
Integrative Analysis of the Role of TP53 in Human Pan-Cancer


Abstract
:1. Introduction
2. Material and Methods
2.1. Identifying Pan-Cancer Expression of TP53
2.2. Survival Prognosis, Mutation, and mRNA Expression Analysis
2.3. Pan-Cancer Clinical Value of TP53
2.4. Analysis of Immune Infiltration
2.5. Tumor Stemness of TP53
2.6. Analysis of Single-Cell Sequencing Data
2.7. Enrichment Analysis of TP53-Related Genes
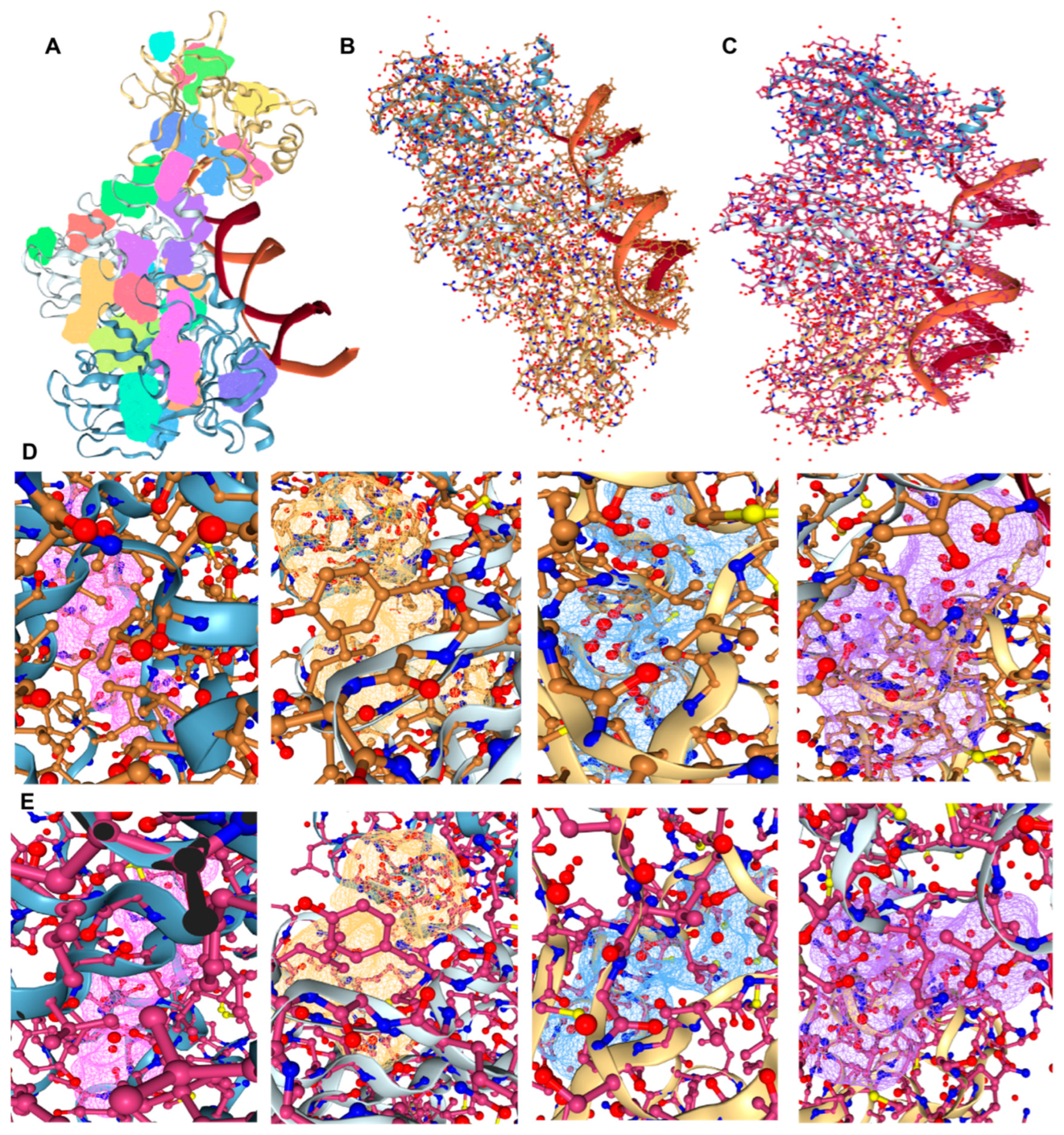
2.8. Drug Screening and Molecular Docking
2.9. Statistical Analysis
3. Results
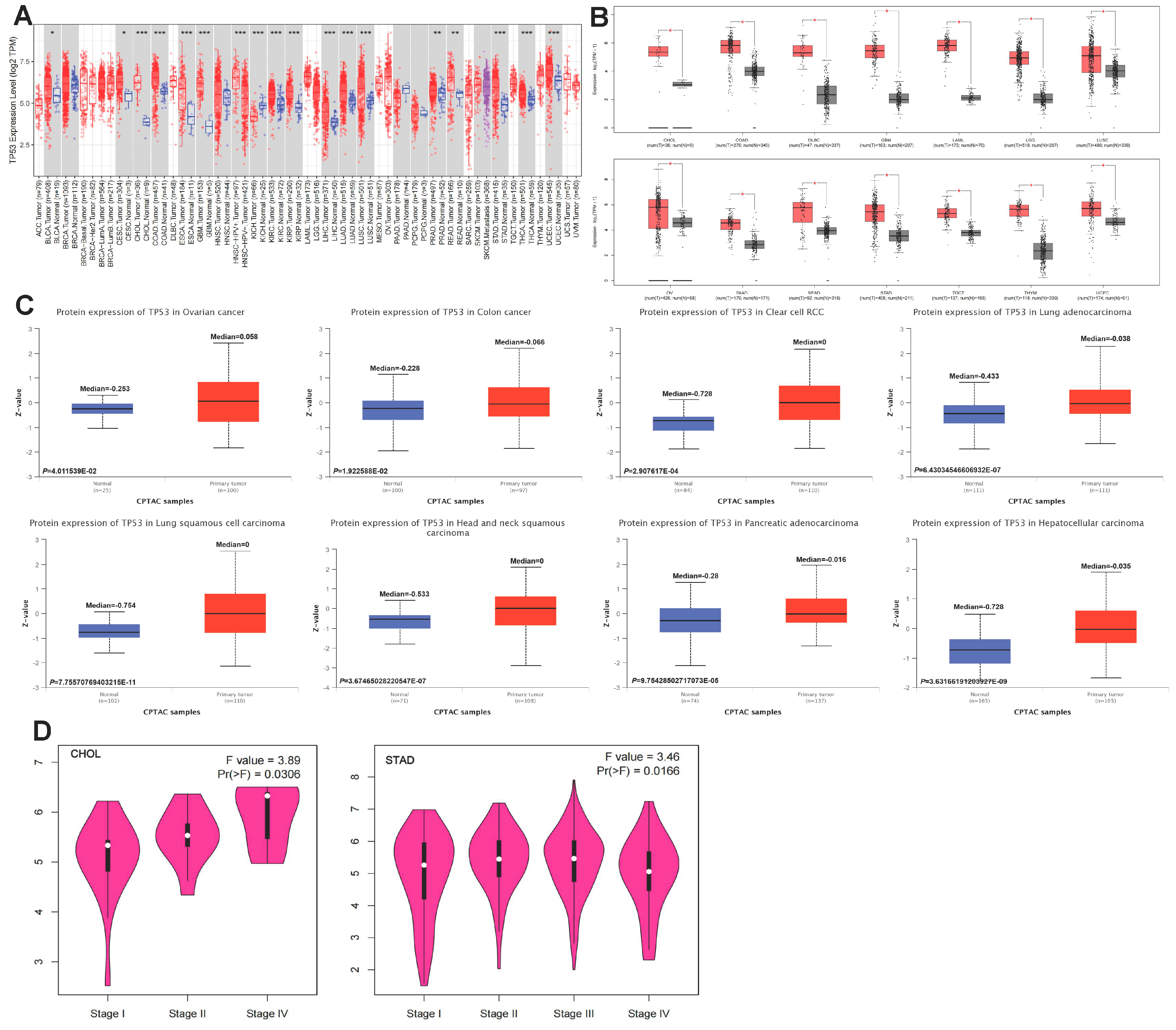
3.1. Aberrant Pan-Cancer Expression of TP53
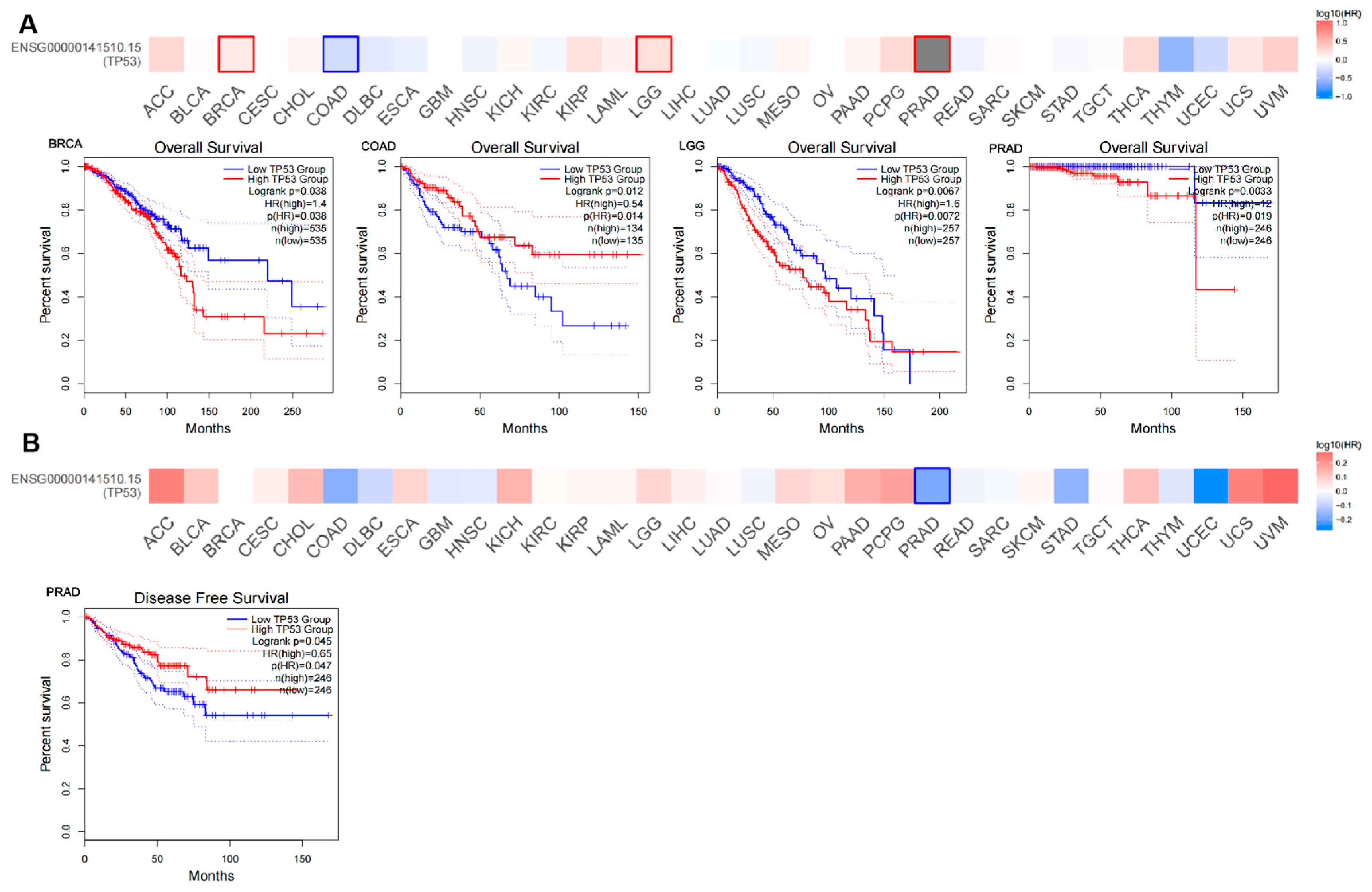
3.2. Survival Analysis of TP53 Expression in Pan Cancer
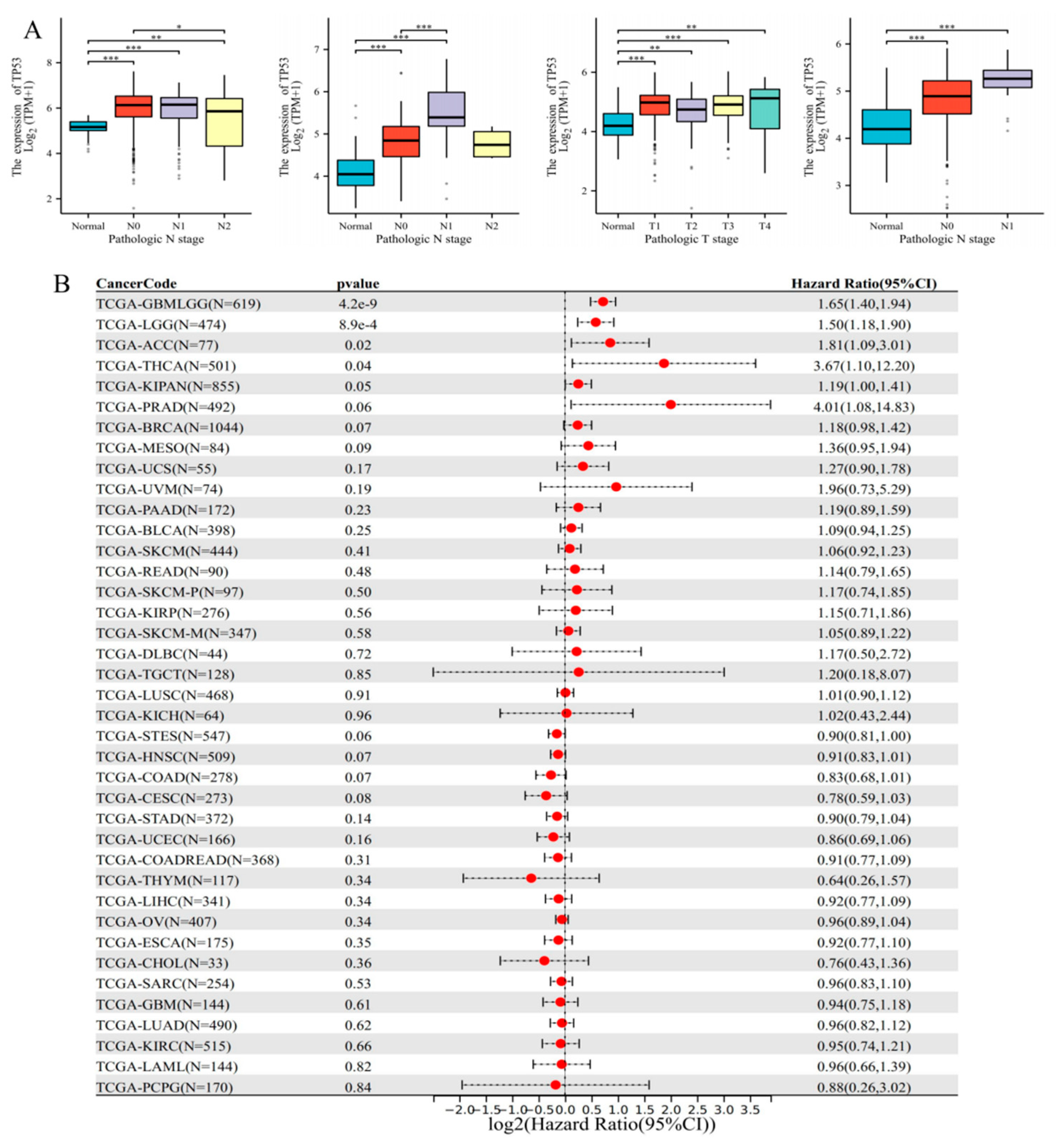
3.3. Clinical Significance and Prognostic Analysis of TP53
3.4. Diagnostic ROC Analysis of TP53
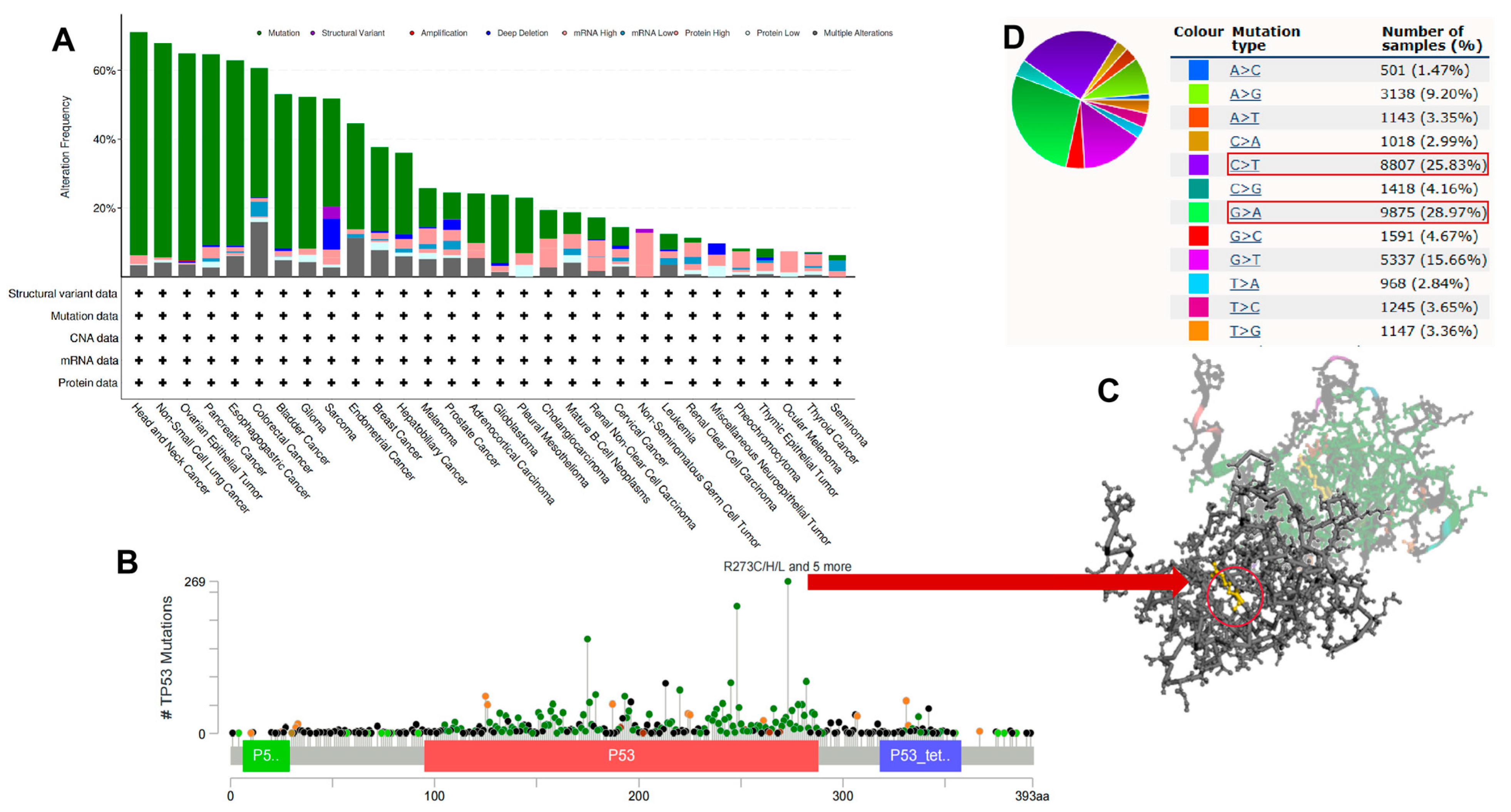
3.5. Pan-Cancer Genetic Alterations of TP53
3.6. Pan-Cancer mRNA Expression of TP53
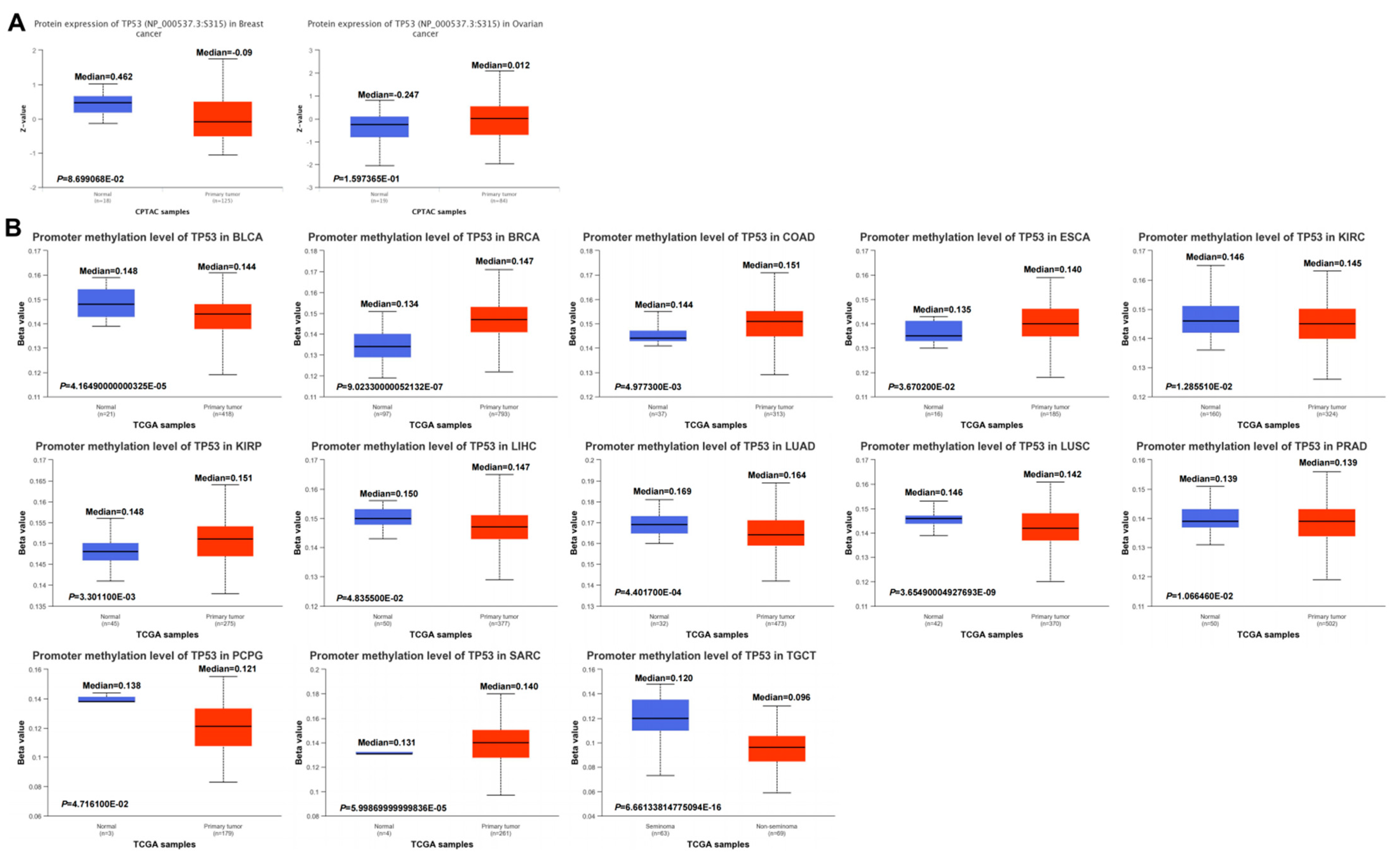
3.7. Pan-Cancer DNA Methylation of TP53
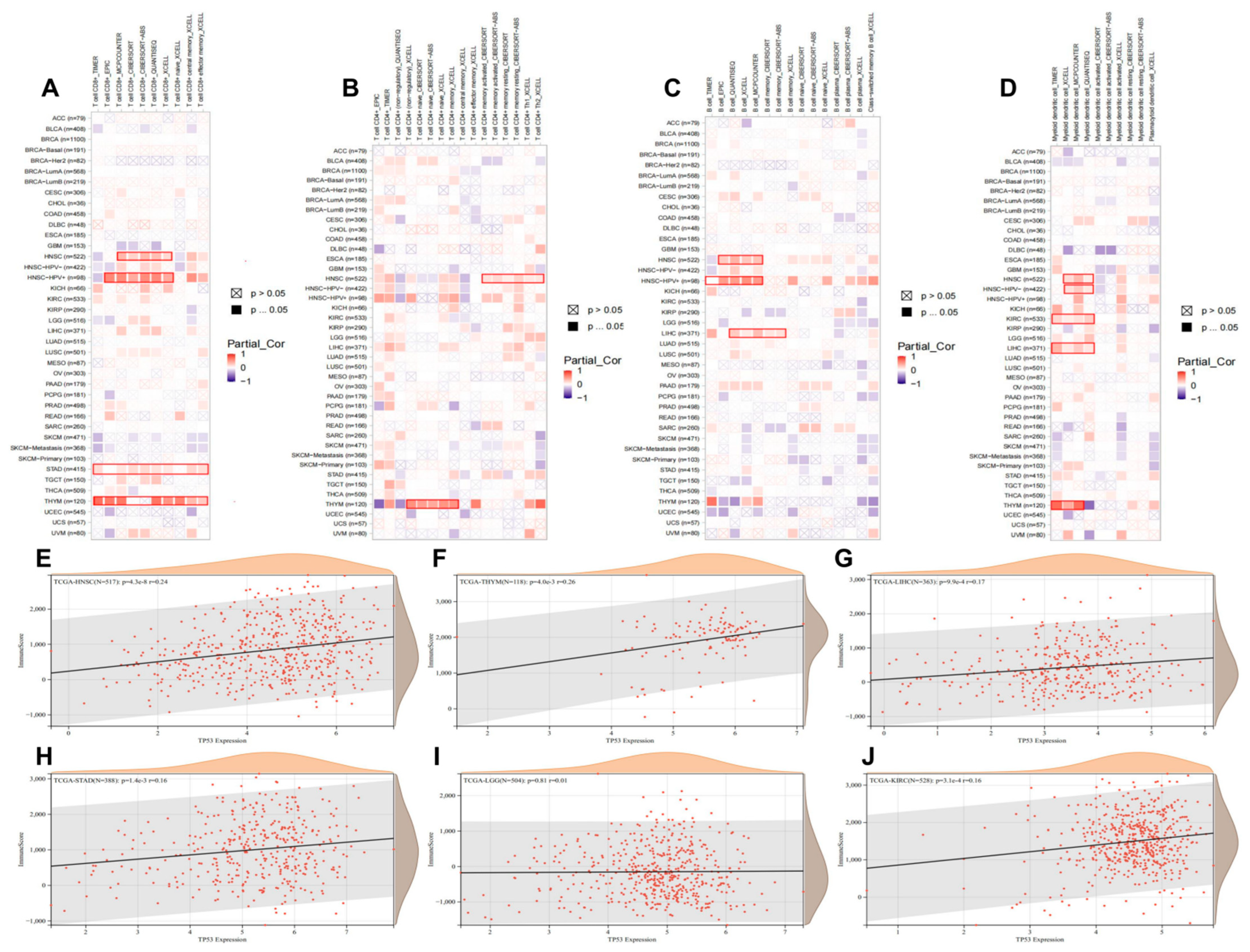
3.8. Pan-Cancer Roles of TP53 in Immune Infiltration
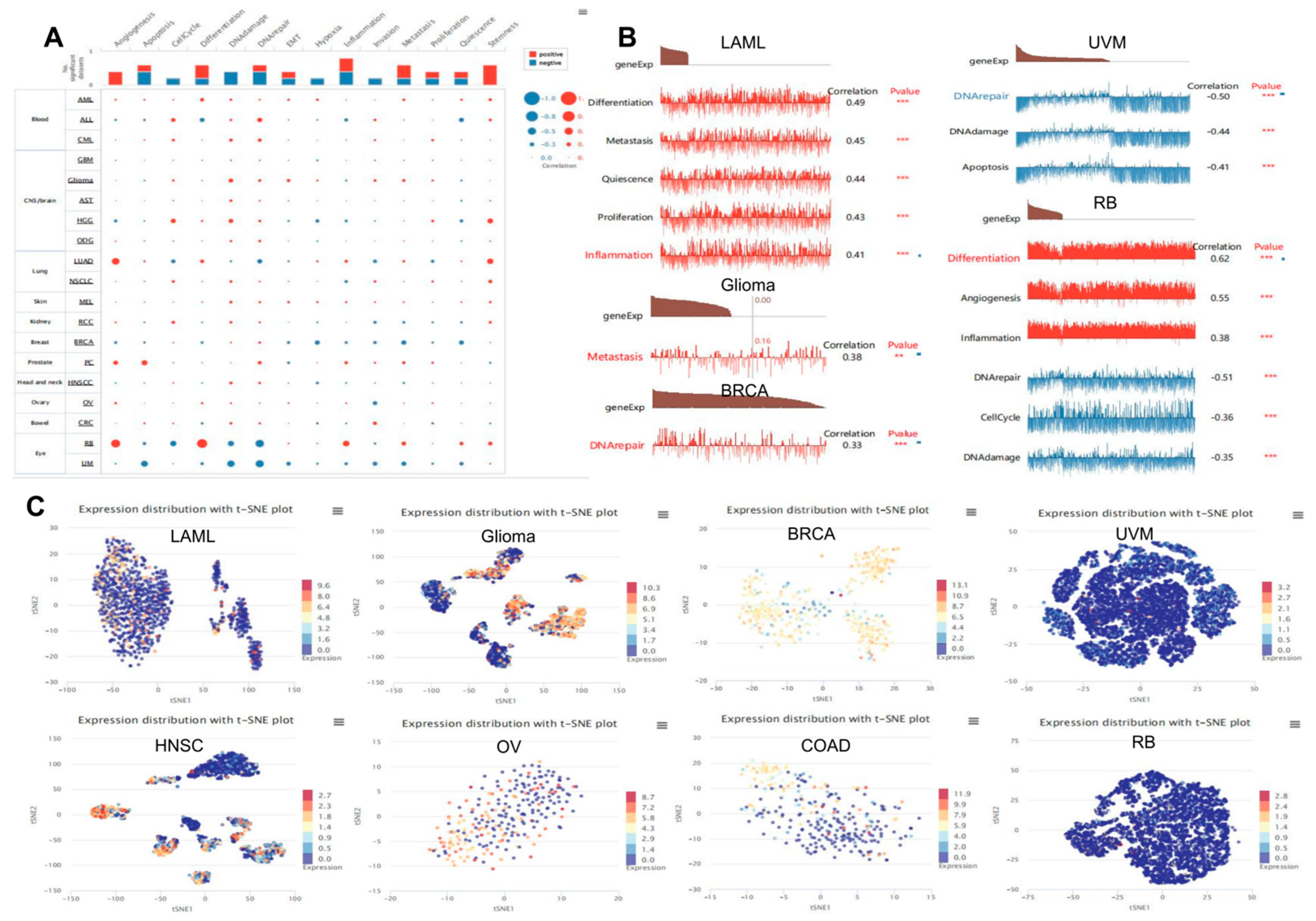
3.9. Analysis of Tumor Stemness and Immune Checkpoints
3.10. Expression Pattern of TP53 in a Single Cell and Its Relationship with Cancer Functional Status
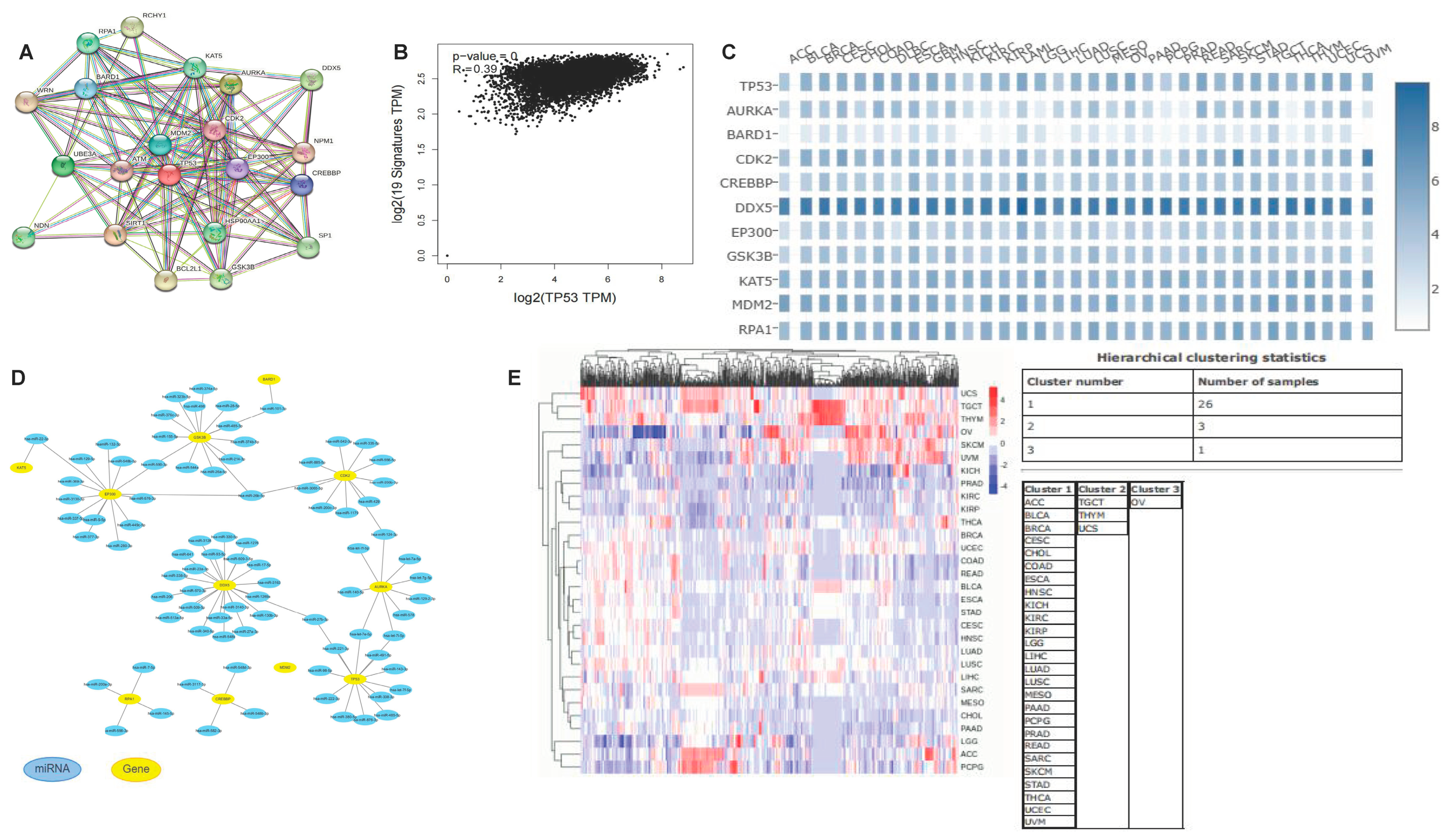
3.11. Co-Expression Network of TP53
3.12. TP53-Related Transcription Factors
3.13. Therapeutic Drugs and Molecular Docking
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Strasser, A.; Kelly, G.L. Should mutant TP53 be targeted for cancer therapy? Cell Death Differ. 2022, 29, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Koifman, G.; Aloni-Grinstein, R.; Rotter, V. p53 balances between tissue hierarchy and anarchy. J. Mol. Cell Biol. 2019, 11, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant p53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef]
- Silwal-Pandit, L.; Langerød, A.; Børresen-Dale, A.L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar] [CrossRef]
