
OPA1 Dominant Optic Atrophy: Diagnostic Approach in the Pediatric Population

 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Results
2.1. Ophthalmological Examination
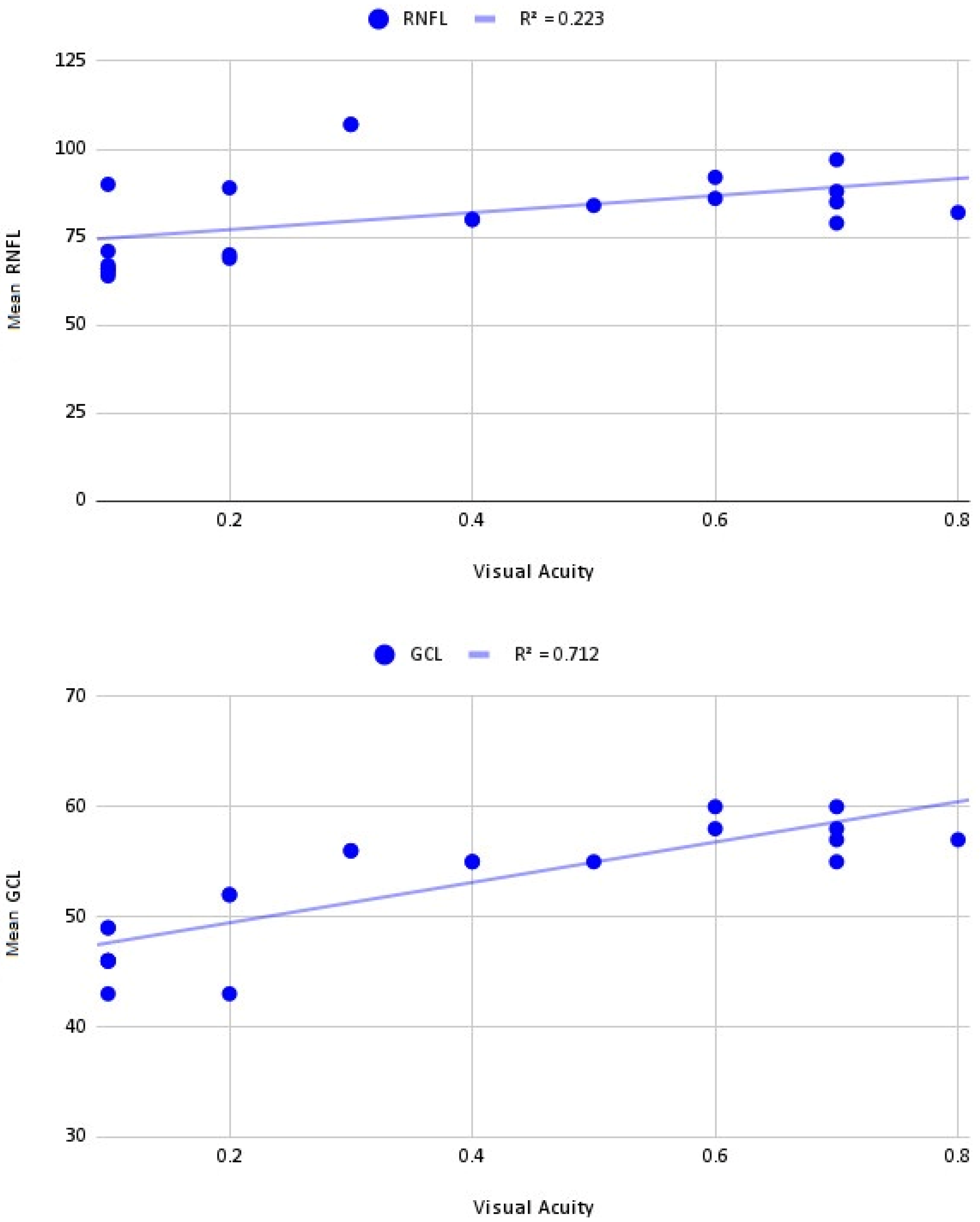
2.2. Optic Coherence Tomography
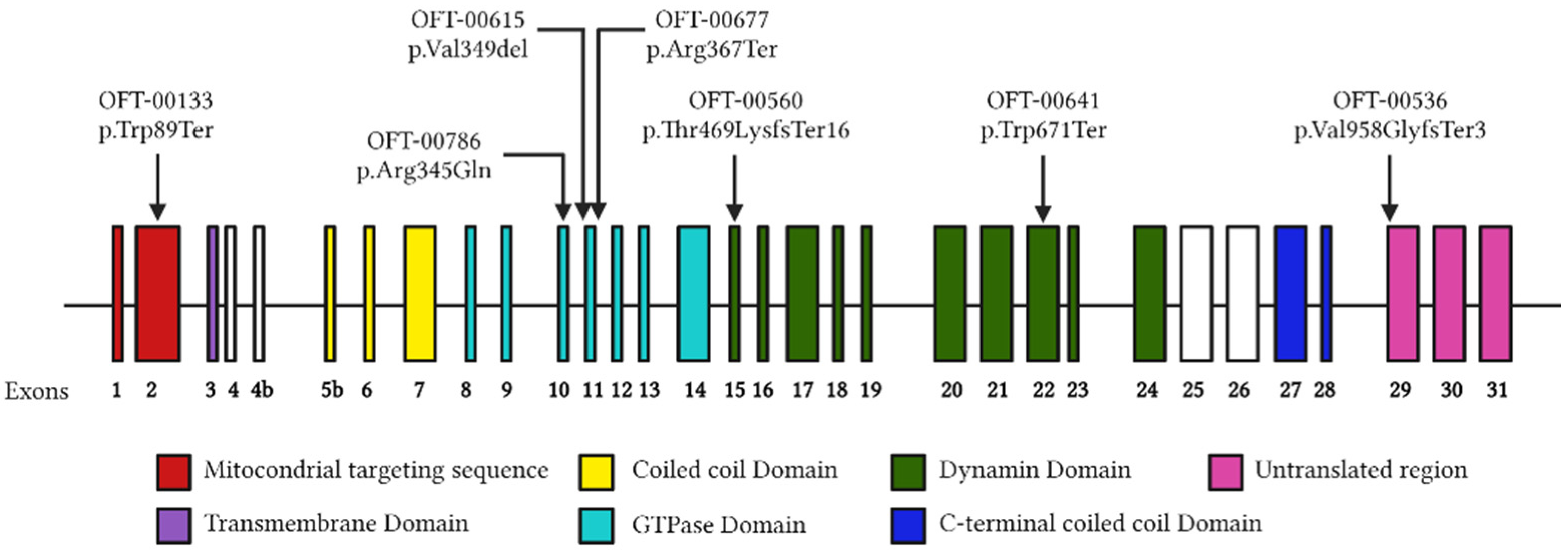
2.3. Molecular Genetics
2.3.1. OFT-00133
2.3.2. OFT-00536
2.3.3. OFT-00560
2.3.4. OFT-00615
2.3.5. OFT-00641
2.3.6. OFT-00677
2.3.7. OFT-00786

{kind=link}
{kind=link}
| Family ID | Individual ID | Sex | Relationship | Mutation | Location | ACMG Criteria * | ACMG Result | Coding Impact | Zygosity | Segregation Analysis Performed | De Novo/ Inherited | Reported by |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OFT-00133 | 2620605 | M | Proband | NM_130837.3:c.267G>A (p.Trp89Ter) | Exon 2 | PVS1, PM2, PP1 | P | Nonsense | Hetero | Yes | Maternal | (Le Roux, 2019) [17] |
| OFT-00536 | 2624905 | M | Proband | NM_130837.3:c.2873_2876del (p.Val958GlyfsTer3) | Intron 28 | PVS1, PP2, PP5 | P | Frameshift | Hetero | Yes | Unknown | (Delettre, 2000) [9] (Wang, 2019) [18] |
| 2235284 | M | Brother | ||||||||||
| OFT-00560 | 3338850 | F | Proband | NM_130837.3:c.1406_1407del (p.Thr469LysfsTer16) | Exon 15 | PVS1, PM2, PP1 | P | Frameshift | Hetero | Yes | Paternal | Novel ** |
| 3338857 | F | Brother | Hetero | |||||||||
| OFT-00615 | 3370622 | M | Proband | NM_130837.3:c.1041_1043del (p.Val349del) | Exon 11 | PM1, PM2, PM4, PP1 | LP | In-frame | Hetero | Yes | Maternal | (Gallus, 2012) [19] (Kamakari, 2014) [20] |
| 3395589 | M | Brother | Hetero | |||||||||
| OFT-00641 | 3346308 | M | Proband | NM_130837.3:c.2013G>A (p.Trp671Ter) | Exon 22 | PVS1, PM2, PP1 | P | Nonsense | Hetero | Yes | Paternal | (Ferré, 2009) [21] |
| 3366708 | M | Brother | Hetero | Paternal | ||||||||
| OFT-00677 | 3408438 | F | Proband | NM_130837.3:c.1099C>T (p.Arg367Ter) | Exon 11 | PVS1, PP5, PM2 | P | Nonsense | Hetero | No | Unknown | (Cardaioli, 2006) [22] (Chen, 2014) [23] (Pretegiani, 2017) [24] |
| OFT-00786 | 2854654 | F | Proband | NM_130837.3:c.1034G>A (p.Arg345Gln) | Exon 10 | PP5, PS3, PM2, PP3, PM1, PM5, PP2 | P | Missense | Hetero | No | Unknown | (Pretegiani, 2017) [24] (Liskova, 2016) [25] |
3. Discussion

4. Materials and Methods
4.1. Genetic Studies
4.2. Ophthalmological Studies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A
| Code | Description |
|---|---|
| PVS1 | Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where loss of function is a known mechanism of disease. |
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change. |
| PS2 | De novo (both maternity and paternity confirmed) in a patient with the disease and no family history. |
| PS3 | Well-established in vitro or in vivo functional studies supporting a damaging effect on the gene or gene product. |
