The Roles of Caloric Restriction Mimetics in Central Nervous System Demyelination and Remyelination


Abstract
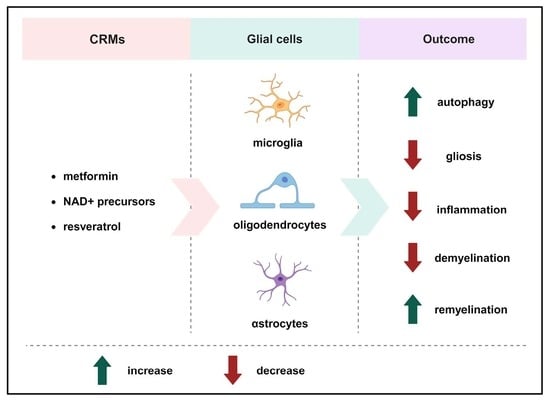
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
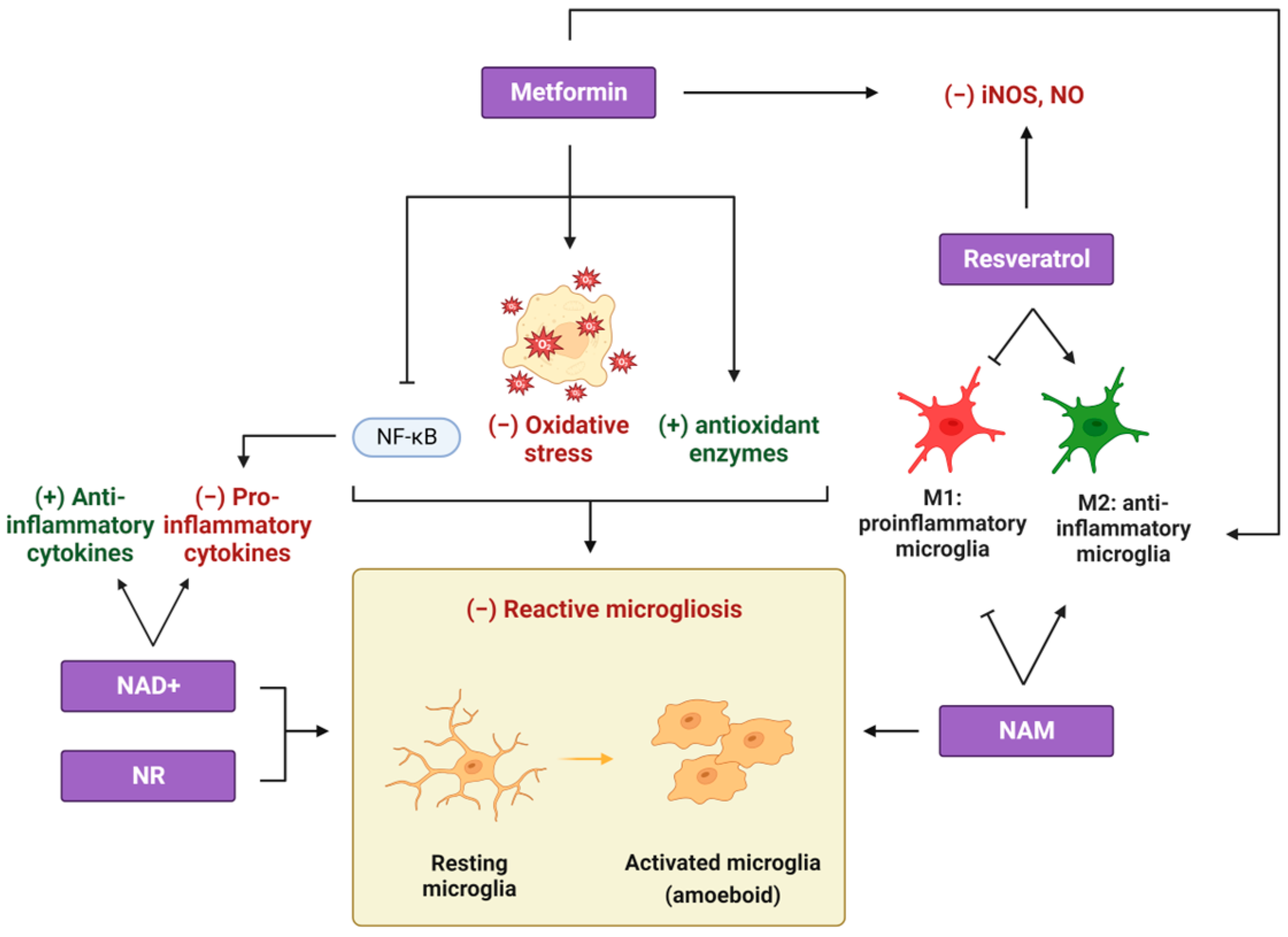
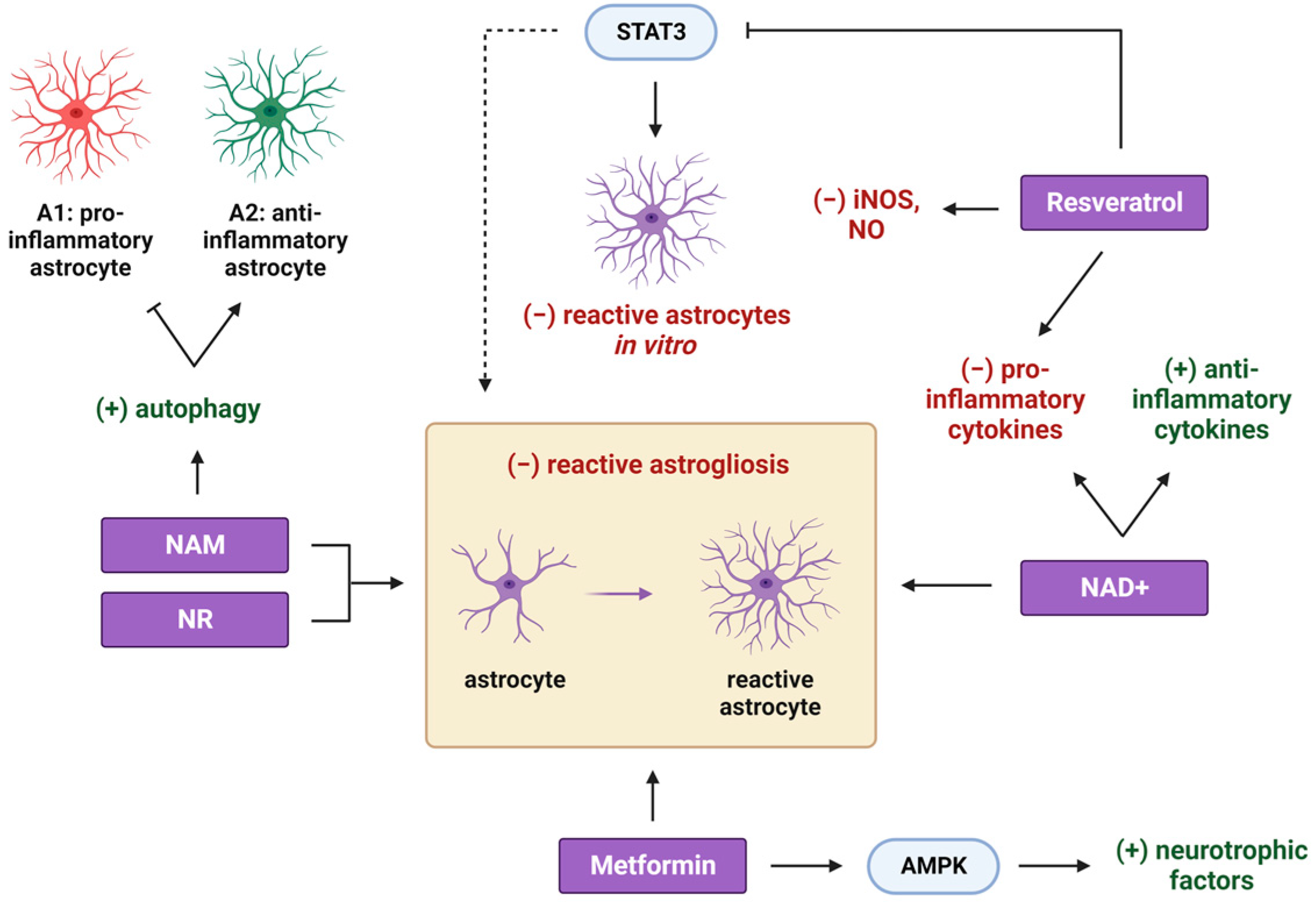
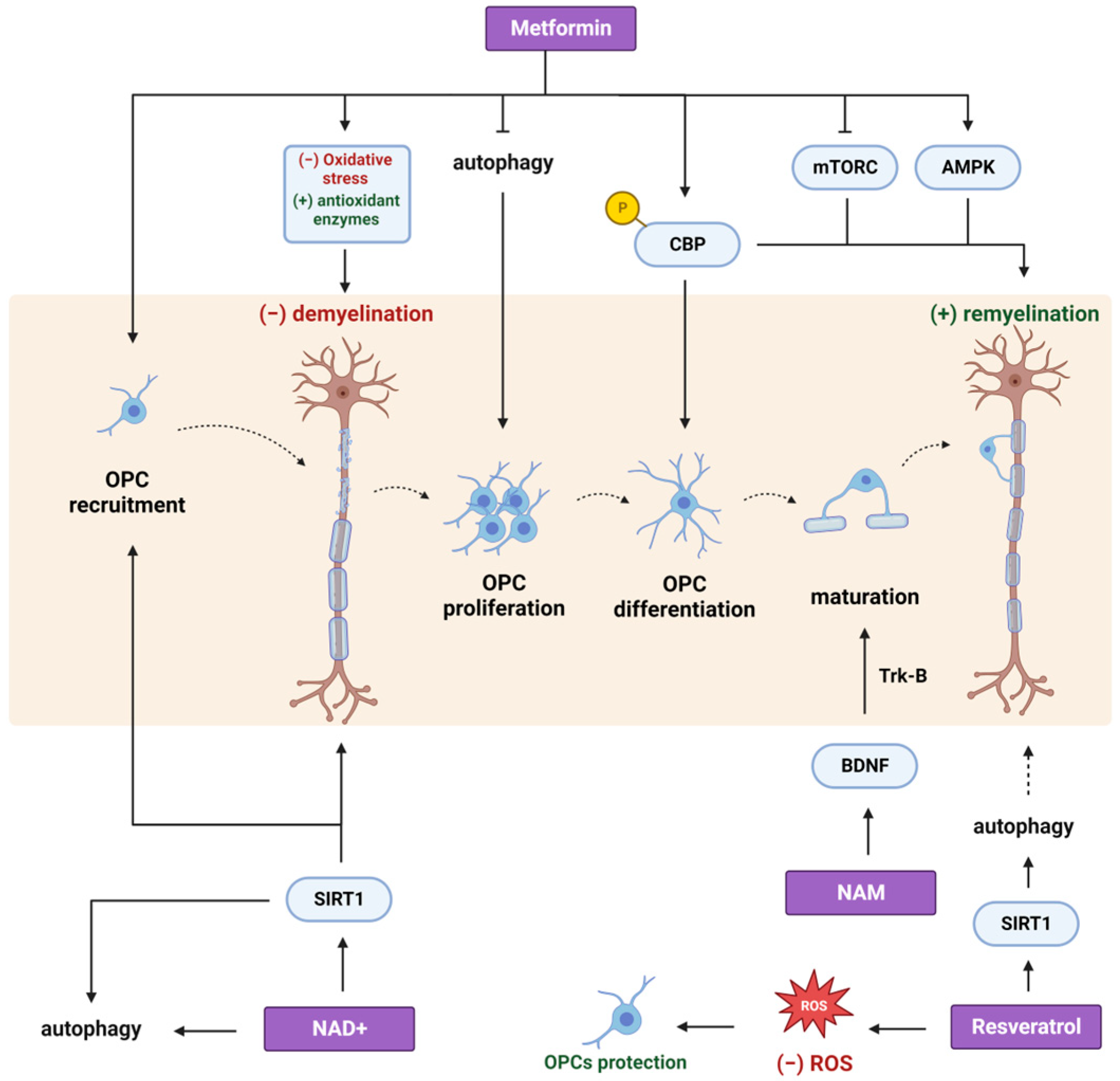
2. Metformin
3. NAD+ Precursors
4. Resveratrol
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, K.A.; Deber, C.M. The structure and function of central nervous system myelin. Crit. Rev. Clin. Lab. Sci. 1993, 30, 29–64. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Poliak, S.; Peles, E. The local differentiation of myelinated axons at nodes of Ranvier. Nat. Rev. Neurosci. 2003, 4, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Wang, F.; Huang, N.X.; Xiao, L.; Mei, F. Oligodendrocytes and myelin: Active players in neurodegenerative brains? Dev. Neurobiol. 2022, 82, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Neely, S.A.; Williamson, J.M.; Klingseisen, A.; Zoupi, L.; Early, J.J.; Williams, A.; Lyons, D.A. New oligodendrocytes exhibit more abundant and accurate myelin regeneration than those that survive demyelination. Nat. Neurosci. 2022, 25, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Baaklini, C.S.; Rawji, K.S.; Duncan, G.J.; Ho, M.F.S.; Plemel, J.R. Central Nervous System Remyelination: Roles of Glia and Innate Immune Cells. Front. Mol. Neurosci. 2019, 12, 225. [Google Scholar] [CrossRef]
- Djannatian, M.; Radha, S.; Weikert, U.; Safaiyan, S.; Wrede, C.; Deichsel, C.; Kislinger, G.; Rhomberg, A.; Ruhwedel, T.; Campbell, D.S.; et al. Myelination generates aberrant ultrastructure that is resolved by microglia. J. Cell Biol. 2023, 222, e202204010. [Google Scholar] [CrossRef]
- Nave, K.A.; Werner, H.B. Myelination of the nervous system: Mechanisms and functions. Annu. Rev. Cell Dev. Biol. 2014, 30, 503–533. [Google Scholar] [CrossRef]
- Yamasaki, R.; Lu, H.; Butovsky, O.; Ohno, N.; Rietsch, A.M.; Cialic, R.; Wu, P.M.; Doykan, C.E.; Lin, J.; Cotleur, A.C.; et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 2014, 211, 1533–1549. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Miron, V.E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 2019, 15, 447–458. [Google Scholar] [CrossRef]
- Miron, V.E. Microglia-driven regulation of oligodendrocyte lineage cells, myelination, and remyelination. J. Leukoc. Biol. 2017, 101, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.M.; Sánchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, S.; Yong, V.W. The extracellular matrix as modifier of neuroinflammation and remyelination in multiple sclerosis. Brain A J. Neurol. 2021, 144, 1958–1973. [Google Scholar] [CrossRef] [PubMed]
- Kalafatakis, I.; Karagogeos, D. Oligodendrocytes and Microglia: Key Players in Myelin Development, Damage and Repair. Biomolecules 2021, 11, 1058. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Skripuletz, T.; Hackstette, D.; Bauer, K.; Gudi, V.; Pul, R.; Voss, E.; Berger, K.; Kipp, M.; Baumgärtner, W.; Stangel, M. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain A J. Neurol. 2013, 136, 147–167. [Google Scholar] [CrossRef]
- Pu, A.; Stephenson, E.L.; Yong, V.W. The extracellular matrix: Focus on oligodendrocyte biology and targeting CSPGs for remyelination therapies. Glia 2018, 66, 1809–1825. [Google Scholar] [CrossRef]
- Bankston, A.N.; Forston, M.D.; Howard, R.M.; Andres, K.R.; Smith, A.E.; Ohri, S.S.; Bates, M.L.; Bunge, M.B.; Whittemore, S.R. Autophagy is essential for oligodendrocyte differentiation, survival, and proper myelination. Glia 2019, 67, 1745–1759. [Google Scholar] [CrossRef]
- Aber, E.R.; Griffey, C.J.; Davies, T.; Li, A.M.; Yang, Y.J.; Croce, K.R.; Goldman, J.E.; Grutzendler, J.; Canman, J.C.; Yamamoto, A. Oligodendroglial macroautophagy is essential for myelin sheath turnover to prevent neurodegeneration and death. Cell Rep. 2022, 41, 111480. [Google Scholar] [CrossRef]
- Ktena, N.; Kaplanis, S.I.; Kolotuev, I.; Georgilis, A.; Kallergi, E.; Stavroulaki, V.; Nikoletopoulou, V.; Savvaki, M.; Karagogeos, D. Autophagic degradation of CNS myelin maintains axon integrity. Cell Stress 2022, 6, 93–107. [Google Scholar] [CrossRef]
- Piccio, L.; Stark, J.L.; Cross, A.H. Chronic calorie restriction attenuates experimental autoimmune encephalomyelitis. J. Leukoc. Biol. 2008, 84, 940–948. [Google Scholar] [CrossRef]
- Mojaverrostami, S.; Pasbakhsh, P.; Madadi, S.; Nekoonam, S.; Zarini, D.; Noori, L.; Shiri, E.; Salama, M.; Zibara, K.; Kashani, I.R. Calorie restriction promotes remyelination in a Cuprizone-Induced demyelination mouse model of multiple sclerosis. Metab. Brain Dis. 2020, 35, 1211–1224. [Google Scholar] [CrossRef]
- Masoro, E.J. Overview of caloric restriction and ageing. Mech. Ageing Dev. 2005, 126, 913–922. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef]
