
DNA Double-Strand Break Repair Inhibitors: YU238259, A12B4C3 and DDRI-18 Overcome the Cisplatin Resistance in Human Ovarian Cancer Cells, but Not under Hypoxia Conditions

Abstract
:
1. Introduction
2. Materials and Methods
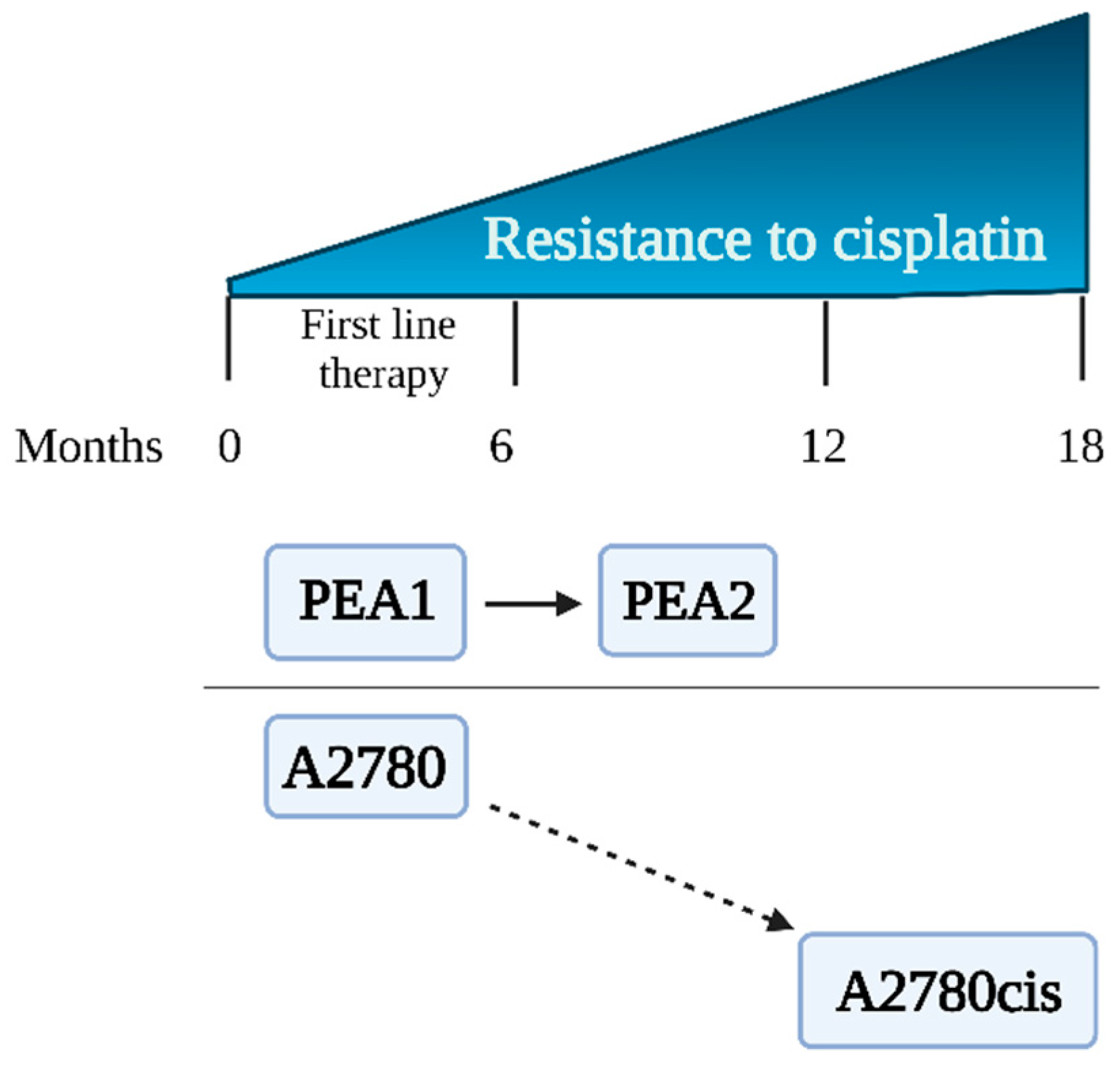
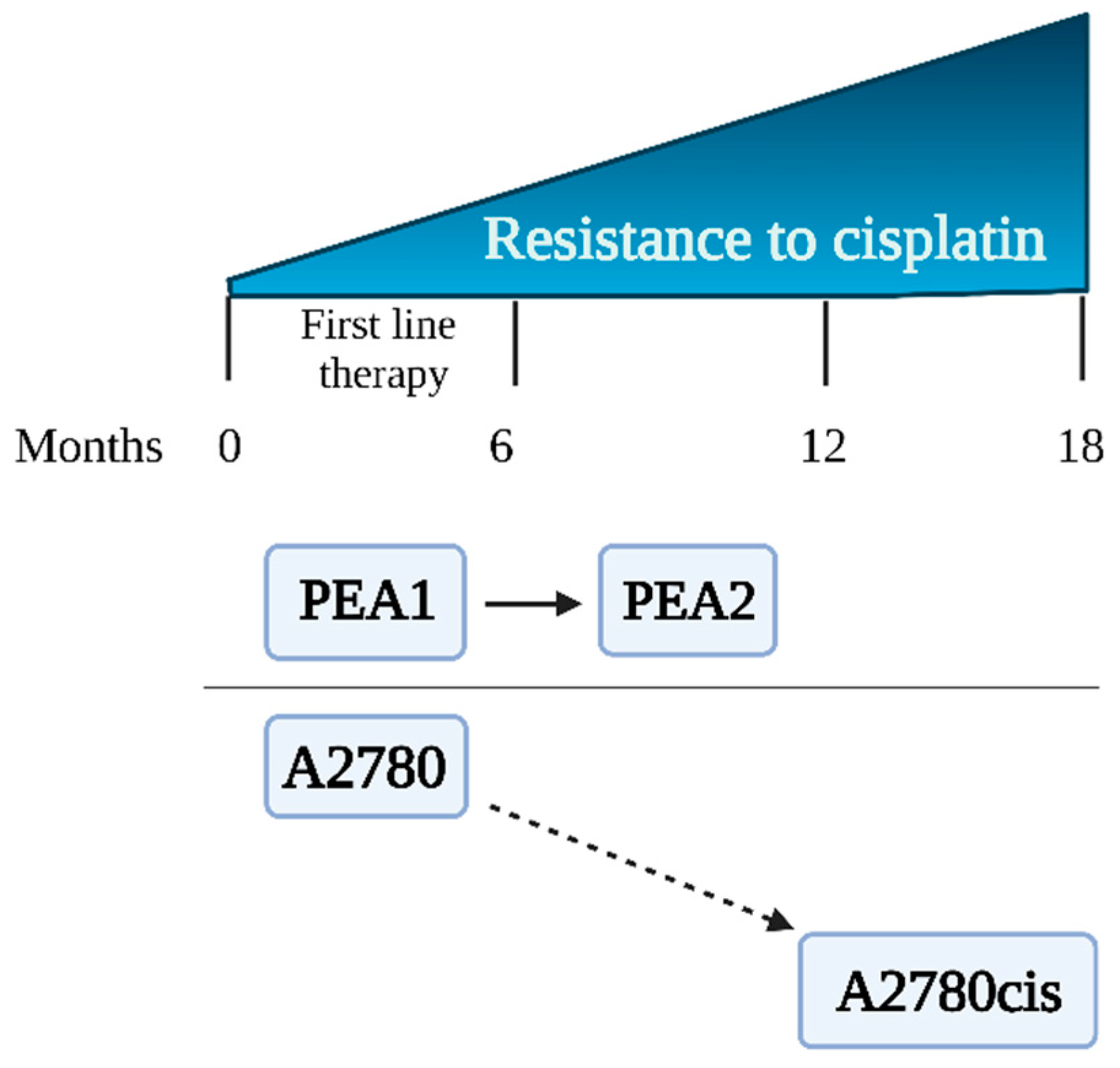
2.1. Cell Lines
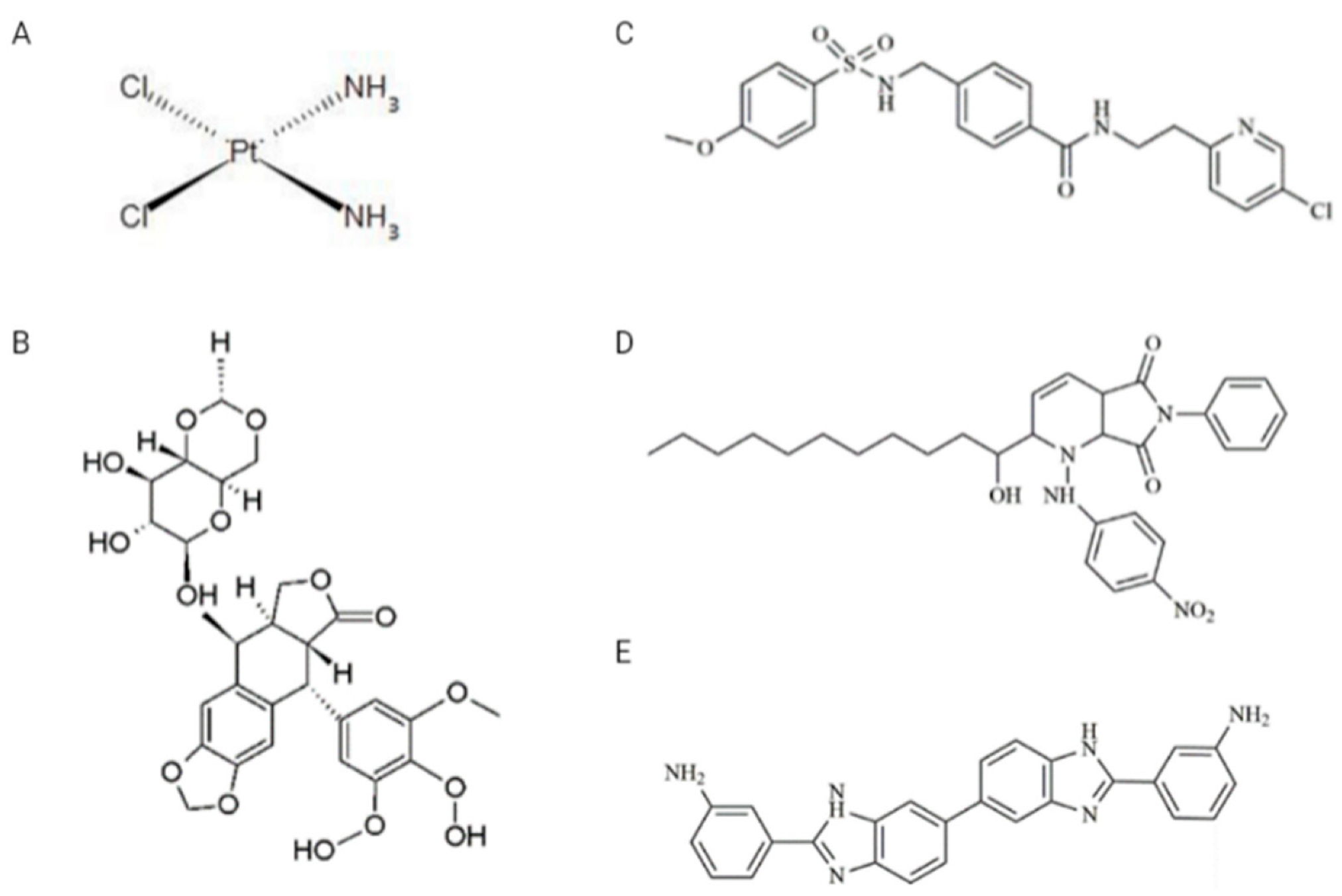
2.2. Chemicals
2.3. Cell Culture in Normoxia and Hypoxia Conditions
2.4. Viability Assay
2.5. Comparison of the Influence of DRIs on the Effect of CDDP and VP-16
2.6. Comet Assay
2.7. H2AX Histone Phosphorylation
2.8. DNA Damage and Repair Analysis
2.9. Cell Cycle Analysis
2.10. Detection of Apoptosis
2.11. Measurement of HIF1A Level from Total Protein
2.12. Gene Expression
2.13. Data Analysis
3. Results
3.1. Pretreatment with DRIs Enhances the Cytotoxic Effect of CDDP/VP-16 in Combined Treatment in Ovarian Cancer Cells at Normoxia Conditions
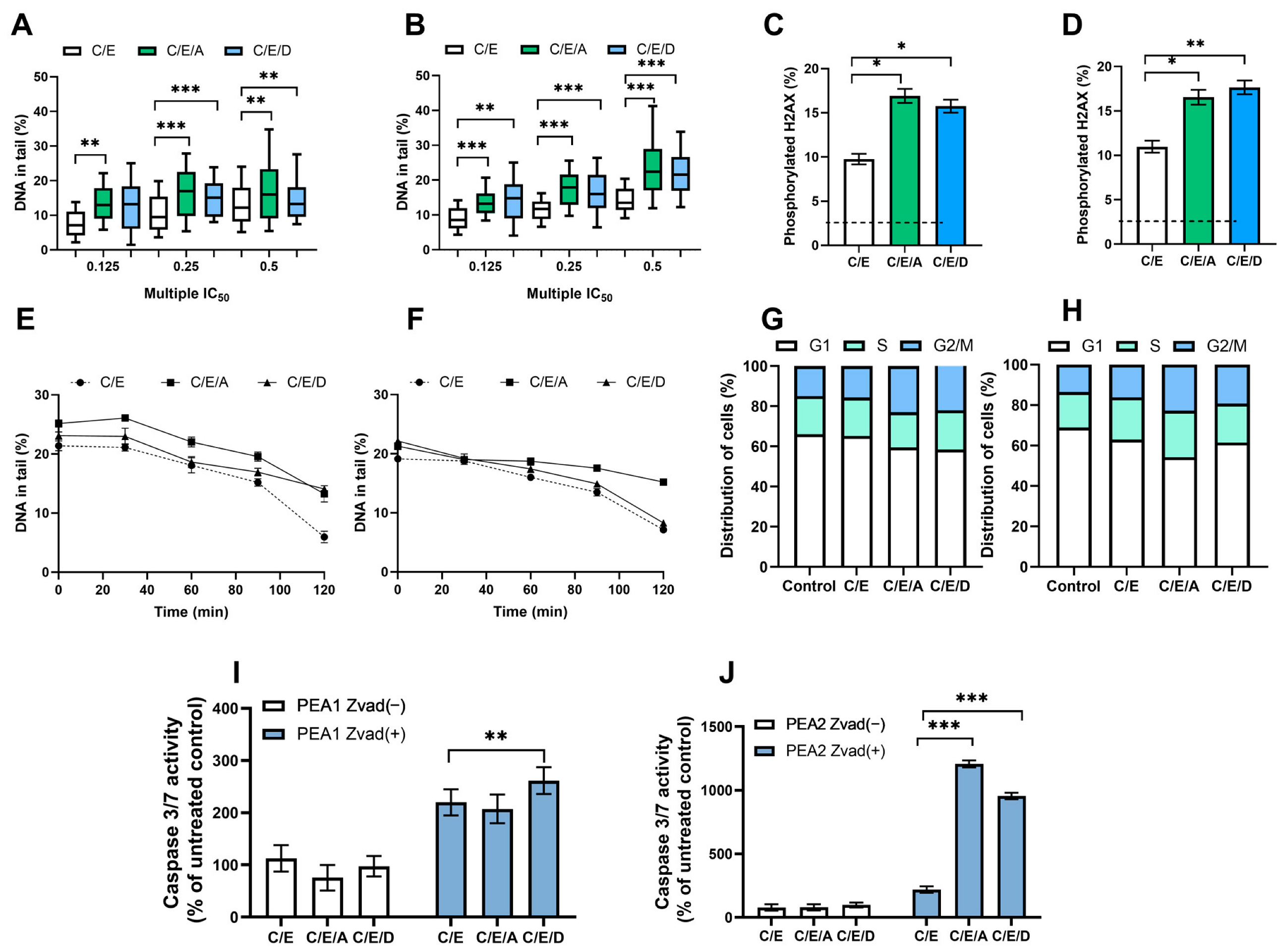
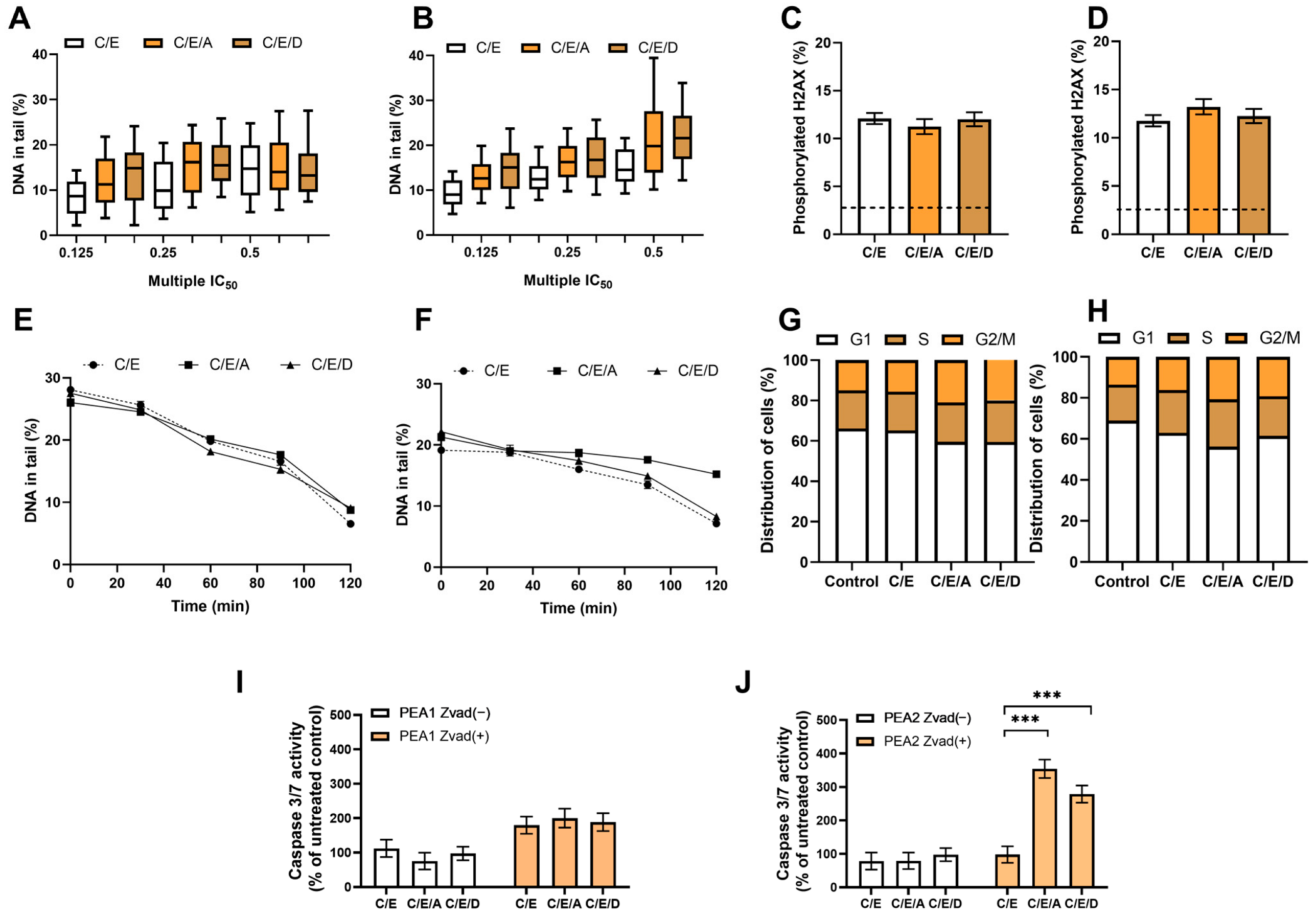
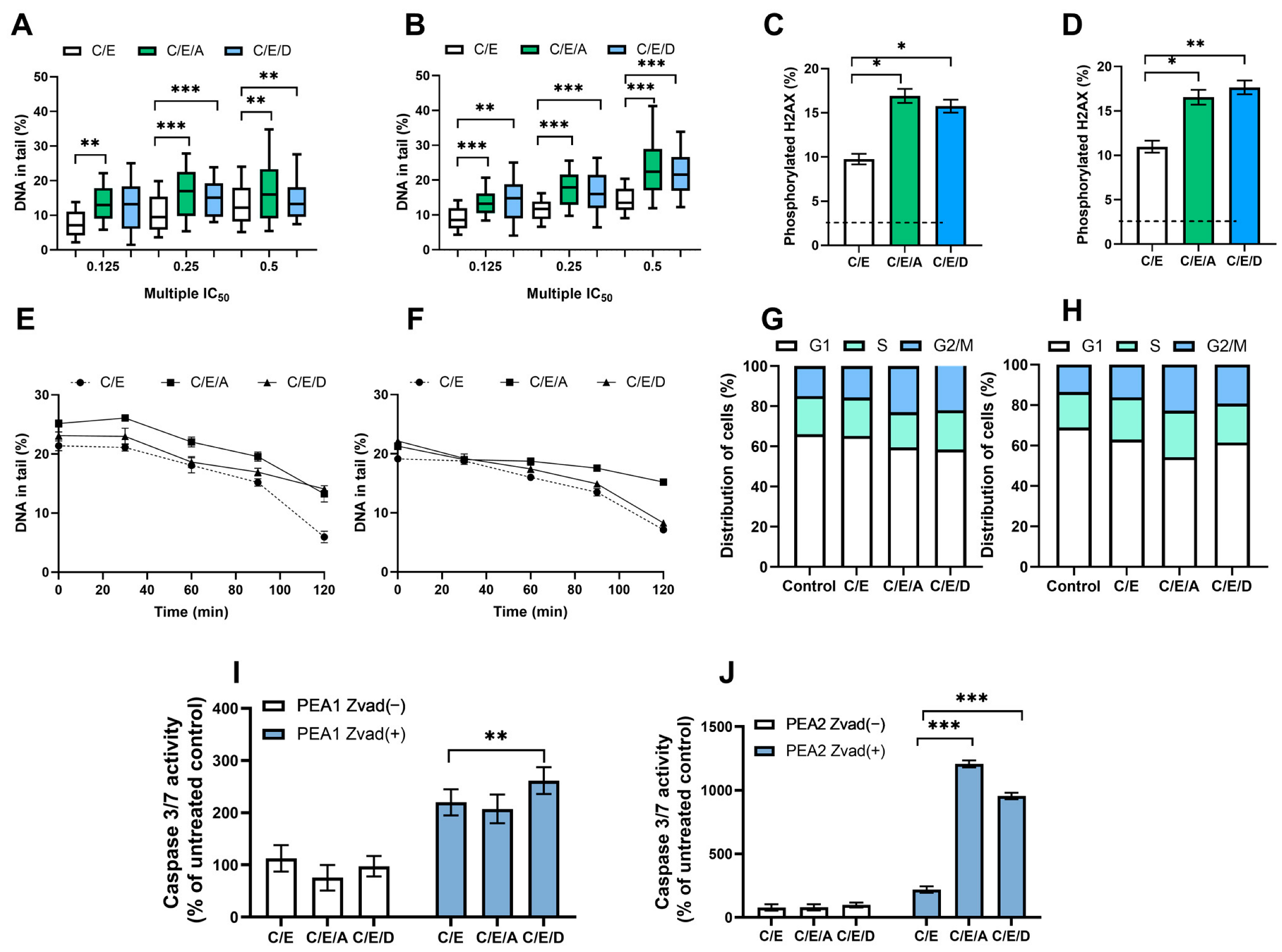
3.2. Pretreatment with DRIs Causes an Increase in the Level of DNA Damage and the Phosphorylated H2AX Accumulation in Ovarian Cancer Cells Treated with CDDP/VP-16 at Normoxia Conditions
3.3. Pretreatment with DRIs Decreases the Efficiency of DNA Damage Repair in Ovarian Cancer Cells Treated with CDDP/VP-16 at Normoxia Conditions
3.4. Pretreatment with DRIs Causes an Accumulation of Cells at the G2/M Phase of the Cell Cycle in Ovarian Cancer Cells Treated with CDDP/VP-16 at Normoxia Conditions
3.5. Pretreatment with DRIs Induce Apoptosis in Ovarian Cancer Cells Treated with CDDP/VP-16 at Normoxia Conditions
3.6. Cell Culture in Hypoxia Conditions Induces Increase in the Level of Transcription Factor Hif1A in Ovarian Cancer Cells
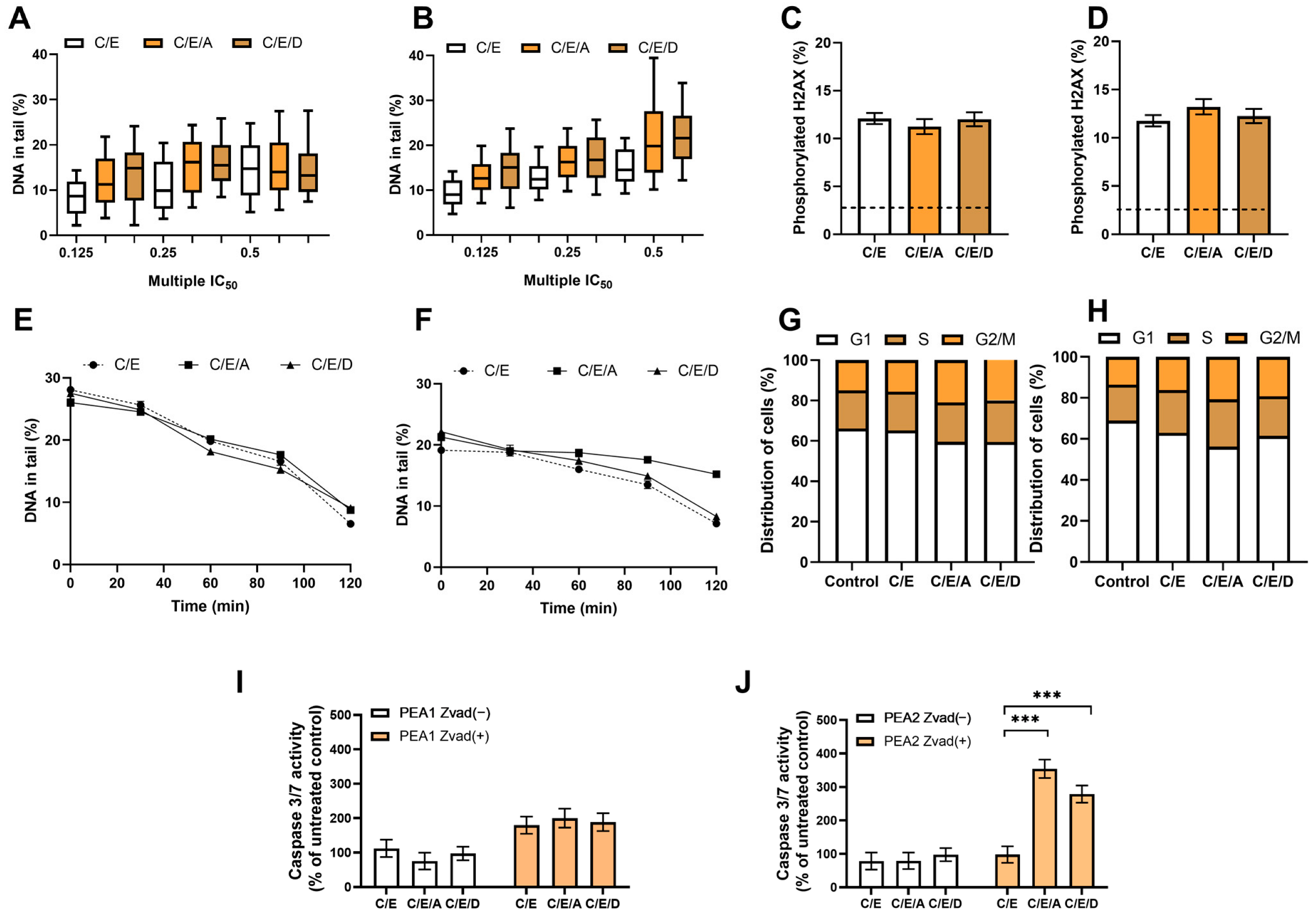
3.7. Pretreatment with DRIs Does Not Enhance the Cytotoxic Effect of CDDP/VP-16 in Combined Treatment in Ovarian Cancer Cells at Hypoxia Conditions
3.8. Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Huang, J.; Chan, W.C.; Ngai, C.H.; Lok, V.; Zhang, L.; Lucero-Prisno, D.E.; Xu, W.; Zheng, Z.-J.; Elcarte, E.; Withers, M.; et al. Worldwide Burden, Risk Factors, and Temporal Trends of Ovarian Cancer: A Global Study. Cancers 2022, 14, 2230. [Google Scholar] [CrossRef]
- Chornokur, G.; Amankwah, E.K.; Schildkraut, J.M.; Phelan, C.M. Global ovarian cancer health disparities. Gynecol. Oncol. 2013, 129, 258–264. [Google Scholar] [CrossRef]
