
Autonomic Nervous System Regulation of Epicardial Adipose Tissue: Potential Roles for Regulator of G Protein Signaling-4



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Sympathetic Nervous System (SNS) of the Heart
3. Parasympathetic Nervous System (PNS) of the Heart
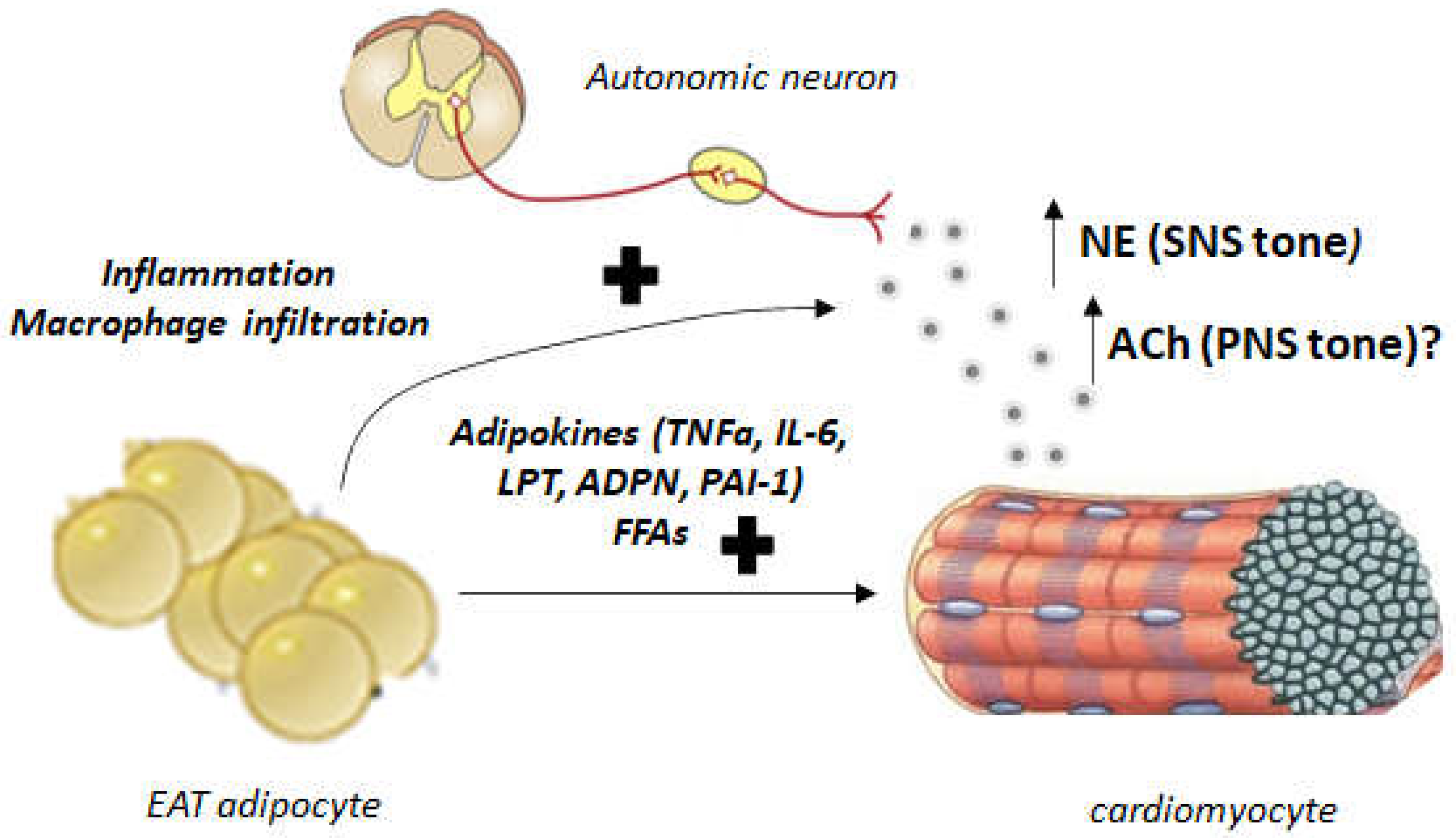
4. Autonomic Dysregulation and EAT: Implications for Human AFib and Heart Failure
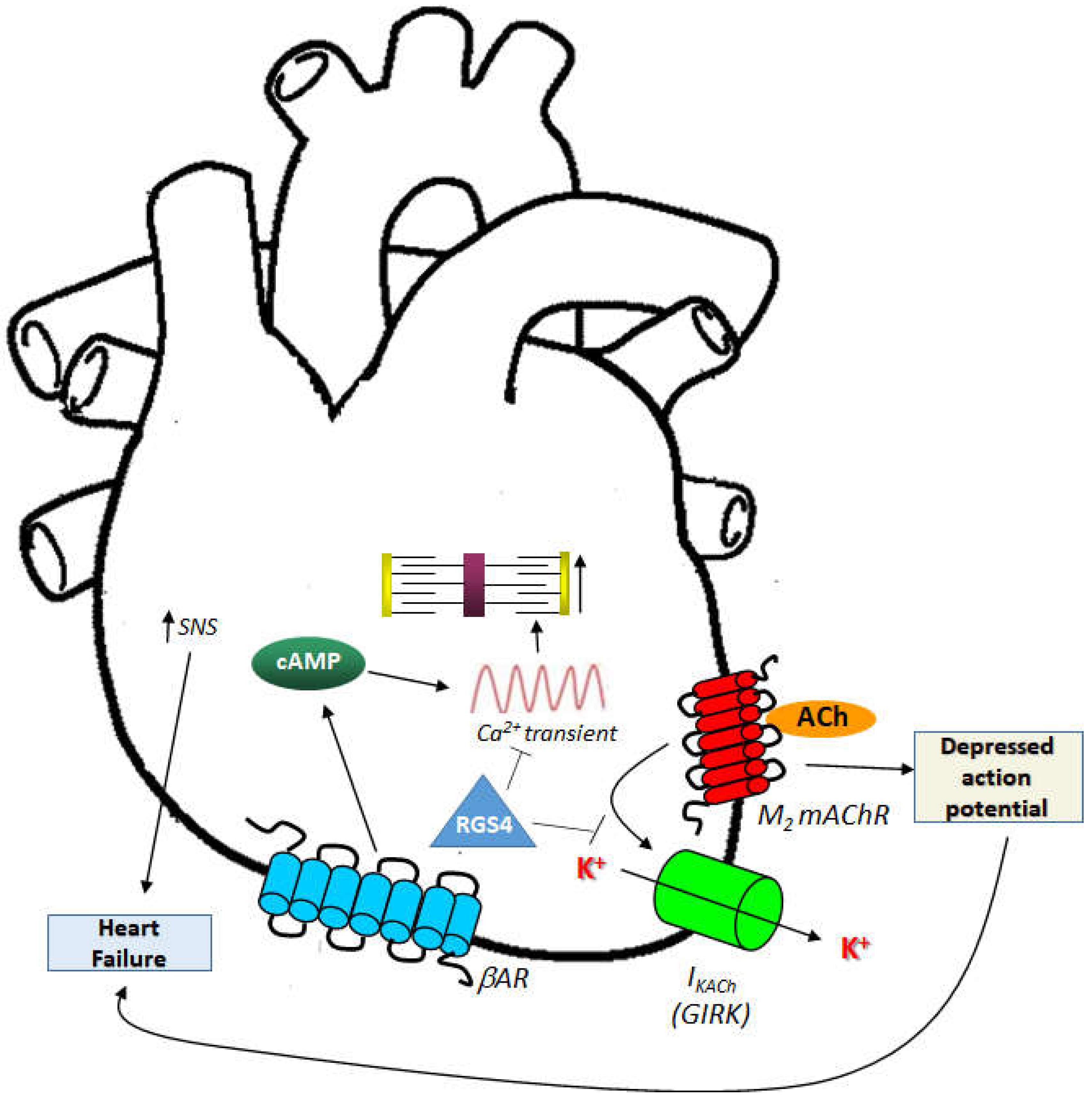
5. RGS4 and EAT Regulation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wong, C.X.; Ganesan, A.N.; Selvanayagam, J.B. Epicardial fat and atrial fibrillation: Current evidence, potential mechanisms, clinical implications, and future directions. Eur. Heart J. 2017, 38, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Ernault, A.C.; Meijborg, V.M.F.; Coronel, R. Modulation of Cardiac Arrhythmogenesis by Epicardial Adipose Tissue: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1730–1745. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Cora, N.; Maning, J.; Brill, A.R.; Sizova, A. Signaling and function of cardiac autonomic nervous system receptors: Insights from the GPCR signalling universe. FEBS J. 2021, 288, 2645–2659. [Google Scholar] [CrossRef] [PubMed]
- Brodde, O.E.; Michel, M.C. Adrenergic and muscarinic receptors in the human heart. Pharmacol. Rev. 1999, 51, 651–690. [Google Scholar] [PubMed]
- Lymperopoulos, A. Arrestins in the Cardiovascular System: An Update. Prog. Mol. Biol. Transl. Sci. 2018, 159, 27–57. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef]
- Xu, X.; Kaindl, J.; Clark, M.J.; Hübner, H.; Hirata, K.; Sunahara, R.K.; Gmeiner, P.; Kobilka, B.K.; Liu, X. Binding pathway determines norepinephrine selectivity for the human β1AR over β2AR. Cell Res. 2021, 31, 569–579. [Google Scholar] [CrossRef]
- Capote, L.A.; Mendez Perez, R.; Lymperopoulos, A. GPCR signaling and cardiac function. Eur. J. Pharmacol. 2015, 763, 143–148. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Garcia, D.; Walklett, K. Pharmacogenetics of cardiac inotropy. Pharmacogenomics 2014, 15, 1807–1821. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Bathgate, A. Pharmacogenomics of the heptahelical receptor regulators G-protein-coupled receptor kinases and arrestins: The known and the unknown. Pharmacogenomics 2012, 13, 323–341. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenal adrenoceptors in heart failure: Fine-tuning cardiac stimulation. Trends Mol. Med. 2007, 13, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A. Ischemic emergency? endothelial cells have their own “adrenaline shot” at hand. Hypertension 2012, 60, 12–14. [Google Scholar] [CrossRef]
- Bylund, D.B.; Eikenberg, D.C.; Hieble, J.P.; Langer, S.Z.; Lefkowitz, R.J.; Minneman, K.P.; Molinoff, P.B.; Ruffolo, R.R., Jr.; Trendelenburg, U. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol. Rev. 1994, 46, 121–136. [Google Scholar] [PubMed]