- Mariño, G.; Pietrocola, F.; Eisenberg, T.; Kong, Y.; Malik, S.A.; Andryushkova, A.; Schroeder, S.; Pendl, T.; Harger, A.; Niso-Santano, M.; et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell 2014, 53, 710–725. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Moon, S.; Hur, S.C.; Jeong, S.M. Fatty acid oxidation regulates cellular senescence by modulating the autophagy-SIRT1 axis. BMB Rep. 2023, 5938. [Google Scholar] [CrossRef]
- Lv, S.; Zhang, Y.; Lin, Y.; Fang, W.; Wang, Y.; Li, Z.; Lin, A.; Dai, X.; Ye, Q.; Zhang, J.; et al. ApoE4 exacerbates the senescence of hippocampal neurons and spatial cognitive impairment by downregulating acetyl-CoA level. Aging Cell 2023, 22, e13932. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, Y. Targeting Autophagy in Aging and Aging-Related Cardiovascular Diseases. Trends Pharmacol. Sci. 2018, 39, 1064–1076. [Google Scholar] [CrossRef]
- Bosi, E. Metformin--the gold standard in type 2 diabetes: What does the evidence tell us? Diabetes Obes. Metab. 2009, 11 (Suppl. S2), 3–8. [Google Scholar] [CrossRef]
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Kroemer, G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab. 2019, 29, 592–610. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef]
- Wang, J.C.; Li, G.Y.; Wang, B.; Han, S.X.; Sun, X.; Jiang, Y.N.; Shen, Y.W.; Zhou, C.; Feng, J.; Lu, S.Y.; et al. Metformin inhibits metastatic breast cancer progression and improves chemosensitivity by inducing vessel normalization via PDGF-B downregulation. J. Exp. Clin. Cancer Res. 2019, 38, 235. [Google Scholar] [CrossRef]
- Sugiura, K.; Okabayashi, K.; Seishima, R.; Ishida, T.; Shigeta, K.; Tsuruta, M.; Hasegawa, H.; Kitagawa, Y. Metformin inhibits the development and metastasis of colorectal cancer. Med. Oncol. 2022, 39, 136. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, L.; Shi, X.; Yang, L.; Hua, F.; Ma, J.; Zhu, W.; Liu, X.; Xuan, R.; Shen, Y.; et al. Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging 2020, 12, 24270–24287. [Google Scholar] [CrossRef]
- Yerevanian, A.; Soukas, A.A. Metformin: Mechanisms in Human Obesity and Weight Loss. Curr. Obes. Rep. 2019, 8, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Du, M.R.; Gao, Q.Y.; Liu, C.L.; Bai, L.Y.; Li, T.; Wei, F.L. Exploring the Pharmacological Potential of Metformin for Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 838173. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Gan, D.; Lin, S.; Zhong, Y.; Chen, M.; Zou, X.; Shao, Z.; Xiao, G. Metformin in aging and aging-related diseases: Clinical applications and relevant mechanisms. Theranostics 2022, 12, 2722–2740. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Guo, Y. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Duca, F.A.; Côté, C.D.; Rasmussen, B.A.; Zadeh-Tahmasebi, M.; Rutter, G.A.; Filippi, B.M.; Lam, T.K. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 2015, 21, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Oh, M.A.; Kim, W.H.; Sohn, H.Y.; Park, S.I. AMP-activated protein kinase inhibits TGF-β-induced fibrogenic responses of hepatic stellate cells by targeting transcriptional coactivator p300. J. Cell. Physiol. 2012, 227, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Caton, P.W.; Nayuni, N.K.; Kieswich, J.; Khan, N.Q.; Yaqoob, M.M.; Corder, R. Metformin suppresses hepatic gluconeogenesis through induction of SIRT1 and GCN5. J. Endocrinol. 2010, 205, 97–106. [Google Scholar] [CrossRef]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y. AMPK and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 85–108. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, J.; Guo, F.L.; Gao, X.; Xie, X.; Liu, S.; Yang, X.; Yang, X.; Zhang, L.; Ye, Y.; et al. Metformin Ameliorates Synaptic Defects in a Mouse Model of AD by Inhibiting Cdk5 Activity. Front. Cell. Neurosci. 2020, 14, 170. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, S.; Fan, Z.; Li, Z.; Zhu, Y.; Shen, T.; Li, K.; Yan, Y.; Tian, J.; Liu, Z.; et al. Metformin attenuates plaque-associated tau pathology and reduces amyloid-β burden in APP/PS1 mice. Alzheimers Res. Ther. 2021, 13, 40. [Google Scholar] [CrossRef]
- Saewanee, N.; Praputpittaya, T.; Malaiwong, N.; Chalorak, P.; Meemon, K. Neuroprotective effect of metformin on dopaminergic neurodegeneration and α-synuclein aggregation in C. elegans model of Parkinson’s disease. Neurosci. Res. 2019, 162, 13–21. [Google Scholar] [CrossRef]
- Sanchis, A.; García-Gimeno, M.A.; Cañada-Martínez, A.J.; Sequedo, M.D.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zQ175 mouse model of Huntington disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Samaras, K.; Makkar, S.; Crawford, J.D.; Kochan, N.A.; Wen, W.; Draper, B.; Trollor, J.N.; Brodaty, H.; Sachdev, P.S. Metformin Use Is Associated With Slowed Cognitive Decline and Reduced Incident Dementia in Older Adults With Type 2 Diabetes: The Sydney Memory and Ageing Study. Diabetes Care 2020, 43, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the Insulin Sensitizer Metformin in Alzheimer Disease: Pilot Data From a Randomized Placebo-controlled Crossover Study. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef]
