Mult. Scler. Relat. Disord. 2020, 45, 102377. [Google Scholar] [CrossRef]
- de Ruijter, N.S.; Kramer, G.; Gons, R.A.R.; Hengstman, G.J.D. Neuromyelitis optica spectrum disorder after presumed coronavirus (COVID-19) infection: A case report. Mult. Scler. Relat. Disord. 2020, 46, 102474. [Google Scholar] [CrossRef]
- Toscano, G.; Palmerini, F.; Ravaglia, S.; Ruiz, L.; Invernizzi, P.; Cuzzoni, M.G.; Franciotta, D.; Baldanti, F.; Daturi, R.; Postorino, P.; et al. Guillain-Barré Syndrome Associated with SARS-CoV-2. N. Engl. J. Med. 2020, 382, 2574–2576. [Google Scholar] [CrossRef]
- Sriwastava, S.; Tandon, M.; Kataria, S.; Daimee, M.; Sultan, S. New onset of ocular myasthenia gravis in a patient with COVID-19: A novel case report and literature review. J. Neurol. 2021, 268, 2690–2696. [Google Scholar] [CrossRef]
- Root-Bernstein, R. COVID-19 coagulopathies: Human blood proteins mimic SARS-CoV-2 virus, vaccine proteins and bacterial coinfections inducing autoimmunity: Combinations of bacteria and SARS-CoV-2 synergise to induce autoantibodies targeting cardiolipin, cardiolipin-binding proteins, platelet factor 4, prothrombin, and coagulation factors. Bioessays 2021, 43, e2100158. [Google Scholar] [PubMed]
- Schwarz, M.; Mzoughi, S.; Lozano-Ojalvo, D.; Tan, A.T.; Bertoletti, A.; Guccione, E. T cell immunity is key to the pan-demic endgame: How to measure and monitor it. Curr. Res. Immunol. 2022, 3, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.P.; Chiñas, M.; Julé, A.M.; Taylor, M.; Ohashi, M.; Benamar, M.; Crestani, E.; Son, M.B.F.; Chou, J.; Gebhart, C.; et al. SARS-CoV-2-specific T cell responses in patients with multisystem inflammatory syndrome in children. Clin. Immunol. 2022, 243, 109106. [Google Scholar] [CrossRef] [PubMed]
- Tappe, B.; Lauruschkat, C.D.; Strobel, L.; Pantaleón García, J.; Kurzai, O.; Rebhan, S.; Kraus, S.; Pfeuffer-Jovic, E.; Bussemer, L.; Possler, L.; et al. COVID-19 patients share common, corticosteroid-independent features of impaired host immunity to pathogenic molds. Front. Immunol. 2022, 13, 954985. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.E.; Bartlett, A.W.; Tangye, S.G. Severe COVID-19 represents an undiagnosed primary immunodeficiency in a high proportion of infected individuals. Clin. Transl. Immunol. 2022, 11, e1365. [Google Scholar] [CrossRef]
- Tarhini, H.; Recoing, A.; Bridier-Nahmias, A.; Rahi, M.; Lambert, C.; Martres, P.; Lucet, J.C.; Rioux, C.; Bouzid, D.; Lebourgeois, S.; et al. Long-Term Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infectiousness Among Three Immunocompromised Patients: From Prolonged Viral Shedding to SARS-CoV-2 Superinfection. J. Infect. Dis. 2021, 223, 1522–1527. [Google Scholar] [CrossRef]
- Nakajima, Y.; Ogai, A.; Furukawa, K.; Arai, R.; Anan, R.; Nakano, Y.; Kurihara, Y.; Shimizu, H.; Misaki, T.; Okabe, N.; et al. Prolonged viral shedding of SARS-CoV-2 in an immunocompromised patient. J. Infect. Chemother. 2021, 27, 387–389. [Google Scholar] [CrossRef]