| BS1 | Allele frequency is greater than expected for the disorder. |
| PM1 | Located in a mutational hotspot and/or critical and well-established functional domain (e.g., active site of an enzyme) with no benign variation. |
| PM2 | Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, and Exome Aggregation Consortium. |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant. |
| PM4 | Protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants. |
| PM5 | Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been previously observed. |
| PP1 | Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease. |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease. |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.) |
| PP5 | Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation. |
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, splicing impact, etc.) |
| BP6 | Reputable source recently reports variant as benign, but the evidence is not available to the laboratory to perform an independent evaluation. |
References
- Chun, B.Y.; Rizzo, J.F. Dominant Optic Atrophy and Leber’s Hereditary Optic Neuropathy: Update on Clinical Features and Current Therapeutic Approaches. Semin. Pediatr. Neurol. 2017, 24, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Carelli, V. Mitochondrial Retinopathies. Int. J. Mol. Sci. 2022, 23, 210. [Google Scholar] [CrossRef] [PubMed]
- Weisschuh, N.; Schimpf-Linzenbold, S.; Mazzola, P.; Kieninger, S.; Xiao, T.; Kellner, U.; Neuhann, T.; Kelbsch, C.; Tonagel, F.; Wilhelm, H.; et al. Mutation spectrum of the OPA1 gene in a large cohort of patients with suspected dominant optic atrophy: Identification and classification of 48 novel variants. PLoS ONE 2021, 16, e0253987. [Google Scholar] [CrossRef]
- Del Dotto, V.; Mishra, P.; Vidoni, S.; Fogazza, M.; Maresca, A.; Caporali, L.; McCaffery, J.M.; Cappelletti, M.; Baruffini, E.; Lenaers, G.; et al. OPA1 Isoforms in the Hierarchical Organization of Mitochondrial Functions. Cell Rep. 2017, 19, 2557–2571. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, J.; Jin, Z.; Zhang, Z. Structural and evolutionary characteristics of dynamin-related GTPase OPA1. PeerJ 2019, 7, e7285. [Google Scholar] [CrossRef] [PubMed]
- Maloney, D.M.; Chadderton, N.; Millington-Ward, S.; Palfi, A.; Shortall, C.; O’Byrne, J.J.; Cassidy, L.; Keegan, D.; Humphries, P.; Kenna, P.; et al. Optimized OPA1 Isoforms 1 and 7 Provide Therapeutic Benefit in Models of Mitochondrial Dysfunction. Front. Neurosci. 2020, 14, 571479. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C. Toxic-Metabolic and Hereditary Optic Neuropathies. Contin. Lifelong Learn. Neurol. 2019, 25, 1265–1288. [Google Scholar] [CrossRef] [PubMed]