- Maurer, G.D.; Heller, S.; Wanka, C.; Rieger, J.; Steinbach, J.P. Knockdown of the TP53-Induced Glycolysis and Apoptosis Regulator (TIGAR) Sensitizes Glioma Cells to Hypoxia, Irradiation and Temozolomide. Int. J. Mol. Sci. 2019, 20, 1061. [Google Scholar] [CrossRef]
- Masunaga, S.; Uto, Y.; Nagasawa, H.; Hori, H.; Nagata, K.; Suzuki, M.; Kinashi, Y.; Ono, K. Evaluation of hypoxic cell radio-sensitizers in terms of radio-sensitizing and repair-inhibiting potential. Dependency on p53 status of tumor cells and the effects on intratumor quiescent cells. Anticancer Res. 2006, 26, 1261–1270. [Google Scholar]
- Chiou, Y.H.; Wong, R.H.; Chao, M.R.; Chen, C.Y.; Liou, S.H.; Lee, H. Nickel accumulation in lung tissues is associated with increased risk of p53 mutation in lung cancer patients. Environ. Mol. Mutagen. 2014, 55, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.S.; Ejima, Y.; Tachibana, A.; Yamada, T.; Ishizaki, K.; Shimizu, T.; Nomura, T. DNA damage response pathway in radioadaptive response. Mutat Res. 2002, 504, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Paget, V.; Lechevrel, M.; André, V.; Le Goff, J.; Pottier, D.; Billet, S.; Garçon, G.; Shirali, P.; Sichel, F. Benzo[a]pyrene, aflatoxine B₁ and acetaldehyde mutational patterns in TP53 gene using a functional assay: Relevance to human cancer aetiology. PLoS ONE 2012, 7, e30921. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, A.; Reis, H.M.; Feitelson, M.A. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, M.L.; Buonaguro, L.; Izzo, F.; Buonaguro, F.M. Molecular alterations in hepatocellular carcinoma associated with hepatitis B and hepatitis C infections. Oncotarget 2016, 7, 25087–25102. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Wang, X.W.; Harris, C.C. Hepatitis B virus X protein inhibits nucleotide excision repair. Int. J. Cancer 1999, 80, 875–879. [Google Scholar] [CrossRef]
- Toyooka, S.; Tsuda, T.; Gazdar, A.F. The TP53 gene, tobacco exposure, and lung cancer. Hum. Mutat. 2003, 21, 229–239. [Google Scholar] [CrossRef] [PubMed]