- Brodde, O.E. Beta-adrenoceptors in cardiac disease. Pharmacol. Ther. 1993, 60, 405–430. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Port, J.D.; Asano, K.; Chidiak, P.; Bouvier, M.; Dutcher, D.; Roden, R.L.; Minobe, W.; Tremmel, K.D.; Bristow, M.R. Cardiac adrenergic receptor effects of carvedilol. Eur. Heart J. 1996, 17 (Suppl. B), 8–16. [Google Scholar] [CrossRef]
- Woodcock, E.A.; Du, X.J.; Reichelt, M.E.; Graham, R.M. Cardiac alpha 1-adrenergic drive in pathological remodelling. Cardiovasc. Res. 2008, 77, 452–462. [Google Scholar] [CrossRef]
- Shannon, R.; Chaudhry, M. Effect of alpha1-adrenergic receptors in cardiac pathophysiology. Am. Heart J. 2006, 152, 842–850. [Google Scholar] [CrossRef]
- Philipp, M.; Hein, L. Adrenergic receptor knockout mice: Distinct functions of 9 receptor subtypes. Pharmacol. Ther. 2004, 101, 65–74. [Google Scholar] [CrossRef]
- Philipp, M.; Brede, M.; Hein, L. Physiological significance of alpha(2)-adrenergic receptor subtype diversity: One receptor is not enough. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R287–R295. [Google Scholar] [CrossRef]
- Hein, L.; Altman, J.D.; Kobilka, B.K. Two functionally distinct alpha2-adrenergic receptors regulate sympathetic neurotransmission. Nature 1999, 402, 181–184. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Funakoshi, H.; Eckhart, A.D.; Koch, W.J. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 2007, 13, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Gao, E.; Ebert, S.N.; Dorn, I.I.G.W.; Koch, W.J. Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J. Biol. Chem. 2010, 285, 16378–16386. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Soltys, S.; Koch, W.J. Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of G protein-coupled receptor kinase-2 activity. Mol. Ther. 2008, 16, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef]
- Skeberdis, V.A.; Gendviliene, V.; Zablockaite, D.; Treinys, R.; Macianskiene, R.; Bogdelis, A.; Jurevicius, J.; Fischmeister, R. Beta3-adrenergic receptor activation increases human atrial tissue contractility and stimulates the L-type Ca2+ current. J. Clin. Investig. 2008, 118, 3219–3227. [Google Scholar] [CrossRef]
- Gauthier, C.; Leblais, V.; Kobzik, L.; Trochu, J.N.; Khandoudi, N.; Bril, A.; Balligand, J.-L.; Le Marec, H. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J. Clin. Investig. 1998, 102, 1377–1384. [Google Scholar] [CrossRef]
- Roy, A.; Guatimosim, S.; Prado, V.F. Cholinergic activity as a new target in diseases of the heart. Mol. Med. 2015, 20, 527–537. [Google Scholar] [CrossRef]
- Hartzell, H.C. Regulation of cardiac ion channels by catecholamines, acetylcholine and second messenger systems. Prog. Biophys. Mol. Biol. 1988, 52, 165–247. [Google Scholar] [CrossRef]