- Moore, E.M.; Mander, A.G.; Ames, D.; Kotowicz, M.A.; Carne, R.P.; Brodaty, H.; Woodward, M.; Boundy, K.; Ellis, K.A.; Bush, A.I.; et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care 2013, 36, 2981–2987. [Google Scholar] [CrossRef]
- Dziedzic, A.; Saluk-Bijak, J.; Miller, E.; Bijak, M. Metformin as a Potential Agent in the Treatment of Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 5957. [Google Scholar] [CrossRef]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Largani, S.H.H.; Borhani-Haghighi, M.; Pasbakhsh, P.; Mahabadi, V.P.; Nekoonam, S.; Shiri, E.; Kashani, I.R.; Zendehdel, A. Oligoprotective effect of metformin through the AMPK-dependent on restoration of mitochondrial hemostasis in the cuprizone-induced multiple sclerosis model. J. Mol. Histol. 2019, 50, 263–271. [Google Scholar] [CrossRef]
- Abdi, M.; Pasbakhsh, P.; Shabani, M.; Nekoonam, S.; Sadeghi, A.; Fathi, F.; Abouzaripour, M.; Mohamed, W.; Zibara, K.; Kashani, I.R.; et al. Metformin Therapy Attenuates Pro-inflammatory Microglia by Inhibiting NF-κB in Cuprizone Demyelinating Mouse Model of Multiple Sclerosis. Neurotox. Res. 2021, 39, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, Y.; Itoh, K. Microglial inflammatory reactions regulated by oxidative stress. J. Clin. Biochem. Nutr. 2023, 72, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Nahirnyj, A.; Livne-Bar, I.; Guo, X.; Sivak, J.M. ROS detoxification and proinflammatory cytokines are linked by p38 MAPK signaling in a model of mature astrocyte activation. PLoS ONE 2013, 8, e83049. [Google Scholar] [CrossRef] [PubMed]
- Nath, N.; Khan, M.; Paintlia, M.K.; Singh, I.; Hoda, M.N.; Giri, S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J. Immunol. 2009, 182, 8005–8014. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Q.; Xiong, J.; He, Z.L.; Yuan, Y.; Wang, B.N.; Xu, J.Y.; Wu, M.; Zhang, S.S.; Cai, S.F.; Zhao, J.X.; et al. Metformin promotes microglial cells to facilitate myelin debris clearance and accelerate nerve repairment after spinal cord injury. Acta Pharmacol. Sin. 2022, 43, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Sanadgol, N.; Barati, M.; Houshmand, F.; Hassani, S.; Clarner, T.; Shahlaei, M.; Golab, F. Metformin accelerates myelin recovery and ameliorates behavioral deficits in the animal model of multiple sclerosis via adjustment of AMPK/Nrf2/mTOR signaling and maintenance of endogenous oligodendrogenesis during brain self-repairing period. Pharmacol. Rep. 2020, 72, 641–658. [Google Scholar] [CrossRef] [PubMed]
- Bercury, K.K.; Dai, J.; Sachs, H.H.; Ahrendsen, J.T.; Wood, T.L.; Macklin, W.B. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J. Neurosci. 2014, 34, 4466–4480. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Furusho, M.; Macklin, W.; Bansal, R. Independent and cooperative roles of the Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways during developmental myelination and in adulthood. Glia 2019, 67, 1277–1295. [Google Scholar] [CrossRef]
- Carson, R.P.; Van Nielen, D.L.; Winzenburger, P.A.; Ess, K.C. Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol. Dis. 2012, 45, 369–380. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.E.; Bourne, J.N.; Evangelou, A.V.; Khandker, L.; Macklin, W.B.; Wood, T.L. Loss of Tuberous Sclerosis Complex1 in Adult Oligodendrocyte Progenitor Cells Enhances Axon Remyelination and Increases Myelin Thickness after a Focal Demyelination. J. Neurosci. 2017, 37, 7534–7546. [Google Scholar] [CrossRef]
- Manwani, B.; McCullough, L.D. Function of the master energy regulator adenosine monophosphate-activated protein kinase in stroke. J. Neurosci. Res. 2013, 91, 1018–1029. [Google Scholar] [CrossRef]
- Narine, M.; Azmi, M.A.; Umali, M.; Volz, A.; Colognato, H. The AMPK activator metformin improves recovery from demyelination by shifting oligodendrocyte bioenergetics and accelerating OPC differentiation. Front. Cell. Neurosci. 2023, 17, 1254303. [Google Scholar] [CrossRef]
- Paintlia, A.S.; Paintlia, M.K.; Mohan, S.; Singh, A.K.; Singh, I. AMP-activated protein kinase signaling protects oligodendrocytes that restore central nervous system functions in an experimental autoimmune encephalomyelitis model. Am. J. Pathol. 2013, 183, 526–541. [Google Scholar] [CrossRef]
- Houshmand, F.; Barati, M.; Golab, F.; Ramezani-Sefidar, S.; Tanbakooie, S.; Tabatabaei, M.; Amiri, M.; Sanadgol, N. Metformin-induced AMPK activation stimulates remyelination through induction of neurotrophic factors, downregulation of NogoA and recruitment of Olig2+ precursor cells in the cuprizone murine model of multiple sclerosis. Daru 2019, 27, 583–592. [Google Scholar] [CrossRef]
- Pöyhönen, S.; Er, S.; Domanskyi, A.; Airavaara, M. Effects of Neurotrophic Factors in Glial Cells in the Central Nervous System: Expression and Properties in Neurodegeneration and Injury. Front. Physiol. 2019, 10, 486. [Google Scholar] [CrossRef]
- Kosaraju, J.; Seegobin, M.; Gouveia, A.; Syal, C.; Sarma, S.N.; Lu, K.J.; Ilin, J.; He, L.; Wondisford, F.E.; Lagace, D.; et al. Metformin promotes CNS remyelination and improves social interaction following focal demyelination through CBP Ser436 phosphorylation. Exp. Neurol. 2020, 334, 113454. [Google Scholar] [CrossRef]