- Belkić, K.; Belkić, D. The Challenge of Ovarian Cancer: Steps Toward Early Detection Through Advanced Signal Processing in Magnetic Resonance Spectroscopy. Isr. Med. Assoc. J. 2017, 19, 517–525. [Google Scholar]
- Song, M.; Cui, M.; Liu, K. Therapeutic strategies to overcome cisplatin resistance in ovarian cancer. Eur. J. Med. Chem. 2022, 232, 114205. [Google Scholar] [CrossRef]
- Luvero, D.; Milani, A.; Ledermann, J.A. Treatment options in recurrent ovarian cancer: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2014, 6, 229–239. [Google Scholar] [CrossRef]
- Zoń, A.; Bednarek, I. Cisplatin in Ovarian Cancer Treatment—Known Limitations in Therapy Force New Solutions. Int. J. Mol. Sci. 2023, 24, 7585. [Google Scholar] [CrossRef]
- Arend, R.; Westin, S.N.; Coleman, R.L. Decision analysis for secondline maintenance treatment of platinum sensitive recurrent ovarian cancer: A review. Int. J. Gynecol. Cancer 2020, 30, 684–694. [Google Scholar] [CrossRef]
- Van der Burg, M.E.L.; de Wit, R.; van Putten, W.L.J.; Logmans, A.; Kruit, W.H.J.; Stoter, G.; Verweij, J. Weekly cisplatin and daily oral etoposide is highly effective in platinum pretreated ovarian cancer. Br. J. Cancer 2002, 86, 19–25. [Google Scholar] [CrossRef]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer 2019, 11, 1179299X19860815. [Google Scholar] [CrossRef]
- Stachelek, G.C.; Peterson-Roth, E.; Liu, Y.; Fernandez, R.J.; Pike, L.R.G.; Qian, J.M.; Abriola, L.; Hoyer, D.; Hungerford, W.; Merkel, J.; et al. Yu238259 is a novel inhibitor of homology-dependent dna repair that exhibits synthetic lethality and radiosensitization in repair-deficient tumors. Mol. Cancer Res. 2015, 13, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Zereshkian, A.; Leyton, J.V.; Cai, Z.; Bergstrom, D.; Weinfeld, M.; Reilly, R.M. The human polynucleotide kinase/phosphatase (hPNKP) inhibitor A12B4C3 radiosensitizes human myeloid leukemia cells to Auger electron-emitting anti-CD123 111In-NLS-7G3 radioimmunoconjugates. Nucl. Med. Biol. 2014, 41, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Jun, D.W.; Jeong, Y.S.; Kim, H.J.; Jeong, K.-C.; Kim, S.; Lee, C.-H. Characterization of DDRI-18 (3,3′-(1H,3′H-5,5′-bibenzo[d]imidazole-2,2′-diyl)dianiline), a novel small molecule inhibitor modulating the DNA damage response. Br. J. Pharmacol. 2012, 167, 141–150. [Google Scholar] [CrossRef]
- Behrens, B.C.; Hamilton, T.C.; Masuda, H.; Grotzinger, K.R.; Whang-Peng, J.; Louie, K.G.; Knutsen, T.; McKoy, W.M.; Young, R.C.; Ozols, R.F. Characterization of a cis-diamminedichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res. 1987, 47, 414–418. [Google Scholar]