- Delavari, S.; Abolhassani, H.; Abolnezhadian, F.; Babaha, F.; Iranparast, S.; Ahanchian, H.; Moazzen, N.; Nabavi, M.; Arshi, S.; Fallahpour, M.; et al. Impact of SARS-CoV-2 Pandemic on Patients with Primary Immunodeficiency. J. Clin. Immunol. 2021, 41, 345–355. [Google Scholar] [CrossRef]
- Babaha, F.; Rezaei, N. Primary Immunodeficiency Diseases in COVID-19 Pandemic: A Predisposing or Protective Factor? Am. J. Med. Sci. 2020, 360, 740–741. [Google Scholar] [CrossRef]
- Jacobsen, E.M.; Fabricius, D.; Class, M.; Topfstedt, F.; Lorenzetti, R.; Janowska, I.; Schmidt, F.; Staniek, J.; Zernickel, M.; Stamminger, T.; et al. High antibody levels and reduced cellular response in children up to one year after SARS-CoV-2 infection. Nat. Commun. 2022, 13, 7315. [Google Scholar] [CrossRef]
- Gathmann, B.; Mahlaoui, N.; Ceredih, G.L.; Oksenhendler, E.; Warnatz, K.; Schulze, I.; Kindle, G.; Kuijpers, T.W.; Dutch, W.I.D.; van Beem, R.T.; et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 2014, 134, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinti, I.; Lougaris, V.; Milito, C.; Cinetto, F.; Pecoraro, A.; Mezzaroma, I.; Mastroianni, C.M.; Turriziani, O.; Bondioni, M.P.; Filippini, M.; et al. A possible role for B cells in COVID-19? Lesson from patients with agammaglobulinemia. J. Allergy Clin. Immunol. 2020, 146, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Pulvirenti, F.; Mortari, E.P.; Putotto, C.; Terreri, S.; Fernandez Salinas, A.; Cinicola, B.L.; Cimini, E.; Di Napoli, G.; Sculco, E.; Milito, C.; et al. COVID-19 Severity, Cardiological Outcome, and Immunogenicity of mRNA Vaccine on Adult Patients With 22q11.2 DS. J. Allergy Clin. Immunol. Pract 2022, S2213-2198(22)01052-2. [Google Scholar] [CrossRef] [PubMed]
- Melo, K.M.; Alves, L.M.; Valente, C.F.C.; Tavares, F.S. One-year intravenous immunoglobulin replacement therapy: Efficacy in reducing hospital admissions in pediatric patients with Inborn Errors of Immunity. J. Pediatr. 2022, 98, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Funk, T.; Innocenti, F.; Gomes Dias, J.; Nerlander, L.; Melillo, T.; Gauci, C.; Melillo, J.M.; Lenz, P.; Sebestova, H.; Slezak, P.; et al. Age-specific associations between underlying health conditions and hospitalisation, death and in-hospital death among confirmed COVID-19 cases: A multi-country study based on surveillance data, June to December 2020. Euro Surveill. 2022, 27, 2100883. [Google Scholar] [CrossRef]
- Hensley, M.K.; Bain, W.G.; Jacobs, J.; Nambulli, S.; Parikh, U.; Cillo, A.; Staines, B.; Heaps, A.; Sobolewski, M.D.; Rennick, L.J.; et al. Intractable Coronavirus Disease 2019 (COVID-19) and Prolonged Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Replication in a Chimeric Antigen Receptor-Modified T-Cell Therapy Recipient: A Case Study. Clin. Infect. Dis. 2021, 73, e815–e821. [Google Scholar] [CrossRef]
- van der Made, C.I.; Netea, M.G.; van der Veerdonk, F.L.; Hoischen, A. Clinical implications of host genetic variation and susceptibility to severe or critical COVID-19. Genome Med. 2022, 14, 96. [Google Scholar] [CrossRef]