- Franklin, R.J.; Ffrench-Constant, C.; Edgar, J.M.; Smith, K.J. Neuroprotection and repair in multiple sclerosis. Nat. Rev. Neurol. 2012, 8, 624–634. [Google Scholar] [CrossRef]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J. Neurosci. 2002, 22, 2451–2459. [Google Scholar] [CrossRef]
- Neumann, B.; Baror, R.; Zhao, C.; Segel, M.; Dietmann, S.; Rawji, K.S.; Foerster, S.; McClain, C.R.; Chalut, K.; van Wijngaarden, P.; et al. Metformin Restores CNS Remyelination Capacity by Rejuvenating Aged Stem Cells. Cell Stem Cell 2019, 25, 473–485.e8. [Google Scholar] [CrossRef]
- Najafi, M.; Cheki, M.; Rezapoor, S.; Geraily, G.; Motevaseli, E.; Carnovale, C.; Clementi, E.; Shirazi, A. Metformin: Prevention of genomic instability and cancer: A review. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018, 827, 1–8. [Google Scholar] [CrossRef]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e6. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef]
- Hofer, S.J.; Davinelli, S.; Bergmann, M.; Scapagnini, G.; Madeo, F. Caloric Restriction Mimetics in Nutrition and Clinical Trials. Front. Nutr. 2021, 8, 717343. [Google Scholar] [CrossRef]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD+ Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Mitchell, S.J.; Bernier, M.; Aon, M.A.; Cortassa, S.; Kim, E.Y.; Fang, E.F.; Palacios, H.H.; Ali, A.; Navas-Enamorado, I.; Di Francesco, A.; et al. Nicotinamide Improves Aspects of Healthspan, but Not Lifespan, in Mice. Cell Metab. 2018, 27, 667–676.e4. [Google Scholar] [CrossRef]
- Abdellatif, M.; Trummer-Herbst, V.; Koser, F.; Durand, S.; Adão, R.; Vasques-Nóvoa, F.; Freundt, J.K.; Voglhuber, J.; Pricolo, M.R.; Kasa, M.; et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci. Transl. Med. 2021, 13, eabd7064. [Google Scholar] [CrossRef]
- Wu, K.; Li, B.; Lin, Q.; Xu, W.; Zuo, W.; Li, J.; Liu, N.; Tu, T.; Zhang, B.; Xiao, Y.; et al. Nicotinamide mononucleotide attenuates isoproterenol-induced cardiac fibrosis by regulating oxidative stress and Smad3 acetylation. Life Sci. 2021, 274, 119299. [Google Scholar] [CrossRef]
- Scatozza, F.; Moschella, F.; D’Arcangelo, D.; Rossi, S.; Tabolacci, C.; Giampietri, C.; Proietti, E.; Facchiano, F.; Facchiano, A. Nicotinamide inhibits melanoma in vitro and in vivo. J. Exp. Clin. Cancer Res. 2020, 39, 211. [Google Scholar] [CrossRef]
- Jung, M.; Lee, K.M.; Im, Y.; Seok, S.H.; Chung, H.; Kim, D.Y.; Han, D.; Lee, C.H.; Hwang, E.H.; Park, S.Y.; et al. Nicotinamide (niacin) supplement increases lipid metabolism and ROS-induced energy disruption in triple-negative breast cancer: Potential for drug repositioning as an anti-tumor agent. Mol. Oncol. 2022, 16, 1795–1815. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD+ in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.T.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Langley, M.R.; Choi, C.I.; Peclat, T.R.; Guo, Y.; Simon, W.L.; Yoon, H.; Kleppe, L.; Lucchinetti, C.F.; Chini, C.C.S.; Chini, E.N.; et al. Critical Role of Astrocyte NAD+ Glycohydrolase in Myelin Injury and Regeneration. J. Neurosci. 2021, 41, 8644–8667. [Google Scholar] [CrossRef] [PubMed]
- Roboon, J.; Hattori, T.; Ishii, H.; Takarada-Iemata, M.; Le, T.M.; Shiraishi, Y.; Ozaki, N.; Yamamoto, Y.; Sugawara, A.; Okamoto, H.; et al. Deletion of CD38 Suppresses Glial Activation and Neuroinflammation in a Mouse Model of Demyelination. Front. Cell. Neurosci. 2019, 13, 258. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef]
- Veto, S.; Acs, P.; Bauer, J.; Lassmann, H.; Berente, Z.; Setalo, G., Jr.; Borgulya, G.; Sumegi, B.; Komoly, S.; Gallyas, F., Jr.; et al. Inhibiting poly(ADP-ribose) polymerase: A potential therapy against oligodendrocyte death. Brain A J. Neurol. 2010, 133, 822–834. [Google Scholar] [CrossRef]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Koya, D. Chapter 3—Role of Sirt1 as a Regulator of Autophagy. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: San Diego, CA, USA, 2016; pp. 89–100. [Google Scholar] [CrossRef]
- Li, Y.; Yang, G.; Yang, X.; He, Y.; Wang, W.; Zhang, J.; Li, T.; Zhang, W.; Lin, R. Nicotinic acid inhibits vascular inflammation via the SIRT1-dependent signaling pathway. J. Nutr. Biochem. 2015, 26, 1338–1347. [Google Scholar] [CrossRef]