- Koshkin, V.; De Oliveira, M.B.; Peng, C.; Ailles, L.E.; Liu, G.; Covens, A.; Krylov, S.N. Multi-drug-resistance efflux in cisplatin-naive and cisplatin-exposed A2780 ovarian cancer cells responds differently to cell culture dimensionality. Mol. Clin. Oncol. 2021, 15, 161. [Google Scholar] [CrossRef] [PubMed]
- Viscarra, T.; Buchegger, K.; Jofre, I.; Riquelme, I.; Zanella, L.; Abanto, M.; Parker, A.C.; Piccolo, S.R.; Roa, J.C.; Ili, C.; et al. Functional and transcriptomic characterization of carboplatin-resistant A2780 ovarian cancer cell line. Biol. Res. 2019, 52, 13. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, T.C.; Young, R.C.; Ozols, R.F. Experimental model systems of ovarian cancer: Applications to the design and evaluation of new treatment approaches. Semin. Oncol. 1984, 11, 285–298. [Google Scholar]
- Pavlacky, J.; Polak, J. Technical Feasibility and Physiological Relevance of Hypoxic Cell Culture Models. Front. Endocrinol. 2020, 11, 57. [Google Scholar] [CrossRef]
- Langdon, S.P.; Lawrie, S.S.; Hay, F.G.; Hawkes, M.M.; McDonald, A.; Hayward, I.P.; Schol, D.J.; Hilgers, J.; Leonard, R.C.; Smyth, J.F. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 1988, 48, 6166–6172. [Google Scholar]
- Place, T.L.; Domann, F.E.; Case, A.J. Limitations of oxygen delivery to cells in culture: An underappreciated problem in basic and translational research. Free Radic. Biol. Med. 2017, 113, 311–322. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R. The comet assay for DNA damage and repair: Principles, applications, and limitations. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Lu, Y.; Li, Z.; Li, L.; Niu, D.; Xu, W.; Liu, J.; Fu, L.; Zhou, Z.; Gu, Y.; et al. Ganetespib overcomes resistance to PARP inhibitors in breast cancer by targeting core proteins in the DNA repair machinery. Investig. New Drugs 2017, 35, 251–259. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, S.; Cheng, S.; Yang, J.; Wang, Y. Clinical application of PARP inhibitors in ovarian cancer: From molecular mechanisms to the current status. J. Ovarian Res. 2023, 16, 6. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Tudrej, P.; Kujawa, K.A.; Cortez, A.J.; Lisowska, K.M. Characteristics of in vitro model systems for ovarian cancer studies. Oncol. Clin. Pract. 2019, 15, 246–259. [Google Scholar] [CrossRef]
- DeVries, A.A.; Dennis, J.; Tyrer, J.P.; Peng, P.-C.; Coetzee, S.G.; Reyes, A.L.; Plummer, J.T.; Davis, B.D.; Chen, S.S.; Dezem, F.S.; et al. Copy Number Variants Are Ovarian Cancer Risk Alleles at Known and Novel Risk Loci. J. Natl. Cancer Inst. 2022, 114, 1533–1544. [Google Scholar] [CrossRef]