- Li, D.L.; Liu, B.H.; Sun, L.; Zhao, M.; He, X.; Yu, X.J.; Zang, W.J. Alterations of muscarinic acetylcholine receptors-2, 4 and alpha7-nicotinic acetylcholine receptor expression after ischaemia/reperfusion in the rat isolated heart. Clin. Exp. Pharmacol. Physiol. 2010, 37, 1114–1119. [Google Scholar] [CrossRef]
- Anderson, A.; Kulkarni, K.; Marron Fernandez de Velasco, E.; Carlblom, N.; Xia, Z.; Nakano, A.; Martemyanov, K.A.; Tolkacheva, E.G.; Wickman, K. Expression and relevance of the G protein-gated K+ channel in the mouse ventricle. Sci. Rep. 2018, 8, 1192. [Google Scholar] [CrossRef]
- Giles, W.; Noble, S.J. Changes in membrane currents in bullfrog atrium produced by acetylcholine. J. Physiol. 1976, 261, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Dhein, S.; van Koppen, C.J.; Brodde, O.E. Muscarinic receptors in the mammalian heart. Pharmacol. Res. 2001, 44, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Brodde, O.E.; Bruck, H.; Leineweber, K.; Seyfarth, T. Presence, distribution and physiological function of adrenergic and muscarinic receptor subtypes in the human heart. Basic Res. Cardiol. 2001, 96, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; Norby, F.L.; Whitsel, E.A.; Soliman, E.Z.; Chen, L.Y.; Loehr, L.R.; Fuster, V.; Heiss, G.; Coresh, J.; Alonso, A. Autonomic dysfunction and incidence of atrial fibrillation: Results from 20 years follow-up. J. Am. Coll. Cardiol. 2017, 69, 291–299. [Google Scholar] [CrossRef]
- Jungen, C.; Scherschel, K.; Eickholt, C.; Kuklik, P.; Klatt, N.; Bork, N.; Salzbrunn, T.; Alken, F.; Angendohr, S.; Klene, C.; et al. Disruption of cardiac cholinergic neurons enhances susceptibility to ventricular arrhythmias. Nat. Commun. 2017, 8, 14155. [Google Scholar] [CrossRef]
- González, S.A.; Forcada, P.; de Cavanagh, E.M.; Inserra, F.; Svane, J.C.; Obregón, S.; Castellaro, C.; Olano, D.; Hita, A.; Kotliar, C.V. Sodium intake is associated with parasympathetic tone and metabolic parameters in mild hypertension. Am. J. Hypertens. 2012, 25, 620–624. [Google Scholar] [CrossRef]
- McAreavey, D.; Neilson, J.M.; Ewing, D.J.; Russell, D.C. Cardiac parasympathetic activity during the early hours of acute myocardial infarction. Br. Heart J. 1989, 62, 165–170. [Google Scholar] [CrossRef]
- Lombardi, F.; Sandrone, G.; Spinnler, M.T.; Torzillo, D.; Lavezzaro, G.C.; Brusca, A.; Malliani, A. Heart rate variability in the early hours of an acute myocardial infarction. Am. J. Cardiol. 1996, 77, 1037–1044. [Google Scholar] [CrossRef]
- Grassi, G.; Seravalle, G.; Bertinieri, G.; Turri, C.; Stella, M.L.; Scopelliti, F.; Mancia, G. Sympathetic and reflex abnormalities in heart failure secondary to ischaemic or idiopathic dilated cardiomyopathy. Clin. Sci. 2001, 101, 141–146. [Google Scholar] [CrossRef]
- Saw, E.L.; Kakinuma, Y.; Fronius, M.; Katare, R. The non-neuronal cholinergic system in the heart: A comprehensive review. J. Mol. Cell. Cardiol. 2018, 125, 129–139. [Google Scholar] [CrossRef]