- Asanad, S.; Tian, J.J.; Frousiakis, S.; Jiang, J.P.; Kogachi, K.; Felix, C.M.; Fatemeh, D.; Irvine, A.G.; Ter-Zakarian, A.; Falavarjani, K.G.; et al. Optical Coherence Tomography of the Retinal Ganglion Cell Complex in Leber’s Hereditary Optic Neuropathy and Dominant Optic Atrophy. Curr. Eye Res. 2019, 44, 638–644. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 4. [Google Scholar] [CrossRef]
- Caporali, L.; Maresca, A.; Capristo, M.; Del Dotto, V.; Tagliavini, F.; Valentino, M.L.; La Morgia, C.; Carelli, V. Incomplete penetrance in mitochondrial optic neuropathies. Mitochondrion 2017, 36, 130–137. [Google Scholar] [CrossRef]
- Maeda-Katahira, A.; Nakamura, N.; Hayashi, T.; Katagiri, S.; Shimizu, S.; Ohde, H.; Matsunaga, T.; Kaga, K.; Nakano, T.; Kameya, S.; et al. Autosomal dominant optic atrophy with OPA1 gene mutations accompanied by auditory neuropathy and other systemic complications in a Japanese cohort. Mol. Vis. 2019, 25, 559–573. [Google Scholar] [PubMed]
- Romagnoli, M.; La Morgia, C.; Carbonelli, M.; Di Vito, L.; Amore, G.; Zenesini, C.; Cascavilla, M.L.; Barboni, P.; Carelli, V. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Ann. Clin. Transl. Neurol. 2020, 7, 590–594. [Google Scholar] [CrossRef]
- Barboni, P.; Valentino, M.L.; LA Morgia, C.; Carbonelli, M.; Savini, G.; De Negri, A.; Simonelli, F.; Sadun, F.; Caporali, L.; Maresca, A.; et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain 2013, 136, e231. [Google Scholar] [CrossRef] [PubMed]
- Barrio-Barrio, J.; Noval, S.; Galdós, M.; Ruiz-Canela, M.; Bonet, E.; Capote, M.; Lopez, M. Multicenter Spanish study of spectral-domain optical coherence tomography in normal children. Acta Ophthalmol. 2013, 91, e56–e63. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Maquat, L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Schimpf, S.; Fuhrmann, N.; Schaich, S.; Wissinger, B. Comprehensive cDNA study and quantitative transcript analysis of mutant OPA1 transcripts containing premature termination codons. Hum. Mutat. 2008, 29, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, B.; Lenaers, G.; Zanlonghi, X.; Amati-Bonneau, P.; Chabrun, F.; Foulonneau, T.; Caignard, A.; Leruez, S.; Gohier, P.; Procaccio, V.; et al. OPA1: 516 unique variants and 831 patients registered in an updated centralized Variome database. Orphanet J. Rare Dis. 2019, 14, 214. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, M.; Liu, X.; Huang, Y.; Zhou, Y.; Liu, Q.; Chen, X.; Zhao, C.; Wang, M. Targeted next-generation sequencing extends the mutational spectrums for OPA1 mutations in Chinese families with optic atrophy. Mol. Vis. 2019, 25, 912–920. [Google Scholar]
- Gallus, G.; Cardaioli, E.; Rufa, A.; Collura, M.; Da Pozzo, P.; Pretegiani, E.; Tumino, M.; Pavone, L.; Federico, A. High frequency of OPA1 mutations causing high ADOA prevalence in south-eastern Sicily, Italy. Clin. Genet. 2012, 82, 277–282. [Google Scholar] [CrossRef]