- van Maanen, J.M.; Retèl, J.; de Vries, J.; Pinedo, H.M. Mechanism of action of antitumor drug etoposide: A review. J. Natl. Cancer Inst. 1988, 80, 1526–1533. [Google Scholar] [CrossRef]
- Reyhanoglu, G.; Tadi, P. Etoposide. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Winsey, S.L.; Haldar, N.A.; Marsh, H.P.; Bunce, M.; Marshall, S.E.; Harris, A.L.; Wojnarowska, F.; Welsh, K.I. A variant within the DNA repair gene XRCC3 is associated with the development of melanoma skin cancer. Cancer Res. 2000, 60, 5612–5616. [Google Scholar]
- Ahnesorg, P.; Smith, P.; Jackson, S.P. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis 2010, 31, 955–960. [Google Scholar] [CrossRef] [PubMed]
- da Cunha Colombo Bonadio, R.R.; Fogace, R.N.; Miranda, V.C.; Diz, M. del P.E. Homologous recombination deficiency in ovarian cancer: A review of its epidemiology and management. Clinics 2018, 73, e450s. [Google Scholar] [CrossRef]
- Miller, R.E.; Elyashiv, O.; El-Shakankery, K.H.; Ledermann, J.A. Ovarian Cancer Therapy: Homologous Recombination Deficiency as a Predictive Biomarker of Response to PARP Inhibitors. Oncol. Targets Ther. 2022, 15, 1105–1117. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef]
- Hansen, L.T.; Lundin, C.; Spang-Thomsen, M.; Petersen, L.N.; Helleday, T. The role of RAD51 in etoposide (VP16) resistance in small cell lung cancer. Int. J. Cancer 2003, 105, 472–479. [Google Scholar] [CrossRef]
- Fan, Z.; Luo, H.; Zhou, J.; Wang, F.; Zhang, W.; Wang, J.; Li, S.; Lai, Q.; Xu, Y.; Wang, G.; et al. Checkpoint kinase-1 inhibition and etoposide exhibit a strong synergistic anticancer effect on chronic myeloid leukemia cell line K562 by impairing homologous recombination DNA damage repair. Oncol. Rep. 2020, 44, 2152–2164. [Google Scholar] [CrossRef]
- Murai, J. Targeting DNA repair and replication stress in the treatment of ovarian cancer. Int. J. Clin. Oncol. 2017, 22, 619–628. [Google Scholar] [CrossRef]
- Lheureux, S.; Bowering, V.; Karakasis, K.; Oza, A.M. Safety evaluation of olaparib for treating ovarian cancer. Expert Opin. Drug Saf. 2015, 14, 1305–1316. [Google Scholar] [CrossRef]
- Majsterek, I.; Sliwinski, T.; Poplawski, T.; Pytel, D.; Kowalski, M.; Slupianek, A.; Skorski, T.; Blasiak, J. Imatinib mesylate (STI571) abrogates the resistance to doxorubicin in human K562 chronic myeloid leukemia cells by inhibition of BCR/ABL kinase-mediated DNA repair. Mutat. Res. 2006, 603, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Pastwa, E.; Poplawski, T.; Lewandowska, U.; Somiari, S.B.; Blasiak, J.; Somiari, R.I. Wortmannin