- von Deimling, A.; Eibl, R.H.; Ohgaki, H.; Louis, D.N.; von Ammon, K.; Petersen, I.; Kleihues, P.; Chung, R.Y.; Wiestler, O.D.; Seizinger, B.R. p53 mutations are associated with 17p allelic loss in grade II and grade III astrocytoma. Cancer Res. 1992, 52, 2987–2990. [Google Scholar]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Bale, T.A.; Rosenblum, M.K. The 2021 WHO Classification of Tumors of the Central Nervous System: An update on pediatric low-grade gliomas and glioneuronal tumors. Brain Pathol. 2022, 32, e13060. [Google Scholar] [CrossRef]
- Eibl, R.H.; Schneemann, M. Medulloblastoma: From TP53 Mutations to Molecular Classification and Liquid Biopsy. Biology 2023, 12, 267. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Eibl, R.H.; Wiestler, O.D.; Yasargil, M.G.; Newcomb, E.W.; Kleihues, P. p53 mutations in nonastrocytic human brain tumors. Cancer Res. 1991, 51, 6202–6205. [Google Scholar] [PubMed]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Hu, F.F.; Xia, M.X.; Han, L.; Zhang, Q.; Guo, A.Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Yan, M.; Zhang, G.; Liu, W.; Deng, C.; Liao, G.; Xu, L.; Luo, T.; Yan, H.; Long, Z.; et al. CancerSEA: A cancer single-cell state atlas. Nucleic Acids Res. 2019, 47, D900–D908. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Sarver, A.L.; Sarver, A.E.; Yuan, C.; Subramanian, S. OMCD: OncomiR Cancer Database. BMC Cancer 2018, 18, 1223. [Google Scholar] [CrossRef]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.-W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Badal, V.D.; Kundrotas, P.J.; Vakser, I.A. Text mining for modeling of protein complexes enhanced by machine learning. Bioinformatics 2021, 37, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef]
- Ghatak, D.; Das Ghosh, D.; Roychoudhury, S. Cancer Stemness: p53 at the Wheel. Front Oncol. 2021, 10, 604124. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Peng, M.; Tang, L.; Ouyang, J.; Xiong, F.; Guo, C.; Tang, Y.; Zhou, Y.; Liao, Q.; et al. Single-cell RNA sequencing in cancer research. J. Exp. Clin. Cancer Res. 2021, 40, 81. [Google Scholar] [CrossRef]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef]
- Wang, S.; Jin, S.; Liu, M.-D.; Pang, P.; Wu, H.; Qi, Z.-Z.; Liu, F.-Y.; Sun, C.-F. Hsa-let-7e-5p Inhibits the Proliferation and Metastasis of Head and Neck Squamous Cell Carcinoma Cells by Targeting Chemokine Receptor 7. J. Cancer 2019, 10, 1941–1948. [Google Scholar] [CrossRef]