- Li, Y.; Yang, G.; Yang, X.; Wang, W.; Zhang, J.; He, Y.; Zhang, W.; Jing, T.; Lin, R. Nicotinic acid inhibits NLRP3 inflammasome activation via SIRT1 in vascular endothelial cells. Int. Immunopharmacol. 2016, 40, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, C.; Ying, W. SIRT2 and Akt mediate NAD+-induced and NADH-induced increases in the intracellular ATP levels of BV2 microglia under basal conditions. Neuroreport 2018, 29, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Acklin, S.; Sadhukhan, R.; Du, W.; Patra, M.; Cholia, R.; Xia, F. Nicotinamide riboside alleviates cisplatin-induced peripheral neuropathy via SIRT2 activation. Neurooncol. Adv. 2022, 4, vdac101. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kong, M.; Wang, Y.; Mao, Y.; Xu, H.; He, W.; He, Y.; Gu, J. Nicotinamide mononucleotide improves the Alzheimer’s disease by regulating intestinal microbiota. Biochem. Biophys. Res. Commun. 2023, 670, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Rehman, I.U.; Khan, A.; Ahmad, R.; Choe, K.; Park, H.Y.; Lee, H.J.; Atiq, A.; Park, J.; Hahm, J.R.; Kim, M.O. Neuroprotective Effects of Nicotinamide against MPTP-Induced Parkinson’s Disease in Mice: Impact on Oxidative Stress, Neuroinflammation, Nrf2/HO-1 and TLR4 Signaling Pathways. Biomedicines 2022, 10, 2929. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, A.; Diwan, V.; Kaur, H.; Bhateja, D.; Singh, C.K.; Sharma, S.; Padi, S.S.V. Nicotinamide reverses behavioral impairments and provides neuroprotection in 3-nitropropionic acid induced animal model ofHuntington’s disease: Implication of oxidative stress- poly(ADP- ribose) polymerase pathway. Metab. Brain Dis. 2018, 33, 1911–1921. [Google Scholar] [CrossRef]
- Trammell, S.A.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Vreones, M.; Mustapic, M.; Moaddel, R.; Pucha, K.A.; Lovett, J.; Seals, D.R.; Kapogiannis, D.; Martens, C.R. Oral nicotinamide riboside raises NAD+ and lowers biomarkers of neurodegenerative pathology in plasma extracellular vesicles enriched for neuronal origin. Aging Cell 2023, 22, e13754. [Google Scholar] [CrossRef]
- Brakedal, B.; Dölle, C.; Riemer, F.; Ma, Y.; Nido, G.S.; Skeie, G.O.; Craven, A.R.; Schwarzlmüller, T.; Brekke, N.; Diab, J.; et al. The NADPARK study: A randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 2022, 34, 396–407.e6. [Google Scholar] [CrossRef] [PubMed]
- Nicotinamide as an Early Alzheimer’s Disease Treatment. Available online: https://clinicaltrials.gov/study/NCT03061474 (accessed on 8 November 2023).
- Wang, X.; Li, B.; Liu, L.; Zhang, L.; Ma, T.; Guo, L. Nicotinamide adenine dinucleotide treatment alleviates the symptoms of experimental autoimmune encephalomyelitis by activating autophagy and inhibiting the NLRP3 inflammasome. Int. Immunopharmacol. 2021, 90, 107092. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Li, B.; Wang, J.; Guo, R.; Tian, Y.; Song, S.; Guo, L. Protective effect and mechanism of nicotinamide adenine dinucleotide against optic neuritis in mice with experimental autoimmune encephalomyelitis. Int. Immunopharmacol. 2021, 98, 107846. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Kaplanis, S.I.; Kaffe, D.; Ktena, N.; Lygeraki, A.; Kolliniati, O.; Savvaki, M.; Karagogeos, D. Nicotinamide enhances myelin production after demyelination through reduction of astrogliosis and microgliosis. Front. Cell. Neurosci. 2023, 17, 1201317. [Google Scholar] [CrossRef] [PubMed]
- Roboon, J.; Hattori, T.; Ishii, H.; Takarada-Iemata, M.; Nguyen, D.T.; Heer, C.D.; O’Meally, D.; Brenner, C.; Yamamoto, Y.; Okamoto, H.; et al. Inhibition of CD38 and supplementation of nicotinamide riboside ameliorate lipopolysaccharide-induced microglial and astrocytic neuroinflammation by increasing NAD+. J. Neurochem. 2021, 158, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Liu, Y.; Zhang, G.; Wu, H.; Hou, Y. Progesterone suppresses Aβ(42)-induced neuroinflammation by enhancing autophagy in astrocytes. Int. Immunopharmacol. 2018, 54, 336–343. [Google Scholar] [CrossRef]
- Pais, T.F.; Szegő, É.M.; Marques, O.; Miller-Fleming, L.; Antas, P.; Guerreiro, P.; de Oliveira, R.M.; Kasapoglu, B.; Outeiro, T.F. The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J. 2013, 32, 2603–2616. [Google Scholar] [CrossRef]
- Li, W.; Zhang, B.; Tang, J.; Cao, Q.; Wu, Y.; Wu, C.; Guo, J.; Ling, E.A.; Liang, F. Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J. Neurosci. 2007, 27, 2606–2616. [Google Scholar] [CrossRef]
- Jayasena, T.; Poljak, A.; Braidy, N.; Zhong, L.; Rowlands, B.; Muenchhoff, J.; Grant, R.; Smythe, G.; Teo, C.; Raftery, M.; et al. Application of Targeted Mass Spectrometry for the Quantification of Sirtuins in the Central Nervous System. Sci. Rep. 2016, 6, 35391. [Google Scholar] [CrossRef]
- Ma, X.R.; Zhu, X.; Xiao, Y.; Gu, H.M.; Zheng, S.S.; Li, L.; Wang, F.; Dong, Z.J.; Wang, D.X.; Wu, Y.; et al. Restoring nuclear entry of Sirtuin 2 in oligodendrocyte progenitor cells promotes remyelination during ageing. Nat. Commun. 2022, 13, 1225. [Google Scholar] [CrossRef] [PubMed]