potentiates the combined effect of etoposide and cisplatin in human glioma cells. Int. J. Biochem. Cell Biol. 2014, 53, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Kopa, P.; Macieja, A.; Gulbas, I.; Pastwa, E.; Poplawski, T. Inhibition of DNA-PK potentiates the synergistic effect of NK314 and etoposide combination on human glioblastoma cells. Mol. Biol. Rep. 2019, 47, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Macieja, A.; Kopa, P.; Galita, G.; Pastwa, E.; Majsterek, I.; Poplawski, T. Comparison of the effect of three different topoisomerase II inhibitors combined with cisplatin in human glioblastoma cells sensitized with double strand break repair inhibitors. Mol. Biol. Rep. 2019, 46, 3625–3636. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Robey, R.; Bates, S.; Fojo, T. Pretreatment with DNA-damaging agents permits selective killing of checkpoint-deficient cells by microtubule-active drugs. J. Clin. Investig. 2000, 105, 533–539. [Google Scholar] [CrossRef]
- Toulany, M.; Kasten-Pisula, U.; Brammer, I.; Wang, S.; Chen, J.; Dittmann, K.; Baumann, M.; Dikomey, E.; Rodemann, H.P. Blockage of Epidermal Growth Factor Receptor-Phosphatidylinositol 3-Kinase-AKT Signaling Increases Radiosensitivity of K-RAS Mutated Human Tumor Cells In vitro by Affecting DNA Repair. Clin. Cancer Res. 2006, 12, 4119–4126. [Google Scholar] [CrossRef]
- Chen, S.-H.; Yu, X. Targeting dePARylation selectively suppresses DNA repair–defective and PARP inhibitor–resistant malignancies. Sci. Adv. 2019, 5, eaav4340. [Google Scholar] [CrossRef]
- Squatrito, M.; Brennan, C.W.; Helmy, K.; Huse, J.T.; Petrini, J.H.; Holland, E.C. Loss of ATM/Chk2/p53 Pathway Components Accelerates Tumor Development and Contributes to Radiation Resistance in Gliomas. Cancer Cell 2010, 18, 619–629. [Google Scholar] [CrossRef]
- Riffle, S.; Pandey, R.N.; Albert, M.; Hegde, R.S. Linking hypoxia, DNA damage and proliferation in multicellular tumor spheroids. BMC Cancer 2017, 17, 338. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, L.; Lu, A.; Han, Y.; Colangelo, D.; Bukata, C.; Scibetta, A.; Yousefzadeh, M.J.; Li, X.; Gurkar, A.U.; et al. ATM is a key driver of NF-κB-dependent DNA-damage-induced senescence, stem cell dysfunction and aging. Aging 2020, 12, 4688–4710. [Google Scholar] [CrossRef]