- Kamakari, S.; Koutsodontis, G.; Tsilimbaris, M.; Fitsios, A.; Chrousos, G. First report of OPA1 screening in Greek patients with autosomal dominant optic atrophy and identification of a previously undescribed OPA1 mutation. Mol. Vis. 2014, 20, 691–703. [Google Scholar]
- Ferrã©, M.; Bonneau, D.; Milea, D.; Chevrollier, A.; Verny, C.; Dollfus, H.; Ayuso, C.; Defoort, S.; Vignal, C.; Zanlonghi, X.; et al. Molecular screening of 980 cases of suspected hereditary optic neuropathy with a report on 77 novel OPA1 mutations. Hum. Mutat. 2009, 30, E692–E705. [Google Scholar] [CrossRef] [PubMed]
- Cardaioli, E.; Gallus, G.N.; Da Pozzo, P.; Rufa, A.; Franceschini, R.; Motolese, E.; Caporossi, A.; Dotti, M.T.; Federico, A. A novel mutation producing premature termination codon at the OPA1 gene causes autosomal dominant optic atrophy. J. Neurol. 2006, 253, 672–673. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, K.; Zhang, X.; Jiang, F.; Liu, L.; Dong, B.; Ren, Y.; Li, Y. Mutation Screening of Mitochondrial DNA as Well as OPA1 and OPA3 in a Chinese Cohort With Suspected Hereditary Optic Atrophy. Investig. Opthalmol. Vis. Sci. 2014, 55, 6987–6995. [Google Scholar] [CrossRef][Green Version]
- Pretegiani, E.; Rosini, F.; Rufa, A.; Gallus, G.; Cardaioli, E.; Da Pozzo, P.; Bianchi, S.; Serchi, V.; Collura, M.; Franceschini, R.; et al. Genotype-phenotype and OCT correlations in Autosomal Dominant Optic Atrophy related to OPA1 gene mutations: Report of 13 Italian families. J. Neurol. Sci. 2017, 382, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Liskova, P.; Tesarova, M.; Dudakova, L.; Svecova, S.; Kolarova, H.; Honzik, T.; Seto, S.; Votruba, M. OPA1 analysis in an international series of probands with bilateral optic atrophy. Acta Ophthalmol. 2017, 95, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Jang, J.D.; Jamieson, R.V.; Grigg, J.R. Long-Term Follow-Up Study of Autosomal Dominant Optic Atrophy in an Australian Population. Asia-Pac. J. Ophthalmol. 2012, 1, 88–90. [Google Scholar] [CrossRef]
- Lin, C.-W.; Huang, C.-W.; Luo, A.C.; Chou, Y.-T.; Huang, Y.-S.; Chen, P.-L.; Chen, T.-C. Genetic Spectrum and Characteristics of Hereditary Optic Neuropathy in Taiwan. Genes 2021, 12, 1378. [Google Scholar] [CrossRef]
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 1–12. [Google Scholar] [CrossRef]
- Chun, B.Y.; Rizzo, J.F. Dominant optic atrophy: Updates on the pathophysiology and clinical manifestations of the optic atrophy 1 mutation. Curr. Opin. Ophthalmol. 2016, 27, 475–480. [Google Scholar] [CrossRef]