- Niculae, A.M.; Dobre, M.; Herlea, V.; Manuc, T.E.; Trandafir, B.; Milanesi, E.; Hinescu, M.E. Let-7 microRNAs Are Possibly Associated with Perineural Invasion in Colorectal Cancer by Targeting IGF Axis. Life 2022, 12, 1638. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Z.; Zhao, S.; Song, Y.; Si, L.; Wang, X. Identification key genes, key miRNAs and key transcription factors of lung adenocarcinoma. J. Thorac. Dis. 2020, 12, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Li, M.; Liu, Z.; Li, T.; Zhao, L.; Fu, W. Glycolysis-Related LINC02432/Hsa-miR-98-5p/HK2 Axis Inhibits Ferroptosis and Predicts Immune Infiltration, Tumor Mutation Burden, and Drug Sensitivity in Pancreatic Adenocarcinoma. Front. Pharmacol. 2022, 13, 937413. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Hagn, F.; Klein, C.; Demmer, O.; Marchenko, N.; Vaseva, A.; Moll, U.M.; Kessler, H. BclxL changes conformation upon binding to wild-type but not mutant p53 DNA binding domain. J. Biol. Chem. 2010, 285, 3439–3450. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Nottingham, L.; Duggal, P.; Pernas, F.G.; Yan, B.; Yang, X.P.; Chen, Z.; Van Waes, C. Deficient TP53 expression, function, and cisplatin sensitivity are restored by quinacrine in head and neck cancer. Clin. Cancer Res. 2007, 13 Pt 1, 6568–6578. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Lee, S.; Park, J.Y.; Lee, S.Y.; Kim, H.S. Induction of p53-Dependent Apoptosis by Prostaglandin A2. Biomolecules 2020, 10, 492. [Google Scholar] [CrossRef]
- Elayapillai, S.; Ramraj, S.; Benbrook, D.M.; Bieniasz, M.; Wang, L.; Pathuri, G.; Isingizwe, Z.R.; Kennedy, A.L.; Zhao, Y.D.; Lightfoot, S.; et al. Potential and mechanism of mebendazole for treatment and maintenance of ovarian cancer. Gynecol. Oncol. 2021, 160, 302–311. [Google Scholar] [CrossRef]
- Gong, L.; Zhang, D.; Dong, Y.; Lei, Y.; Qian, Y.; Tan, X.; Han, S.; Wang, J. Integrated Bioinformatics Analysis for Identificating the Therapeutic Targets of Aspirin in Small Cell Lung Cancer. J. Biomed. Inform. 2018, 88, 20–28. [Google Scholar] [CrossRef]