- Cam, H.; Easton, J.B.; High, A.; Houghton, P.J. mTORC1 Signaling under Hypoxic Conditions Is Controlled by ATM-Dependent Phosphorylation of HIF-1α. Mol. Cell 2010, 40, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Likhatcheva, M.; Gieling, R.G.; Brown, J.A.L.; Demonacos, C.; Williams, K.J. A Novel Mechanism of Ataxia Telangiectasia Mutated Mediated Regulation of Chromatin Remodeling in Hypoxic Conditions. Front. Cell Dev. Biol. 2021, 9, 720194. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Thomlinson, R.H.; Gray, L.H. The Histological Structure of Some Human Lung Cancers and the Possible Implications for Radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef]
- Vaupel, P.; Harrison, L. Tumor Hypoxia: Causative Factors, Compensatory Mechanisms, and Cellular Response. Oncologist 2004, 9, 4–9. [Google Scholar] [CrossRef]
- Lloyd, M.C.; Cunningham, J.J.; Bui, M.M.; Gillies, R.J.; Brown, J.S.; Gatenby, R.A. Darwinian Dynamics of Intratumoral Heterogeneity: Not Solely Random Mutations but Also Variable Environmental Selection Forces. Cancer Res. 2016, 76, 3136–3144. [Google Scholar] [CrossRef]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]
- Terry, S.; Faouzi Zaarour, R.; Hassan Venkatesh, G.; Francis, A.; El-Sayed, W.; Buart, S.; Bravo, P.; Thiery, J.; Chouaib, S. Role of Hypoxic Stress in Regulating Tumor Immunogenicity, Resistance and Plasticity. Int. J. Mol. Sci. 2018, 19, 3044. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Scheme 1 (ES 1) | ||
|---|---|---|
| Cell line | A2780 | A2780cis |
| ECACC number | 93112519 | 93112517 |
| Description | Cell lines established from previously untreated patients [17]. | Cell line obtained in vitro after treating parent A2780 cells with increasing concentrations of CDDP [14]. |
| Morphology | Epithelial cells | Epithelial cells |
| Growth mode | Adherent | Adherent |
| Doubling time (hours) | 18 | 20 |
| Sensitivity to CDDP | Sensitive | Resistant |
| Experimental Scheme 2 (ES 2) | ||
| Cell line | PEA1 | PEA2 |
| ECACC number | 10032306 | 10032307 |
| Description | PEA1 was collected prior to treatment with CDDP and prednimustine. Human ovarian cancer, estrogen positive [18]. | Cells collected from the same patient as PEA1 after treatment with CDDP and prednimustine [19]. |
| Morphology | Epithelial; swirling pattern of cells | Epithelial, swirling pattern of cells |
| Growth mode | Adherent | Adherent |
| Doubling time (hours) | 37 | 66 |
| Sensitivity to CDDP | Sensitive | Resistant |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| A | ABL1 | APEX1 | ATM | ATR | ATRIP | ATRX | BARD1 | BAX | BBC3 | BLM | BRCA1 | BRIP1 |
| B | CDC25A | CDC25C | CDK7 | CDKN1A | CHEK1 | CHEK2 | CIB1 | CRY1 | CSNK2A2 | DDB1 | DDB2 | DDIT3 |