- Barboni, P.; Savini, G.; Parisi, V.; Carbonelli, M.; La Morgia, C.; Maresca, A.; Sadun, F.; De Negri, A.M.; Carta, A.; Sadun, A.A.; et al. Retinal Nerve Fiber Layer Thickness in Dominant Optic Atrophy. Ophthalmology 2011, 118, 2076–2080. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Bailie, M.; Atawan, A.; Chinnery, P.F.; Griffiths, P.G. Pattern of retinal ganglion cell loss in dominant optic atrophy due to OPA1 mutations. Eye 2011, 25, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Milea, D.; Sander, B.; Wegener, M.; Jensen, H.; Kjer, B.; Jørgensen, T.M.; Lund-Andersen, H.; Larsen, M. Axonal loss occurs early in dominant optic atrophy. Acta Ophthalmol. 2009, 88, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Corajevic, N.; Larsen, M.; Rönnbäck, C. Thickness mapping of individual retinal layers and sectors by Spectralis SD-OCT in Autosomal Dominant Optic Atrophy. Acta Ophthalmol. 2018, 96, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Votruba, M.; Pesch, U.E.A.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Kushnareva, Y.; Seong, Y.; Andreyev, A.Y.; Kuwana, T.; Kiosses, W.B.; Votruba, M.; Newmeyer, D.D. Mitochondrial dysfunction in an Opa1(Q285STOP) mouse model of dominant optic atrophy results from Opa1 haploinsufficiency. Cell Death Dis. 2016, 7, e2309. [Google Scholar] [CrossRef]
- Kushnareva, Y.E.; Gerencser, A.A.; Bossy, B.; Ju, W.-K.; White, A.D.; Waggoner, J.; Ellisman, M.H.; Perkins, G.; Bossy-Wetzel, E. Loss of OPA1 disturbs cellular calcium homeostasis and sensitizes for excitotoxicity. Cell Death Differ. 2013, 20, 353–365. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; de Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Grau, T.; Burbulla, L.F.; Engl, G.; Delettre, C.; Delprat, B.; Oexle, K.; Leo-Kottler, B.; Roscioli, T.; Krüger, R.; Rapaport, D.; et al. A novel heterozygous OPA3 mutation located in the mitochondrial target sequence results in altered steady-state levels and fragmented mitochondrial network. J. Med. Genet. 2013, 50, 848–858. [Google Scholar] [CrossRef]
- Ham, M.; Han, J.; Osann, K.; Smith, M.; Kimonis, V. Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy. Mitochondrion 2019, 46, 262–269. [Google Scholar] [CrossRef]
- Ahmad, K.E.; Davis, R.L.; Sue, C.M. A novel OPA1 mutation causing variable age of onset autosomal dominant optic atrophy plus in an Australian family. J. Neurol. 2015, 262, 2323–2328. [Google Scholar] [CrossRef]
- Kane, M.S.; Alban, J.; Desquiret-Dumas, V.; Gueguen, N.; Ishak, L.; Ferre, M.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Lenaers, G.; et al. Autophagy controls the pathogenicity of OPA1 mutations in dominant optic atrophy. J. Cell. Mol. Med. 2017, 21, 2284–2297. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Thompson-Lowrey, A.J.; Reiss, A.; Mayorov, V.; Jia, H.; Biousse, V.; Newman, N.J.; Brown, M.D. OPA1 mutations and mitochondrial DNA haplotypes in autosomal dominant optic atrophy. Genet. Med. 2006, 8, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Yu-Wai-Man, P.; Griffiths, P.G.; Caporali, L.; Salomao, S.S.; Berezovsky, A.; Carelli, V.; Zeviani, M.; Chinnery, P.F. Variation in OPA1 does not explain the incomplete penetrance of Leber hereditary optic neuropathy. Mol. Vis. 2010, 16, 2760–2764. [Google Scholar] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]