- He, C.; Qin, H.; Tang, H.; Yang, D.; Li, Y.; Huang, Z.; Zhang, D.; Lv, C. Comprehensive bioinformatics analysis of the TP53 signaling pathway in Wilms’ tumor. Ann. Transl. Med. 2020, 8, 1228. [Google Scholar] [CrossRef]
- Strom, E.; Sathe, S.; Komarov, P.G.; Chernova, O.B.; Pavlovska, I.; Shyshynova, I.; Bosykh, D.A.; Burdelya, L.G.; Macklis, R.M.; Skaliter, R.; et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat. Chem. Biol. 2006, 2, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Szabó, B.; Németh, K.; Mészáros, K.; Krokker, L.; Likó, I.; Saskői, É.; Németh, K.; Szabó, P.T.; Szücs, N.; Czirják, S.; et al. Aspirin Mediates Its Antitumoral Effect Through Inhibiting PTTG1 in Pituitary Adenoma. J. Clin. Endocrinol. Metab. 2022, 107, 3066–3079. [Google Scholar] [CrossRef] [PubMed]
- Shan, B.; Zhao, R.; Zhou, J.; Zhang, M.; Qi, X.; Wang, T.; Gong, J.; Wu, Y.; Zhu, Y.; Yang, W.; et al. AURKA Increase the Chemosensitivity of Colon Cancer Cells to Oxaliplatin by Inhibiting the TP53-Mediated DNA Damage Response Genes. Biomed. Res. Int. 2020, 2020, 8916729. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Subhasree, N.; Jiangjiang, Q.; Kalkunte, S.; Minghai, W.; Ruiwen, Z. BCL-2, TP53 and BAX protein expression in superficial urothelial bladder carcinoma. Cancer Lett. 2007, 250, 292–299. [Google Scholar] [CrossRef]
- Henley, M.J.; Koehler, A.N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug Discov. 2021, 20, 669–688. [Google Scholar] [CrossRef]
- Williams, E.A.; Sharaf, R.; Decker, B.; Werth, A.J.; Toma, H.; Montesion, M.; Sokol, E.S.; Pavlick, D.C.; Shah, N.; Williams, K.J.; et al. CDKN2C-Null Leiomyosarcoma: A Novel, Genomically Distinct Class of TP53/RB1-Wild-Type Tumor With Frequent CIC Genomic Alterations and 1p/19q-Codeletion. JCO Precis. Oncol. 2020, 4, 955–971. [Google Scholar] [CrossRef]
- Weitzel, J.N.; Chao, E.C.; Nehoray, B.; Van Tongeren, L.R.; LaDuca, H.; Blazer, K.R.; Slavin, T.; Pesaran, T.; Rybak, C.; Solomon, I.; et al. Somatic TP53 variants frequently confound germ-line testing results. Genet Med. 2018, 20, 809–816. [Google Scholar] [CrossRef]