| C | ERCC1 | ERCC2 | EXO1 | FANCA | FANCD2 | FANCG | FEN1 | GADD45A | GADD45G | H2AFX | HUS1 | LIG1 |
| D | MAPK12 | MBD4 | MCPH1 | MDC1 | MLH1 | MLH3 | MPG | MRE11A | MSH2 | MSH3 | NBN | NTHL1 |
| E | OGG1 | PARP1 | PCNA | PMS1 | PMS2 | PNKP | PPM1D | PPP1R15A | PRKDC | PRKDC | RAD1 | RAD17 |
| F | RAD21 | RAD50 | RAD51 | RAD51B | RAD9A | RBBP8 | REV1 | RNF168 | RNF8 | RPA1 | SIRT1 | SMC1A |
| G | SUMO1 | TOPBP1 | TP53 | TP53BP1 | TP73 | UNG | XPA | XPC | XRCC1 | XRCC2 | XRCC3 | XRCC6 |
| Experimental Scheme | Cell Lines | IC50 (µM)—Normoxia | |
|---|---|---|---|
| CDDP | VP-16 | ||
| ES 1 | A2780 | 13.35 | 34.48 |
| A2780cis | 45.44 | 31.31 | |
| ES 2 | PEA1 | 18.22 | 35.83 |
| PEA2 | 35.96 | 31.27 | |
| PEA1 | PEA2 | ||||
|---|---|---|---|---|---|
| IC50 (µM) | Rf | IC50 (µM) | Rf | ||
| Drugs | CDDP/VP-16 | 9.49 | - | 11.13 | - |
| Drugs + NHEJ inhibitor | CDDP/VP-16/YU238259 | 8.25 | 1.15 | 10.21 | 1.09 |
| Drugs + HR inhibitor | CDDP/VP-16/ A12B4C3 | 5.45 | 1.74 | 5.22 | 2.13 |
| CDDP/VP-16/ DDRI-18 | 4.65 | 2.04 | 4.38 | 2.54 | |
| Drugs Concentration CDDP/VP-16 (µM) | CI (for Drugs) | CI Drugs + YU238259 | CI Drugs + A12B4C3 | CI Drugs + DDRI-18 |
|---|---|---|---|---|
| PEA1 | ||||
| 6.75 (⅛ + ⅛ IC50) | 2.71 | ↓ 0.18 | ↓ 1.97 | ↓ 0.48 |
| 13.5 (¼ + ¼ IC50) | 2.43 | ↓ 0.41 | ↓ 1.65 | ↓ 0.17 |
| 27 (½ + ½ IC50) | 0.15 | ↑ 0.25 | ↓ 0.12 | ↓ 0.14 |
| 54 (1 + 1 IC50) | 0.65 | ↓ 0.18 | ↓ 0.23 | ↓ 0.23 |
| 108 (2 + 2 IC50) | 2.71 | ↓ 0.18 | ↓ 0.57 | ↓ 0.38 |
| PEA2 | ||||
| 8.4 (⅛ + ⅛ IC50) | 2.95 | ↓ 0.77 | ↓ 0.26 | ↓ 0.14 |
| 16.7 (¼ + ¼ IC50) | 1.01 | ↓ 0.56 | ↑ 0.15 | ↓ 0.18 |
| 33.5 (½ + ½ IC50) | 1.05 | ↓ 1.04 | ↓ 0.37 | ↓ 0.26 |
| 67 (1 + 1 IC50) | 0.67 | ↑ 0.96 | ↑ 0.79 | ↓ 0.41 |
| 134 (2 + 2 IC50) | 2.95 | ↓ 0.77 | ↓ 0.26 | ↓ 0.14 |
| Drugs Concentration CDDP/VP-16 (µM) | CI (for Drugs) | CI Drugs + YU238259 | CI Drugs + A12B4C3 | CI Drugs + DDRI-18 |
|---|---|---|---|---|
| PEA1 | ||||
| 9.49 (¼ + ¼ IC50) | 2.95 | ↓ 0.77 | ↓ 1.26 | ↑ 2.14 |
| 19 (½ + ½ IC50) | 1.01 | ↑ 2.56 | ↑ 1.15 | ↑ 1.18 |
| 38 (1 + 1 IC50) | 1.05 | ↑ 1.24 | ↑ 2.37 | ↑ 1.26 |
| 76 (2 + 2 IC50) | 0.67 | ↑ 0.96 | ↑ 0.79 | ↓ 0.41 |
| PEA2 | ||||
| 11.3 (¼ + ¼ IC50) | 1.14 | ↑ 2.48 | ↑ 2.46 | ↑ 2.44 |
| 22.5 (½ + ½ IC50) | 1.78 | ↑ 2.54 | ↓ 0.96 | ↑ 1.98 |
| 45 (1 + 1 IC50) | 1.16 | ↑ 1.94 | ↑ 2.21 | ↑ 2.58 |
| 90 (2 + 2 IC50) | 2.95 | ↑ 4.43 | ↑ 6.26 | ↑ 7.38 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| A | ABL1 | APEX1 | ATM | ATR | ATRIP | ATRX | BARD1 | BAX | BBC3 | BLM | BRCA1 | BRIP1 |
| B | CDC25A | CDC25C | CDK7 | CDKN1A | CHEK1 | CHEK2 | CIB1 | CRY1 | CSNK2A2 | DDB1 | DDB2 | DDIT3 |