- Rawji, K.S.; Young, A.M.H.; Ghosh, T.; Michaels, N.J.; Mirzaei, R.; Kappen, J.; Kolehmainen, K.L.; Alaeiilkhchi, N.; Lozinski, B.; Mishra, M.K.; et al. Niacin-mediated rejuvenation of macrophage/microglia enhances remyelination of the aging central nervous system. Acta Neuropathol. 2020, 139, 893–909. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Li, W.W.; Franklin, R.J. Differences in the early inflammatory responses to toxin-induced demyelination are associated with the age-related decline in CNS remyelination. Neurobiol. Aging 2006, 27, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Natrajan, M.S.; de la Fuente, A.G.; Crawford, A.H.; Linehan, E.; Nuñez, V.; Johnson, K.R.; Wu, T.; Fitzgerald, D.C.; Ricote, M.; Bielekova, B.; et al. Retinoid X receptor activation reverses age-related deficiencies in myelin debris phagocytosis and remyelination. Brain A J. Neurol. 2015, 138, 3581–3597. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, Y.; Ding, J.; Zhao, Z.; Qian, C.; Luan, Y.; Teng, G.J. Nicotinamide Administration Improves Remyelination after Stroke. Neural Plast. 2017, 2017, 7019803. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Wong, A.W.; Willingham, M.M.; van den Buuse, M.; Kilpatrick, T.J.; Murray, S.S. Brain-derived neurotrophic factor promotes central nervous system myelination via a direct effect upon oligodendrocytes. Neurosignals 2010, 18, 186–202. [Google Scholar] [CrossRef]
- Pirola, L.; Fröjdö, S. Resveratrol: One molecule, many targets. IUBMB Life 2008, 60, 323–332. [Google Scholar] [CrossRef]
- Meng, X.; Zhou, J.; Zhao, C.N.; Gan, R.Y.; Li, H.B. Health Benefits and Molecular Mechanisms of Resveratrol: A Narrative Review. Foods 2020, 9, 340. [Google Scholar] [CrossRef]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef]
- Li, T.; Tan, Y.; Ouyang, S.; He, J.; Liu, L. Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene 2022, 808, 145968. [Google Scholar] [CrossRef]
- Mahjabeen, W.; Khan, D.A.; Mirza, S.A. Role of resveratrol supplementation in regulation of glucose hemostasis, inflammation and oxidative stress in patients with diabetes mellitus type 2: A randomized, placebo-controlled trial. Complement. Ther. Med. 2022, 66, 102819. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhou, Q.M.; Lu, Y.Y.; Zhang, H.; Chen, Q.L.; Zhao, M.; Su, S.B. Resveratrol Inhibits the Migration and Metastasis of MDA-MB-231 Human Breast Cancer by Reversing TGF-β1-Induced Epithelial-Mesenchymal Transition. Molecules 2019, 24, 1131. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Gao, J.; Ke, W.; Wang, J.; Li, D.; Liu, R.; Jia, Y.; Wang, X.; Chen, X.; Chen, F.; et al. Resveratrol reduces obesity in high-fat diet-fed mice via modulating the composition and metabolic function of the gut microbiota. Free Radic. Biol. Med. 2020, 156, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Yang, H.; Xie, Y.; Ding, Y.; Kong, D.; Yu, H. Research Progress on Alzheimer’s Disease and Resveratrol. Neurochem. Res. 2020, 45, 989–1006. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, M.G.; Schimith, L.E.; André-Miral, C.; Muccillo-Baisch, A.L.; Arbo, B.D.; Hort, M.A. Neuroprotective Effects of Resveratrol in In vivo and In vitro Experimental Models of Parkinson’s Disease: A Systematic Review. Neurotox. Res. 2022, 40, 319–345. [Google Scholar] [CrossRef] [PubMed]
- Ginés, C.; Cuesta, S.; Kireev, R.; García, C.; Rancan, L.; Paredes, S.D.; Vara, E.; Tresguerres, J.A.F. Protective effect of resveratrol against inflammation, oxidative stress and apoptosis in pancreas of aged SAMP8 mice. Exp. Gerontol. 2017, 90, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Luo, Z.; Jin, M.; Sheng, W.; Wang, H.T.; Long, X.; Wu, Y.; Hu, P.; Xu, H.; Zhang, X. Exploration of age-related mitochondrial dysfunction and the anti-aging effects of resveratrol in zebrafish retina. Aging 2019, 11, 3117–3137. [Google Scholar] [CrossRef]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef]
- Huang, Y.; Lu, J.; Zhan, L.; Wang, M.; Shi, R.; Yuan, X.; Gao, X.; Liu, X.; Zang, J.; Liu, W.; et al. Resveratrol-induced Sirt1 phosphorylation by LKB1 mediates mitochondrial metabolism. J. Biol. Chem. 2021, 297, 100929. [Google Scholar] [CrossRef]
- Um, J.H.; Park, S.J.; Kang, H.; Yang, S.; Foretz, M.; McBurney, M.W.; Kim, M.K.; Viollet, B.; Chung, J.H. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 2010, 59, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.C.; Nicol, C.J.; Cheng, Y.C. Resveratrol activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against amyloid-beta-induced inflammation and oxidative stress. Neurochem. Int. 2018, 115, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Nicol, C.J.B.; Cheng, Y.C.; Yen, C.; Wang, Y.S.; Chiang, M.C. Neuroprotective effects of resveratrol against oxygen glucose deprivation induced mitochondrial dysfunction by activation of AMPK in SH-SY5Y cells with 3D gelatin scaffold. Brain Res. 2020, 1726, 146492. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.; Xiao, D.; Muhammed, A.; Deng, J.; Chen, L.; He, J. Anti-Inflammatory Action and Mechanisms of Resveratrol. Molecules 2021, 26, 229. [Google Scholar] [CrossRef]