- Scheinberg, T.; Goodwin, A.; Ip, E.; Linton, A.; Mak, B.; Smith, D.P.; Stockler, M.R.; Strach, M.C.; Tran, B.; Young, A.L.; et al. Evaluation of a Mainstream Model of Genetic Testing for Men With Prostate Cancer. JCO Oncol. Pract. 2021, 17, e204–e216. [Google Scholar] [CrossRef]
- Kennedy, M.C.; Lowe, S.W. Mutant p53: It’s not all one and the same. Cell Death Differ. 2022, 29, 983–987. [Google Scholar] [CrossRef]
- Sun, H.; Tang, Q.; Chen, Y.; Tang, C.; Zou, S.; Qiu, F. Effects of dendritic cells transfected with full length wild type P53 and modified by gastric cancer lysate on immune response. J. Huazhong Univ. Sci. Technol. Med. Sci. 2004, 24, 460–463. [Google Scholar] [CrossRef]
- Morimoto, I.; Sasaki, Y.; Ishida, S.; Imai, K.; Tokino, T. Identification of the osteopontin gene as a direct target of TP53. Genes Chromosomes Cancer 2002, 33, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Biton, J.; Mansuet-Lupo, A.; Pécuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti-PD-1 in Lung Adenocarcinoma. Clin. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, M.; Wang, X. Cancer type-dependent correlations between TP53 mutations and antitumor immunity. DNA Repair 2020, 88, 102785. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef]
- Nishikawa, S.; Iwakuma, T. Drugs Targeting p53 Mutations with FDA Approval and in Clinical Trials. Cancers 2023, 15, 429. [Google Scholar] [CrossRef]
- Melichar, B.; Adenis, A.; Lockhart, A.C.; Bennouna, J.; Dees, E.C.; Kayaleh, O.; Obermannova, R.; DeMichele, A.; Zatloukal, P.; Zhang, B.; et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: A five-arm phase 2 study. Lancet Oncol. 2015, 16, 395–405. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor | Characteristics | Low Expression of TP53 | High Expression of TP53 | p-Value | Tumor | Characteristics | Low Expression of TP53 | High Expression of TP53 | p-Value | Tumor | Characteristics | Low Expression of TP53 | High Expression of TP53 | p-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | n | 39 | 40 | BLCA | n | 206 | 206 | BRCA | n | 543 | 544 | |||
| Pathologic T stage, n (%) | 0.135197547 | Pathologic T stage, n (%) | 0.596 | Pathologic T stage, n (%) | 0.741 | |||||||||
| T1 | 7 (9.1%) | 2 (2.6%) | T1 | 3 (0.8%) | 2 (0.5%) | T1 | 138 (12.7%) | 140 (12.9%) | ||||||