- Lara, A.; Damasceno, D.D.; Pires, R.; Gros, R.; Gomes, E.R.; Gavioli, M.; Lima, R.; Guimarães, D.; Lima, P.; Bueno, C.R.; et al. Dysautonomia due to reduced cholinergic neurotransmission causes cardiac remodeling and heart failure. Mol. Cell. Biol. 2010, 30, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Gerber, Y.; Weston, S.A.; Enriquez-Sarano, M.; Manemann, S.M.; Chamberlain, A.M.; Jiang, R.; Roger, V.L. Atherosclerotic burden and heart failure after myocardial infarction. JAMA Cardiol. 2016, 1, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Xhaet, O.; DE Roy, L.; Floria, M.; Deceuninck, O.; Blommaert, D.; Dormal, F.; Ballant, E.; LA Meir, M. Integrity of the Ganglionated Plexi Is Essential to Parasympathetic Innervation of the Atrioventricular Node by the Right Vagus Nerve. J. Cardiovasc. Electrophysiol. 2017, 28, 432–437. [Google Scholar] [CrossRef]
- Pabon, M.A.; Manocha, K.; Cheung, J.W.; Lo, J.C. Linking arrhythmias and adipocytes: Insights, mechanisms, and future directions. Front. Physiol. 2018, 9, 1752. [Google Scholar] [CrossRef] [PubMed]
- Coumel, P. Autonomic influences in atrial tachyarrhythmias. J. Cardiovasc. Electrophysiol. 1996, 7, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Zipes, D.P.; Knope, R.F. Electrical properties of the thoracic veins. Am. J. Cardiol. 1972, 29, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.D.; Belevych, A.E. Muscarinic regulation of cardiac ion channels. Br. J. Pharmacol. 2003, 139, 1074–1084. [Google Scholar] [CrossRef]
- Pollard, C.M.; Desimine, V.L.; Wertz, S.L.; Perez, A.; Parker, B.M.; Maning, J.; McCrink, K.A.; Shehadeh, L.A.; Lymperopoulos, A. Deletion of Osteopontin Enhances β₂-Adrenergic Receptor-Dependent Anti-Fibrotic Signaling in Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 1396. [Google Scholar] [CrossRef]
- Rosenshtraukh, L.V.; Zaitsev, A.V.; Fast, V.G.; Pertsov, A.M.; Krinsky, V.I. Vagally induced block and delayed conduction as a mechanism for circus movement tachycardia in frog atria. Circ. Res. 1989, 64, 213–226. [Google Scholar] [CrossRef]
- Francis, G.S. Should asymptomatic ventricular arrhythmias in patients with congestive heart failure be treated with antiarrhythmic drugs? J. Am. Coll. Cardiol. 1988, 12, 274–283. [Google Scholar] [CrossRef]
- Wu, C.K.; Tsai, H.Y.; Su, M.Y.; Wu, Y.F.; Hwang, J.J.; Tseng, W.Y.; Lin, J.L.; Lin, L.Y. Pericardial fat is associated with ventricular tachyarrhythmia and mortality in patients with systolic heart failure. Atherosclerosis 2015, 241, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.S.; Chen, L.S.; Fishbein, M.C.; Lin, S.F.; Nattel, S. Role of the autonomic nervous system in atrial fibrillation: Pathophysiology and therapy. Circ. Res. 2014, 114, 1500–1515. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Zhang, Y.; Bibevski, S.; Marrouche, N.F.; Natale, A.; Mazgalev, T.N. Vagal denervation and atrial fibrillation inducibility: Epicardial fat pad ablation does not have long-term effects. Heart Rhythm 2006, 3, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Scherlag, B.J.; Patterson, E.; Ikeda, A.; Lockwood, D.; Jackman, W.M. Pathophysiologic basis of autonomic ganglionated plexus ablation in patients with atrial fibrillation. Heart Rhythm 2009, 6 (Suppl. 12), S26–S34. [Google Scholar] [CrossRef] [PubMed]