- Broderick, T.L.; Rasool, S.; Li, R.; Zhang, Y.; Anderson, M.; Al-Nakkash, L.; Plochocki, J.H.; Geetha, T.; Babu, J.R. Neuroprotective Effects of Chronic Resveratrol Treatment and Exercise Training in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7337. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.Y.; Dong, Q.X.; Zhu, J.; Sun, X.; Zhang, L.F.; Qiu, M.; Yu, X.L.; Liu, R.T. Resveratrol Rescues Tau-Induced Cognitive Deficits and Neuropathology in a Mouse Model of Tauopathy. Curr. Alzheimer Res. 2019, 16, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.F.; Yu, X.L.; Ji, M.; Liu, S.Y.; Wu, X.L.; Wang, Y.J.; Liu, R.T. Resveratrol alleviates motor and cognitive deficits and neuropathology in the A53T α-synuclein mouse model of Parkinson’s disease. Food Funct. 2018, 9, 6414–6426. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Wang, J.; Yang, B.; Weng, Q.; He, Q. Targeting Microglia and Macrophages: A Potential Treatment Strategy for Multiple Sclerosis. Front. Pharmacol. 2019, 10, 286. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Lee, W.H.; Suk, K. Functional polarization of neuroglia: Implications in neuroinflammation and neurological disorders. Biochem. Pharmacol. 2016, 103, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef] [PubMed]
- Plemel, J.R.; Stratton, J.A.; Michaels, N.J.; Rawji, K.S.; Zhang, E.; Sinha, S.; Baaklini, C.S.; Dong, Y.; Ho, M.; Thorburn, K.; et al. Microglia response following acute demyelination is heterogeneous and limits infiltrating macrophage dispersion. Sci. Adv. 2020, 6, eaay6324. [Google Scholar] [CrossRef]
- Lombardi, M.; Parolisi, R.; Scaroni, F.; Bonfanti, E.; Gualerzi, A.; Gabrielli, M.; Kerlero de Rosbo, N.; Uccelli, A.; Giussani, P.; Viani, P.; et al. Detrimental and protective action of microglial extracellular vesicles on myelin lesions: Astrocyte involvement in remyelination failure. Acta Neuropathol. 2019, 138, 987–1012. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xu, S.; Qian, Y.; Xiao, Q. Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav. Immun. 2017, 64, 162–172. [Google Scholar] [CrossRef]
- Ma, S.; Fan, L.; Li, J.; Zhang, B.; Yan, Z. Resveratrol promoted the M2 polarization of microglia and reduced neuroinflammation after cerebral ischemia by inhibiting miR-155. Int. J. Neurosci. 2020, 130, 817–825. [Google Scholar] [CrossRef]
- Zheng, X.; Sun, K.; Liu, Y.; Yin, X.; Zhu, H.; Yu, F.; Zhao, W. Resveratrol-loaded macrophage exosomes alleviate multiple sclerosis through targeting microglia. J. Control. Release 2023, 353, 675–684. [Google Scholar] [CrossRef]
- Quan, L.; Uyeda, A.; Muramatsu, R. Central nervous system regeneration: The roles of glial cells in the potential molecular mechanism underlying remyelination. Inflamm. Regen. 2022, 42, 7. [Google Scholar] [CrossRef]
- Lu, X.; Ma, L.; Ruan, L.; Kong, Y.; Mou, H.; Zhang, Z.; Wang, Z.; Wang, J.M.; Le, Y. Resveratrol differentially modulates inflammatory responses of microglia and astrocytes. J. Neuroinflamm. 2010, 7, 46. [Google Scholar] [CrossRef]
- Fan, Y.Y.; Huo, J. A1/A2 astrocytes in central nervous system injuries and diseases: Angels or devils? Neurochem. Int. 2021, 148, 105080. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wang, L.; Li, F.; Hu, R.; Ma, J.; Zhang, K.; Cheng, X. Resveratrol Downregulates STAT3 Expression and Astrocyte Activation in Primary Astrocyte Cultures of Rat. Neurochem. Res. 2020, 45, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Ghaiad, H.R.; Nooh, M.M.; El-Sawalhi, M.M.; Shaheen, A.A. Resveratrol Promotes Remyelination in Cuprizone Model of Multiple Sclerosis: Biochemical and Histological Study. Mol. Neurobiol. 2017, 54, 3219–3229. [Google Scholar] [CrossRef] [PubMed]
- Spaas, J.; van Veggel, L.; Schepers, M.; Tiane, A.; van Horssen, J.; Wilson, D.M., 3rd; Moya, P.R.; Piccart, E.; Hellings, N.; Eijnde, B.O.; et al. Oxidative stress and impaired oligodendrocyte precursor cell differentiation in neurological disorders. Cell. Mol. Life Sci. 2021, 78, 4615–4637. [Google Scholar] [CrossRef] [PubMed]
- Samy, D.M.; Zaki, E.I.; Hassaan, P.S.; Abdelmonsif, D.A.; Mohamed, D.Y.; Saleh, S.R. Neurobehavioral, biochemical and histological assessment of the effects of resveratrol on cuprizone-induced demyelination in mice: Role of autophagy modulation. J. Physiol. Biochem. 2023, 79, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Stettner, M.; Wolffram, K.; Mausberg, A.K.; Albrecht, P.; Derksen, A.; Methner, A.; Dehmel, T.; Hartung, H.P.; Dietrich, H.; Kieseier, B.C. Promoting myelination in an in vitro mouse model of the peripheral nervous system: The effect of wine ingredients. PLoS ONE 2013, 8, e66079. [Google Scholar] [CrossRef]
- Zhang, J.; Ren, J.; Liu, Y.; Huang, D.; Lu, L. Resveratrol regulates the recovery of rat sciatic nerve crush injury by promoting the autophagy of Schwann cells. Life Sci. 2020, 256, 117959. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, Y.; Wu, S.; Zhao, M.Y. Review: Myelin clearance is critical for regeneration after peripheral nerve injury. Front. Neurol. 2022, 13, 908148. [Google Scholar] [CrossRef]
- Rosa, P.M.; Martins, L.A.M.; Souza, D.O.; Quincozes-Santos, A. Glioprotective Effect of Resveratrol: An Emerging Therapeutic Role for Oligodendroglial Cells. Mol. Neurobiol. 2018, 55, 2967–2978. [Google Scholar] [CrossRef]
- Yu, P.; Wang, L.; Tang, F.; Guo, S.; Liao, H.; Fan, C.; Yang, Q. Resveratrol-mediated neurorestoration after cerebral ischemic injury—Sonic Hedgehog signaling pathway. Life Sci. 2021, 280, 119715. [Google Scholar] [CrossRef]