| T2 | 21 (27.3%) | 21 (27.3%) | T2 | 54 (14.3%) | 64 (16.9%) | T2 | 322 (29.7%) | 309 (28.5%) | ||||||
| T3 | 2 (2.6%) | 6 (7.8%) | T3 | 103 (27.2%) | 93 (24.6%) | T3 | 65 (6%) | 75 (6.9%) | ||||||
| T4 | 7 (9.1%) | 11 (14.3%) | T4 | 32 (8.5%) | 27 (7.1%) | T4 | 16 (1.5%) | 19 (1.8%) | ||||||
| Pathologic N stage, n (%) | 1 | Pathologic N stage, n (%) | 0.212 | Pathologic N stage, n (%) | 0.627 | |||||||||
| N0 | 33 (42.9%) | 35 (45.5%) | N0 | 110 (29.9%) | 128 (34.8%) | N0 | 268 (25.1%) | 248 (23.2%) | ||||||
| N1 | 4 (5.2%) | 5 (6.5%) | N1 | 24 (6.5%) | 22 (6%) | N1 | 174 (16.3%) | 185 (17.3%) | ||||||
| Clinical M stage, n (%) | 0.486682972 | N2 | 46 (12.5%) | 31 (8.4%) | N2 | 55 (5.1%) | 61 (5.7%) | |||||||
| M0 | 31 (40.3%) | 31 (40.3%) | N3 | 3 (0.8%) | 4 (1.1%) | N3 | 36 (3.4%) | 41 (3.8%) | ||||||
| M1 | 6 (7.8%) | 9 (11.7%) | Pathologic M stage, n (%) | 0.138 | Pathologic M stage, n (%) | 0.395 | ||||||||
| CESC | n | 153 | 153 | M0 | 92 (43.4%) | 109 (51.4%) | M0 | 449 (48.5%) | 456 (49.3%) | |||||
| Pathologic T stage, n (%) | 0.996 | M1 | 2 (0.9%) | 9 (4.2%) | M1 | 8 (0.9%) | 12 (1.3%) | |||||||
| T1 | 66 (27.2%) | 74 (30.5%) | CHOL | n | 17 | 18 | COAD | n | 239 | 239 | ||||
| T2 | 35 (14.4%) | 37 (15.2%) | Pathologic T stage, n (%) | 0.103 | Pathologic T stage, n (%) | 0.603 | ||||||||
| T3 | 10 (4.1%) | 11 (4.5%) | T1 | 12 (34.3%) | 6 (17.1%) | T1 | 6 (1.3%) | 5 (1%) | ||||||
| T4 | 5 (2.1%) | 5 (2.1%) | T2 | 4 (11.4%) | 8 (22.9%) | T3 | 168 (35.2%) | 155 (32.5%) | ||||||
| Pathologic N stage, n (%) | 0.567 | T3 | 1 (2.9%) | 4 (11.4%) | T4 | 28 (5.9%) | 32 (6.7%) | |||||||
| N0 | 60 (30.8%) | 74 (37.9%) | T4 | 0 (0%) | 0 (0%) | T2 | 37 (7.8%) | 46 (9.6%) | ||||||
| N1 | 30 (15.4%) | 31 (15.9%) | Pathologic N stage, n (%) | 0.33 | Pathologic N stage, n (%) | 0.017 | ||||||||
| Pathologic M stage, n (%) | 0.915 | N0 | 14 (46.7%) | 11 (36.7%) | N0 | 134 (28%) | 150 (31.4%) | |||||||
| M0 | 57 (22.3%) | 59 (23%) | N1 | 1 (3.3%) | 4 (13.3%) | N1 | 50 (10.5%) | 58 (12.1%) | ||||||
| M1 | 6 (2.3%) | 5 (2%) | Pathologic M stage, n (%) | 1 | N2 | 55 (11.5%) | 31 (6.5%) | |||||||