- Pokushalov, E.; Kozlov, B.; Romanov, A.; Strelnikov, A.; Bayramova, S.; Sergeevichev, D.; Bogachev-Prokophiev, A.; Zheleznev, S.; Shipulin, V.; Lomivorotov, V.V.; et al. Long-term suppression of atrial fibrillation by botulinum toxin injection into epicardial fat pads in patients undergoing cardiac surgery: One year follow up of a randomized pilot study. Circ. Arrhythmia Electrophysiol. 2015, 8, 1334–1341. [Google Scholar] [CrossRef]
- Couselo-Seijas, M.; López-Canoa, J.N.; Agra-Bermejo, R.M.; Díaz-Rodriguez, E.; Fernandez, A.L.; Martinez-Cereijo, J.M.; Durán-Muñoz, D.; Bravo, S.B.; Velo, A.; González-Melchor, L.; et al. Cholinergic activity regulates the secretome of epicardial adipose tissue: Association with atrial fibrillation. J. Cell. Physiol. 2019, 234, 10512–10522. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Inden, Y.; Mizutani, Y.; Fujii, A.; Kamikubo, Y.; Kanzaki, Y.; Ando, M.; Funabiki, J.; Murase, Y.; Takenaka, M.; et al. Vagal response in cryoballoon ablation of atrial fibrillation and autonomic nervous system: Utility of epicardial adipose tissue location. Arrhythm 2017, 33, 275–282. [Google Scholar] [CrossRef]
- Balcioğlu, A.S.; Çiçek, D.; Akinci, S.; Eldem, H.O.; Bal, U.A.; Okyay, K.; Müderrisoğlu, H. Arrhythmogenic evidence for epicardial adipose tissue: Heart rate variability and turbulence are influenced by epicardial fat thickness. Pacing Clin. Electrophysiol. 2015, 38, 99–106. [Google Scholar] [CrossRef]
- Kim, M.K.; Tanaka, K.; Kim, M.J.; Matsuo, T.; Tomita, T.; Ohkubo, H.; Maeda, S.; Ajisaka, R. Epicardial fat tissue: Relationship with cardiorespiratory fitness in men. Med. Sci. Sports Exerc. 2010, 42, 463–469. [Google Scholar] [CrossRef]
- Quan, K.J.; Lee, J.H.; Van Hare, G.F.; Biblo, L.A.; Mackall, J.A.; Carlson, M.D. Identification and characterization of atrioventricular parasympathetic innervation in humans. J. Cardiovasc. Electrophysiol. 2002, 13, 735–739. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, L.; Wang, K.; Xu, X.; Ji, M.; Zhang, F.; Wang, H.; Hou, Y. Effect of interconnection between cervical vagus trunk, epicardial fat pad on sinus node function, and atrial fibrillation. Pacing Clin. Electrophysiol. 2014, 37, 356–363. [Google Scholar] [CrossRef] [PubMed]