| C | ERCC1 | ERCC2 | EXO1 | FANCA | FANCD2 | FANCG | FEN1 | GADD45A | GADD45G | H2AFX | HUS1 | LIG1 |
| D | MAPK12 | MBD4 | MCPH1 | MDC1 | MLH1 | MLH3 | MPG | MRE11A | MSH2 | MSH3 | NBN | NTHL1 |
| E | OGG1 | PARP1 | PCNA | PMS1 | PMS2 | PNKP | PPM1D | PPP1R15A | PRKDC | PRKDC | RAD1 | RAD17 |
| F | RAD21 | RAD50 | RAD51 | RAD51B | RAD9A | RBBP8 | REV1 | RNF168 | RNF8 | RPA1 | SIRT1 | SMC1A |
| G | SUMO1 | TOPBP1 | TP53 | TP53BP1 | TP73 | UNG | XPA | XPC | XRCC1 | XRCC2 | XRCC3 | XRCC6 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| A | ABL1 | APEX1 | ATM | ATR | ATRIP | ATRX | BARD1 | BAX | BBC3 | BLM | BRCA1 | BRIP1 |
| B | CDC25A | CDC25C | CDK7 | CDKN1A | CHEK1 | CHEK2 | CIB1 | CRY1 | CSNK2A2 | DDB1 | DDB2 | DDIT3 |
| C | ERCC1 | ERCC2 | EXO1 | FANCA | FANCD2 | FANCG | FEN1 | GADD45A | GADD45G | H2AFX | HUS1 | LIG1 |
| D | MAPK12 | MBD4 | MCPH1 | MDC1 | MLH1 | MLH3 | MPG | MRE11A | MSH2 | MSH3 | NBN | NTHL1 |
| E | OGG1 | PARP1 | PCNA | PMS1 | PMS2 | PNKP | PPM1D | PPP1R15A | PRKDC | PRKDC | RAD1 | RAD17 |
| F | RAD21 | RAD50 | RAD51 | RAD51B | RAD9A | RBBP8 | REV1 | RNF168 | RNF8 | RPA1 | SIRT1 | SMC1A |
| G | SUMO1 | TOPBP1 | TP53 | TP53BP1 | TP73 | UNG | XPA | XPC | XRCC1 | XRCC2 | XRCC3 | XRCC6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macieja, A.; Gulbas, I.; Popławski, T. DNA Double-Strand Break Repair Inhibitors: YU238259, A12B4C3 and DDRI-18 Overcome the Cisplatin Resistance in Human Ovarian Cancer Cells, but Not under Hypoxia Conditions. Curr. Issues Mol. Biol. 2023, 45, 7915-7932. https://doi.org/10.3390/cimb45100500
Macieja A, Gulbas I, Popławski T. DNA Double-Strand Break Repair Inhibitors: YU238259, A12B4C3 and DDRI-18 Overcome the Cisplatin Resistance in Human Ovarian Cancer Cells, but Not under Hypoxia Conditions. Current Issues in Molecular Biology. 2023; 45(10):7915-7932. https://doi.org/10.3390/cimb45100500
Chicago/Turabian StyleMacieja, Anna, Izabela Gulbas, and Tomasz Popławski. 2023. "DNA Double-Strand Break Repair Inhibitors: YU238259, A12B4C3 and DDRI-18 Overcome the Cisplatin Resistance in Human Ovarian Cancer Cells, but Not under Hypoxia Conditions" Current Issues in Molecular Biology 45, no. 10: 7915-7932. https://doi.org/10.3390/cimb45100500
APA StyleMacieja, A., Gulbas, I., & Popławski, T. (2023). DNA Double-Strand Break Repair Inhibitors: YU238259, A12B4C3 and DDRI-18 Overcome the Cisplatin Resistance in Human Ovarian Cancer Cells, but Not under Hypoxia Conditions. Current Issues in Molecular Biology, 45(10), 7915-7932. https://doi.org/10.3390/cimb45100500