- Elhabyan, A.; Elyaacoub, S.; Sanad, E.; Abukhadra, A.; Elhabyan, A.; Dinu, V. The role of host genetics in susceptibility to severe viral infections in humans and insights into host genetics of severe COVID-19: A systematic review. Virus Res. 2020, 289, 198163. [Google Scholar] [CrossRef]
- Asano, T.; Boisson, B.; Onodi, F.; Matuozzo, D.; Moncada-Velez, M.; Maglorius Renkilaraj, M.R.L.; Zhang, P.; Meertens, L.; Bolze, A.; Materna, M.; et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 2021, 6, eabl4348. [Google Scholar] [CrossRef]
- Knight, J.S.; Caricchio, R.; Casanova, J.L.; Combes, A.J.; Diamond, B.; Fox, S.E.; Hanauer, D.A.; James, J.A.; Kanthi, Y.; Ladd, V.; et al. The intersection of COVID-19 and autoimmunity. J. Clin. Investig. 2021, 131, e154886. [Google Scholar] [CrossRef]
- Milota, T.; Sobotkova, M.; Smetanova, J.; Bloomfield, M.; Vydlakova, J.; Chovancova, Z.; Litzman, J.; Hakl, R.; Novak, J.; Malkusova, I.; et al. Risk Factors for Severe COVID-19 and Hospital Admission in Patients With Inborn Errors of Immunity—Results From a Multicenter Nationwide Study. Front. Immunol. 2022, 13, 835770. [Google Scholar] [CrossRef] [PubMed]
- Shields, A.M.; Tadros, S.; Al-Hakim, A.; Neil, J.M.; Lin, M.M.N.; Chan, M.; Goddard, S.; Dempster, J.; Dziadzio, M.; Patel, S.Y.; et al. Impact of vaccination on hospitalisation and mortality from COVID-19 in patients with primary and secondary immunodeficiency: The United Kingdom experience. Front. Immunol. 2022, 13, 984376. [Google Scholar] [CrossRef] [PubMed]
- OPons, S.; Uhel, F.; Frapy, E.; Sérémé, Y.; Zafrani, L.; Aschard, H.; Skurnik, D. How Protective are Antibodies to SARS-CoV-2, the Main Weapon of the B-Cell Response? Stem Cell Rev. Rep. 2022, 1–16. [Google Scholar] [CrossRef]
- Lliaro, P.; Torreele, E.; Vaillant, M. COVID-19 vaccine efficacy and effectiveness-the elephant (not) in the room. Lancet Microbe 2021, 2, e279–e280. [Google Scholar]
- de Lemos Rieper, C.; Galle, P.; Hansen, M.B. Characterization and potential clinical applications of autoantibodies against cytokines. Cytokine Growth Factor Rev. 2009, 20, 61–75. [Google Scholar] [CrossRef]
- Knight, V. Immunodeficiency and Autoantibodies to Cytokines. J. Appl. Lab. Med. 2022, 7, 151–164. [Google Scholar] [CrossRef]
- Puel, A.; Bastard, P.; Bustamante, J.; Casanova, J.L. Human autoantibodies underlying infectious diseases. J. Exp. Med. 2022, 219, e20211387. [Google Scholar] [CrossRef]
- Chen, Z.M.; Yang, X.Y.; Li, Z.T.; Guan, W.J.; Qiu, Y.; Li, S.Q.; Zhan, Y.Q.; Lei, Z.Y.; Liu, J.; Zhang, J.Q.; et al. Anti-Interferon-γ Autoantibodies Impair T-Lymphocyte Responses in Patients with Talaromyces marneffei Infections. Infect. Drug Resist. 2022, 15, 3381–3393. [Google Scholar] [CrossRef]
- Van der Wijst, M.G.P.; Vazquez, S.E.; Hartoularos, G.C.; Bastard, P.; Grant, T.; Bueno, R.; Lee, D.S.; Greenland, J.R.; Sun, Y.; Perez, R.; et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci. Transl. Med. 2021, 13, eabh2624. [Google Scholar] [CrossRef]
- Casanova, J.L.; Abel, L. From rare disorders of immunity to common determinants of infection: Following the mechanistic thread. Cell 2022, 185, 3086–3103. [Google Scholar] [CrossRef]