- White, C.M.; Sander, S.; Coleman, C.I.; Gallagher, R.; Takata, H.; Humphrey, C.; Henyan, N.; Gillespie, E.L.; Kluger, J. Impact of epicardial anterior fat pad retention on postcardiothoracic surgery atrial fibrillation incidence: The AFIST-III Study. J. Am. Coll. Cardiol. 2007, 49, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Burgeiro, A.; Fuhrmann, A.; Cherian, S.; Espinoza, D.; Jarak, I.; Carvalho, R.A.; Loureiro, M.; Patrício, M.; Antunes, M.; Carvalho, E. Glucose uptake and lipid metabolism are impaired in epicardial adipose tissue from heart failure patients with or without diabetes. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E550–E564. [Google Scholar] [CrossRef] [PubMed]
- Katlandur, H.; Ozbek, K.; Keser, A. Letter to the Editor: The effect of autonomic nervous system on the impairment of glucose uptake and lipid metabolism in epicardial adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E862. [Google Scholar] [CrossRef][Green Version]
- White, A. Cardiac sympathetic denervation in the failing heart: A role for epicardial adipose tissue. Circ. Res. 2016, 118, 1189–1191. [Google Scholar] [CrossRef]
- Parisi, V.; Rengo, G.; Perrone-Filardi, P.; Pagano, G.; Femminella, G.D.; Paolillo, S.; Petraglia, L.; Gambino, G.; Caruso, A.; Grimaldi, M.G.; et al. Increased epicardial adipose tissue volume correlates with cardiac sympathetic denervation in patients with heart failure. Circ. Res. 2016, 118, 1244–1253. [Google Scholar] [CrossRef]
- Iacobellis, G.; Leonetti, F.; Singh, N.; Sharma, A.M. Relationship of epicardial adipose tissue with atrial dimensions and diastolic function in morbidly obese subjects. Int. J. Cardiol. 2007, 115, 272–273. [Google Scholar] [CrossRef]
- Iacobellis, G.; Singh, N.; Wharton, S.; Sharma, A.M. Substantial changes in epicardial fat thickness after weight loss in severely obese subjects. Obesity 2008, 16, 1693–1697. [Google Scholar] [CrossRef]
- Packer, M. Leptin-aldosterone-neprilysin axis: Identification of its distinctive role in the pathogenesis of the three phenotypes of heart failure in people with obesity. Circulation 2018, 137, 1614–1631. [Google Scholar] [CrossRef]
- Bansal, G.; Druey, K.M.; Xie, Z. R4 RGS proteins: Regulation of G-protein signaling and beyond. Pharmacol. Ther. 2007, 116, 473–495. [Google Scholar] [CrossRef]
- O’Brien, J.B.; Wilkinson, J.C.; Roman, D.L. Regulator of G-protein signaling (RGS) proteins as drug targets: Progress and future potentials. J. Biol. Chem. 2019, 294, 18571–18585. [Google Scholar] [CrossRef]
- Squires, K.E.; Montañez-Miranda, C.; Pandya, R.R.; Torres, M.P.; Hepler, J.R. Genetic Analysis of Rare Human Variants of Regulators of G Protein Signaling Proteins and Their Role in Human Physiology and Disease. Pharmacol. Rev. 2018, 70, 446–474. [Google Scholar] [CrossRef] [PubMed]