| Family ID | Individual ID | Sex | Race | Age at Diagnosis, Years | Present Complaint | Family History | Color Vision |
|---|---|---|---|---|---|---|---|
| OFT-00133 | 2620605 | M | Caucasian | 6 | Reduced VA | Yes | Normal |
| OFT-00536 | 2624905 | M | Asian | 9 | Esotropia | Yes | Normal |
| OFT-00536 | 2235284 | M | Asian | 14 | Reduced VA | Yes | Normal |
| OFT-00560 | 3338850 | F | Caucasian | 9 | Reduced VA | No | Normal |
| OFT-00560 | 3338857 | F | Caucasian | 15 | Asymptomatic | Yes | Normal |
| OFT-00615 | 3370622 | M | Caucasian | 8 | Reduced VA | Yes | Normal |
| OFT-00615 | 3395589 | M | Caucasian | 15 | Reduced VA | Yes | Normal |
| OFT-00641 | 3346308 | M | Caucasian | 4 | Reduced VA | No | Tritanopia |
| OFT-00641 | 3366708 | M | Caucasian | 2 | Brother DOA | Yes | No cooperationn |
| OFT-00677 | 3408438 | F | Caucasian | 14 | Reduced VA | No | Normal |
| OFT-00786 | 2854654 | F | Caucasian | 7 | Reduced VA | Yes | Normal |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arruti, N.; Rodríguez-Solana, P.; Nieves-Moreno, M.; Guerrero-Carretero, M.; del Pozo, Á.; Montaño, V.E.F.; Santos-Simarro, F.; Rikeros-Orozco, E.; Delgado-Mora, L.; Vallespín, E.; et al. OPA1 Dominant Optic Atrophy: Diagnostic Approach in the Pediatric Population. Curr. Issues Mol. Biol. 2023, 45, 465-478. https://doi.org/10.3390/cimb45010030
Arruti N, Rodríguez-Solana P, Nieves-Moreno M, Guerrero-Carretero M, del Pozo Á, Montaño VEF, Santos-Simarro F, Rikeros-Orozco E, Delgado-Mora L, Vallespín E, et al. OPA1 Dominant Optic Atrophy: Diagnostic Approach in the Pediatric Population. Current Issues in Molecular Biology. 2023; 45(1):465-478. https://doi.org/10.3390/cimb45010030
Chicago/Turabian StyleArruti, Natalia, Patricia Rodríguez-Solana, María Nieves-Moreno, Marta Guerrero-Carretero, Ángela del Pozo, Victoria E. F. Montaño, Fernando Santos-Simarro, Emi Rikeros-Orozco, Luna Delgado-Mora, Elena Vallespín, and et al. 2023. "OPA1 Dominant Optic Atrophy: Diagnostic Approach in the Pediatric Population" Current Issues in Molecular Biology 45, no. 1: 465-478. https://doi.org/10.3390/cimb45010030
APA StyleArruti, N., Rodríguez-Solana, P., Nieves-Moreno, M., Guerrero-Carretero, M., del Pozo, Á., Montaño, V. E. F., Santos-Simarro, F., Rikeros-Orozco, E., Delgado-Mora, L., Vallespín, E., & Noval, S. (2023). OPA1 Dominant Optic Atrophy: Diagnostic Approach in the Pediatric Population. Current Issues in Molecular Biology, 45(1), 465-478. https://doi.org/10.3390/cimb45010030