- Fonseca-Kelly, Z.; Nassrallah, M.; Uribe, J.; Khan, R.S.; Dine, K.; Dutt, M.; Shindler, K.S. Resveratrol neuroprotection in a chronic mouse model of multiple sclerosis. Front. Neurol. 2012, 3, 84. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Han, H.; Cao, P.; Yu, W.; Yang, C.; Gao, Y.; Yuan, W. Resveratrol improves neuron protection and functional recovery through enhancement of autophagy after spinal cord injury in mice. Am. J. Transl. Res. 2017, 9, 4607–4616. [Google Scholar] [PubMed]
- Jablonska, B.; Gierdalski, M.; Chew, L.J.; Hawley, T.; Catron, M.; Lichauco, A.; Cabrera-Luque, J.; Yuen, T.; Rowitch, D.; Gallo, V. Sirt1 regulates glial progenitor proliferation and regeneration in white matter after neonatal brain injury. Nat. Commun. 2016, 7, 13866. [Google Scholar] [CrossRef] [PubMed]
- Rafalski, V.A.; Ho, P.P.; Brett, J.O.; Ucar, D.; Dugas, J.C.; Pollina, E.A.; Chow, L.M.; Ibrahim, A.; Baker, S.J.; Barres, B.A.; et al. Expansion of oligodendrocyte progenitor cells following SIRT1 inactivation in the adult brain. Nat. Cell Biol. 2013, 15, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Prozorovski, T.; Ingwersen, J.; Lukas, D.; Göttle, P.; Koop, B.; Graf, J.; Schneider, R.; Franke, K.; Schumacher, S.; Britsch, S.; et al. Regulation of sirtuin expression in autoimmune neuroinflammation: Induction of SIRT1 in oligodendrocyte progenitor cells. Neurosci. Lett. 2019, 704, 116–125. [Google Scholar] [CrossRef] [PubMed]
- A Phase I Double Blind Study of Metformin Acting on Endogenous Neural Progenitor Cells in Children with Multiple Sclerosis. Available online: https://clinicaltrials.gov/study/NCT04121468 (accessed on 8 November 2023).
- Metformin Treatment in Progressive Multiple Sclerosis. Available online: https://www.clinicaltrials.gov/study/NCT05349474 (accessed on 8 November 2023).
- Nicotinamide Riboside Supplementation in Progressive Multiple Sclerosis. Available online: https://clinicaltrials.gov/study/NCT05740722 (accessed on 8 November 2023).
- La Rosa, G.; Lonardo, M.S.; Cacciapuoti, N.; Muscariello, E.; Guida, B.; Faraonio, R.; Santillo, M.; Damiano, S. Dietary Polyphenols, Microbiome, and Multiple Sclerosis: From Molecular Anti-Inflammatory and Neuroprotective Mechanisms to Clinical Evidence. Int. J. Mol. Sci. 2023, 24, 7247. [Google Scholar] [CrossRef]
- Shamsher, E.; Khan, R.S.; Davis, B.M.; Dine, K.; Luong, V.; Somavarapu, S.; Cordeiro, M.F.; Shindler, K.S. Nanoparticles Enhance Solubility and Neuroprotective Effects of Resveratrol in Demyelinating Disease. Neurotherapeutics 2023, 20, 1138–1153. [Google Scholar] [CrossRef]
- Ruckh, J.M.; Zhao, J.W.; Shadrach, J.L.; van Wijngaarden, P.; Rao, T.N.; Wagers, A.J.; Franklin, R.J. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell 2012, 10, 96–103. [Google Scholar] [CrossRef]
- Shen, S.; Sandoval, J.; Swiss, V.A.; Li, J.; Dupree, J.; Franklin, R.J.; Casaccia-Bonnefil, P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat. Neurosci. 2008, 11, 1024–1034. [Google Scholar] [CrossRef]
- Tutuncu, M.; Tang, J.; Zeid, N.A.; Kale, N.; Crusan, D.J.; Atkinson, E.J.; Siva, A.; Pittock, S.J.; Pirko, I.; Keegan, B.M.; et al. Onset of progressive phase is an age-dependent clinical milestone in multiple sclerosis. Mult. Scler. 2013, 19, 188–198. [Google Scholar] [CrossRef]
- Goldschmidt, T.; Antel, J.; König, F.B.; Brück, W.; Kuhlmann, T. Remyelination capacity of the MS brain decreases with disease chronicity. Neurology 2009, 72, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Strijbis, E.M.M.; Kooi, E.J.; van der Valk, P.; Geurts, J.J.G. Cortical Remyelination Is Heterogeneous in Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2017, 76, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013, 125, 841–859. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Miron, V.; Cui, Q.; Wegner, C.; Antel, J.; Brück, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain A J. Neurol. 2008, 131, 1749–1758. [Google Scholar] [CrossRef]
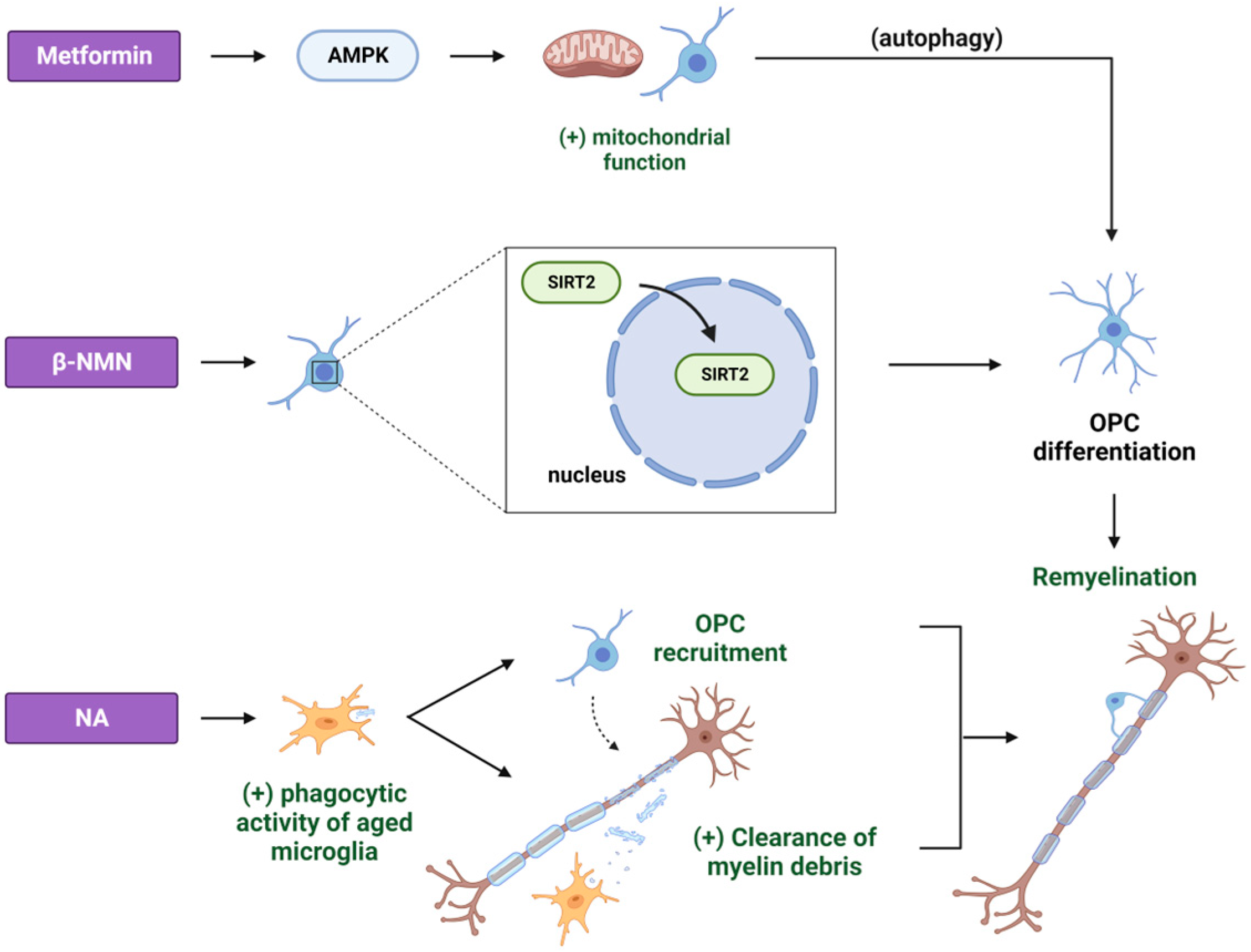
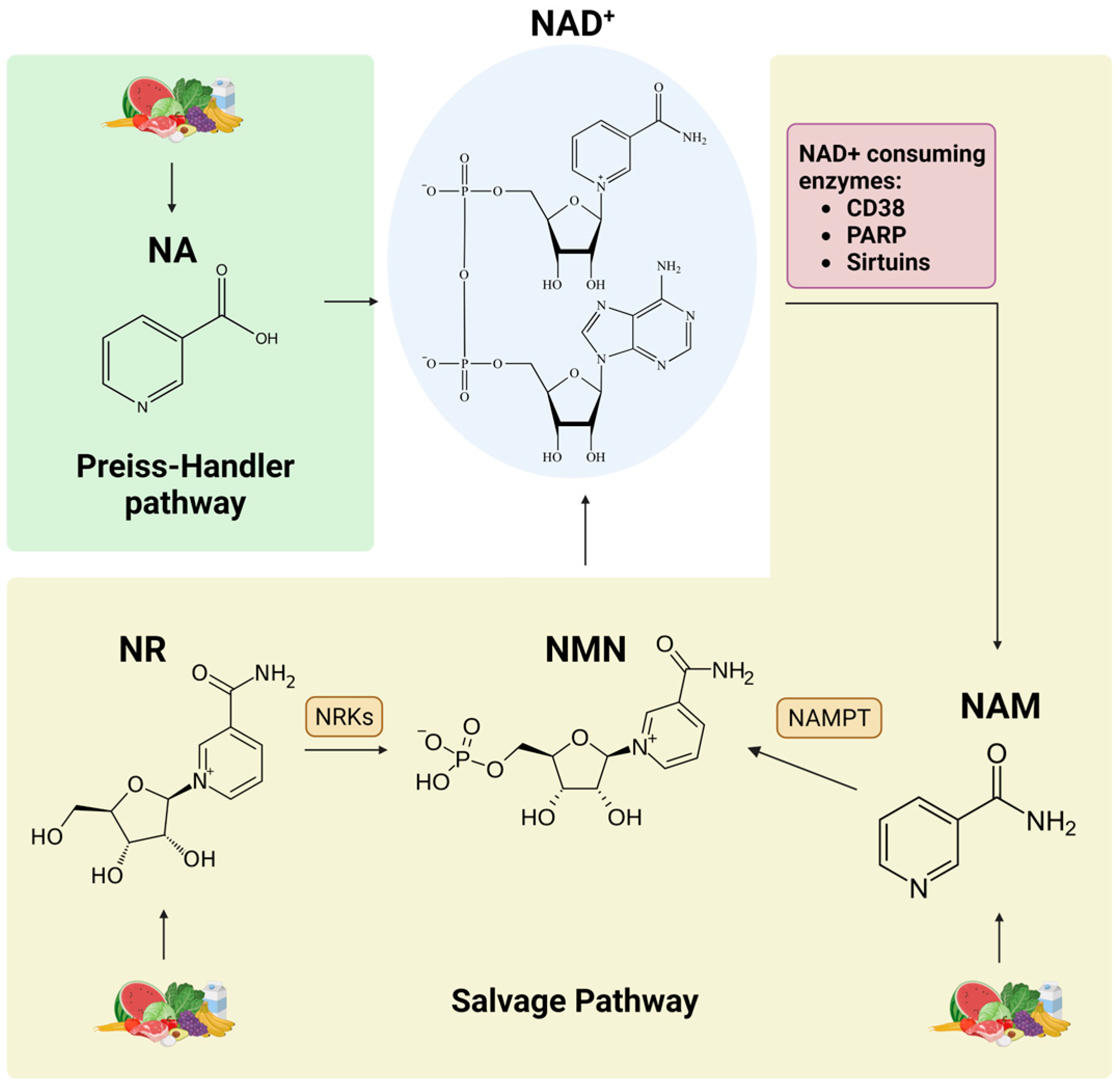
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaffe, D.; Kaplanis, S.I.; Karagogeos, D. The Roles of Caloric Restriction Mimetics in Central Nervous System Demyelination and Remyelination. Curr. Issues Mol. Biol. 2023, 45, 9526-9548. https://doi.org/10.3390/cimb45120596
Kaffe D, Kaplanis SI, Karagogeos D. The Roles of Caloric Restriction Mimetics in Central Nervous System Demyelination and Remyelination. Current Issues in Molecular Biology. 2023; 45(12):9526-9548. https://doi.org/10.3390/cimb45120596
Chicago/Turabian StyleKaffe, Despoina, Stefanos Ioannis Kaplanis, and Domna Karagogeos. 2023. "The Roles of Caloric Restriction Mimetics in Central Nervous System Demyelination and Remyelination" Current Issues in Molecular Biology 45, no. 12: 9526-9548. https://doi.org/10.3390/cimb45120596
APA StyleKaffe, D., Kaplanis, S. I., & Karagogeos, D. (2023). The Roles of Caloric Restriction Mimetics in Central Nervous System Demyelination and Remyelination. Current Issues in Molecular Biology, 45(12), 9526-9548. https://doi.org/10.3390/cimb45120596