- Perschbacher, K.J.; Deng, G.; Fisher, R.A.; Gibson-Corley, K.N.; Santillan, M.K.; Grobe, J.L. Regulators of G protein signaling in cardiovascular function during pregnancy. Physiol. Genom. 2018, 50, 590–604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Mende, U. Regulators of G-protein signaling in the heart and their potential as therapeutic targets. Circ. Res. 2011, 109, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Masuho, I.; Balaji, S.; Muntean, B.S.; Skamangas, N.K.; Chavali, S.; Tesmer, J.J.G.; Babu, M.M.; Martemyanov, K.A. A Global Map of G Protein Signaling Regulation by RGS Proteins. Cell 2020, 183, 503–521.e19. [Google Scholar] [CrossRef]
- Dowal, L.; Elliott, J.; Popov, S.; Wilkie, T.M.; Scarlata, S. Determination of the contact energies between a regulator of G protein signaling and G protein subunits and phospholipase C beta 1. Biochemistry 2001, 40, 414–421. [Google Scholar] [CrossRef]
- Huang, J.; Zhou, H.; Mahavadi, S.; Sriwai, W.; Murthy, K.S. Inhibition of Galphaq-dependent PLC-beta1 activity by PKG and PKA is mediated by phosphorylation of RGS4 and GRK2. Am. J. Physiol. Cell Physiol. 2007, 292, C200–C208. [Google Scholar] [CrossRef]
- Cifelli, C.; Rose, R.A.; Zhang, H.; Voigtlaender-Bolz, J.; Bolz, S.S.; Backx, P.H.; Heximer, S.P. RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ. Res. 2008, 103, 527–535. [Google Scholar] [CrossRef]
- Stewart, A.; Huang, J.; Fisher, R.A. RGS Proteins in Heart: Brakes on the Vagus. Front. Physiol. 2012, 3, 95. [Google Scholar] [CrossRef]
- Rogers, J.H.; Tamirisa, P.; Kovacs, A.; Weinheimer, C.; Courtois, M.; Blumer, K.J.; Kelly, D.P.; Muslin, A.J. RGS4 causes increased mortality and reduced cardiac hypertrophy in response to pressure overload. J. Clin. Investig. 1999, 104, 567–576. [Google Scholar] [CrossRef][Green Version]
- Tamirisa, P.; Blumer, K.J.; Muslin, A.J. RGS4 inhibits G-protein signaling in cardiomyocytes. Circulation 1999, 99, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.H.; Tsirka, A.; Kovacs, A.; Blumer, K.J.; Dorn, G.W., 2nd; Muslin, A.J. RGS4 reduces contractile dysfunction and hypertrophic gene induction in Galpha q overexpressing mice. J. Mol. Cell. Cardiol. 2001, 33, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Watson, N.; Zahner, J.; Rottman, J.N.; Blumer, K.J.; Muslin, A.J. RGS3 and RGS4 are GTPase activating proteins in the heart. J. Mol. Cell. Cardiol. 1998, 30, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Owen, V.J.; Burton, P.B.; Mullen, A.J.; Birks, E.J.; Barton, P.; Yacoub, M.H. Expression of RGS3, RGS4 and Gi alpha 2 in acutely failing donor hearts and end-stage heart failure. Eur. Heart J. 2001, 22, 1015–1020. [Google Scholar] [CrossRef]
- Mittmann, C.; Chung, C.H.; Höppner, G.; Michalek, C.; Nose, M.; Schüler, C.; Schuh, A.; Eschenhagen, T.; Weil, J.; Pieske, B.; et al. Expression of ten RGS proteins in human myocardium: Functional characterization of an upregulation of RGS4 in heart failure. Cardiovasc. Res. 2002, 55, 778–786. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Opel, A.; Nobles, M.; Montaigne, D.; Finlay, M.; Anderson, N.; Breckenridge, R.; Tinker, A. Absence of the Regulator of G-protein Signaling, RGS4, Predisposes to Atrial Fibrillation and Is Associated with Abnormal Calcium Handling. J. Biol. Chem. 2015, 290, 19233–19244. [Google Scholar] [CrossRef]