- Kastritis, E.; Kitas, G.D.; Vassilopoulos, D.; Giannopoulos, G.; Dimopoulos, M.A.; Sfikakis, P.P. Systemic autoimmune diseases, anti-rheumatic therapies, COVID-19 infection risk and patient outcomes. Rheumatol. Int. 2020, 40, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.Y.; Tiew, P.Y.; Koh, M.S. Managing adult asthma during the COVID-19 pandemic: A 2022 review and current recommendations. Ann. Acad. Med. Singap. 2022, 51, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Ventura-López, C.; Cervantes-Luevano, K.; Aguirre-Sánchez, J.S.; Flores-Caballero, J.C.; Alvarez-Delgado, C.; Bernaldez-Sarabia, J.; Sánchez-Campos, N.; Lugo-Sánchez, L.A.; Rodríguez-Vázquez, I.C.; Sander-Padilla, J.G.; et al. Treatment with metformin glycinate reduces SARS-CoV-2 viral load: An in vitro model and randomised, double-blind, Phase IIb clinical trial. Biomed. Pharmacother. 2022, 152, 113223. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Obata, Y.; Maruo, Y.; Yamaguchi, H.; Kosugi, M.; Irie, Y.; Hazama, Y.; Yasuda, T. Metformin-associated Lactic Acidosis with Hypoglycemia during the COVID-19 Pandemic. Intern. Med. 2022, 61, 2333–2337. [Google Scholar] [CrossRef]
- Liu, J.; Dong, J.; Yu, Y.; Yang, X.; Shu, J.; Bao, H. Corticosteroids showed more efficacy in treating hospitalised patients with COVID-19 than standard care but the effect is minimal: A systematic review and meta-analysis. Front. Public Health. 2022, 10, 847695. [Google Scholar] [CrossRef]
- Takeshita, Y.; Terada, J.; Hirasawa, Y.; Kinoshita, T.; Tajima, H.; Koshikawa, K.; Kinouchi, T.; Isaka, Y.; Shionoya, Y.; Fujikawa, A.; et al. Development of a novel score model to predict hyperinflammation in COVID-19 as a forecast of optimal steroid administration timing. Front. Med. 2022, 9, 935255. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Wide Spectrum Antibodies | Specific Antibodies |
|---|---|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garmendia, J.V.; García, A.H.; De Sanctis, C.V.; Hajdúch, M.; De Sanctis, J.B. Autoimmunity and Immunodeficiency in Severe SARS-CoV-2 Infection and Prolonged COVID-19. Curr. Issues Mol. Biol. 2023, 45, 33-50. https://doi.org/10.3390/cimb45010003
Garmendia JV, García AH, De Sanctis CV, Hajdúch M, De Sanctis JB. Autoimmunity and Immunodeficiency in Severe SARS-CoV-2 Infection and Prolonged COVID-19. Current Issues in Molecular Biology. 2023; 45(1):33-50. https://doi.org/10.3390/cimb45010003
Chicago/Turabian StyleGarmendia, Jenny Valentina, Alexis Hipólito García, Claudia Valentina De Sanctis, Marián Hajdúch, and Juan Bautista De Sanctis. 2023. "Autoimmunity and Immunodeficiency in Severe SARS-CoV-2 Infection and Prolonged COVID-19" Current Issues in Molecular Biology 45, no. 1: 33-50. https://doi.org/10.3390/cimb45010003
APA StyleGarmendia, J. V., García, A. H., De Sanctis, C. V., Hajdúch, M., & De Sanctis, J. B. (2023). Autoimmunity and Immunodeficiency in Severe SARS-CoV-2 Infection and Prolonged COVID-19. Current Issues in Molecular Biology, 45(1), 33-50. https://doi.org/10.3390/cimb45010003