- Posokhova, E.; Wydeven, N.; Allen, K.L.; Wickman, K.; Martemyanov, K.A. RGS6/Gβ5 complex accelerates IKACh gating kinetics in atrial myocytes and modulates parasympathetic regulation of heart rate. Circ. Res. 2010, 107, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, J.; Maity, B.; Gao, Z.; Lorca, R.A.; Gudmundsson, H.; Li, J.; Stewart, A.; Swaminathan, P.D.; Ibeawuchi, S.R.; et al. RGS6, a modulator of parasympathetic activation in heart. Circ. Res. 2010, 107, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.M.; Borges, J.I.; Suster, M.S.; Sizova, A.; Cora, N.; Desimine, V.L.; Lymperopoulos, A. Regulator of G-Protein Signaling-4 Attenuates Cardiac Adverse Remodeling and Neuronal Norepinephrine Release-Promoting Free Fatty Acid Receptor FFAR3 Signaling. Int. J. Mol. Sci. 2022, 23, 5803. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Suster, M.S.; Borges, J.I. Short-Chain Fatty Acid Receptors and Cardiovascular Function. Int. J. Mol. Sci. 2022, 23, 3303. [Google Scholar] [CrossRef]
- Conte, M.; Petraglia, L.; Cabaro, S.; Valerio, V.; Poggio, P.; Pilato, E.; Attena, E.; Russo, V.; Ferro, A.; Formisano, P.; et al. Epicardial Adipose Tissue and Cardiac Arrhythmias: Focus on Atrial Fibrillation. Front. Cardiovasc. Med. 2022, 9, 932262. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Nattel, S.; Lip, G.Y.H.; Ren, J. Inflammasome Signaling in Atrial Fibrillation: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 2349–2366. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbone, A.M.; Del Calvo, G.; Nagliya, D.; Sharma, K.; Lymperopoulos, A. Autonomic Nervous System Regulation of Epicardial Adipose Tissue: Potential Roles for Regulator of G Protein Signaling-4. Curr. Issues Mol. Biol. 2022, 44, 6093-6103. https://doi.org/10.3390/cimb44120415
Carbone AM, Del Calvo G, Nagliya D, Sharma K, Lymperopoulos A. Autonomic Nervous System Regulation of Epicardial Adipose Tissue: Potential Roles for Regulator of G Protein Signaling-4. Current Issues in Molecular Biology. 2022; 44(12):6093-6103. https://doi.org/10.3390/cimb44120415
Chicago/Turabian StyleCarbone, Alexandra M., Giselle Del Calvo, Deepika Nagliya, Karina Sharma, and Anastasios Lymperopoulos. 2022. "Autonomic Nervous System Regulation of Epicardial Adipose Tissue: Potential Roles for Regulator of G Protein Signaling-4" Current Issues in Molecular Biology 44, no. 12: 6093-6103. https://doi.org/10.3390/cimb44120415
APA StyleCarbone, A. M., Del Calvo, G., Nagliya, D., Sharma, K., & Lymperopoulos, A. (2022). Autonomic Nervous System Regulation of Epicardial Adipose Tissue: Potential Roles for Regulator of G Protein Signaling-4. Current Issues in Molecular Biology, 44(12), 6093-6103. https://doi.org/10.3390/cimb44120415