| MX | 62 (24.2%) | 67 (26.2%) | M0 | 13 (40.6%) | 14 (43.8%) | Pathologic M stage, n (%) | 0.905 | |||||||
| DLBC | n | 24 | 24 | M1 | 2 (6.2%) | 3 (9.4%) | M0 | 177 (42.7%) | 172 (41.4%) | |||||
| Clinical stage, n (%) | 0.081 | ESCA | n | 81 | 82 | M1 | 34 (8.2%) | 32 (7.7%) | ||||||
| Stage I | 4 (9.5%) | 4 (9.5%) | Pathologic T stage, n (%) | 0.518 | HNSC | n | 252 | 252 | ||||||
| Stage II | 6 (14.3%) | 11 (26.2%) | T1 | 16 (11%) | 11 (7.6%) | Pathologic T stage, n (%) | 0.488 | |||||||
| Stage III | 5 (11.9%) | 0 (0%) | T2 | 19 (13.1%) | 18 (12.4%) | T1 | 23 (5.1%) | 22 (4.9%) | ||||||
| Stage IV | 5 (11.9%) | 7 (16.7%) | T3 | 36 (24.8%) | 41 (28.3%) | T2 | 64 (14.3%) | 71 (15.8%) | ||||||
| KICH | n | 32 | 33 | T4 | 1 (0.7%) | 3 (2.1%) | T3 | 47 (10.5%) | 49 (10.9%) | |||||
| Pathologic T stage, n (%) | 0.338 | Pathologic N stage, n (%) | 0.398 | T4 | 96 (21.4%) | 76 (17%) | ||||||||
| T1 | 8 (12.3%) | 12 (18.5%) | N0 | 28 (19.4%) | 38 (26.4%) | Pathologic N stage, n (%) | 0.731 | |||||||
| T2 | 14 (21.5%) | 11 (16.9%) | N1 | 35 (24.3%) | 28 (19.4%) | N0 | 94 (22.9%) | 77 (18.7%) | ||||||
| T3 | 10 (15.4%) | 8 (12.3%) | N2 | 5 (3.5%) | 4 (2.8%) | N1 | 33 (8%) | 33 (8%) | ||||||
| T4 | 0 (0%) | 2 (3.1%) | N3 | 2 (1.4%) | 4 (2.8%) | N2 | 83 (20.2%) | 84 (20.4%) | ||||||
| Pathologic N stage, n (%) | 0.675 | Pathologic M stage, n (%) | 1 | N3 | 3 (0.7%) | 4 (1%) | ||||||||
| N0 | 23 (52.3%) | 16 (36.4%) | M0 | 60 (46.5%) | 61 (47.3%) | Pathologic M stage, n (%) | 0.45 | |||||||
| N1 | 1 (2.3%) | 2 (4.5%) | M1 | 4 (3.1%) | 4 (3.1%) | M0 | 104 (55%) | 84 (44.4%) | ||||||
| N2 | 1 (2.3%) | 1 (2.3%) | KIRC | n | 270 | 271 | M1 | 0 (0%) | 1 (0.5%) | |||||
| Pathologic M stage, n (%) | 1 | Pathologic T stage, n (%) | 0.017 | KIRP | n | 145 | 146 | |||||||
| M0 | 19 (52.8%) | 15 (41.7%) | T1 | 128 (23.7%) | 151 (27.9%) | Pathologic T stage, n (%) | 0.232 | |||||||
| M1 | 1 (2.8%) | 1 (2.8%) | T2 | 47 (8.7%) | 24 (4.4%) | T1 | 102 (35.3%) | 91 (31.5%) | ||||||
| LIHC | n | 187 | 187 | T3 | 91 (16.8%) | 89 (16.5%) | T2 | 17 (5.9%) | 17 (5.9%) | |||||
| Pathologic T stage, n (%) | 0.254 | T4 | 4 (0.7%) | 7 (1.3%) | T3 | 25 (8.7%) | 35 (12.1%) | |||||||
| T1 | 98 (26.4%) | 85 (22.9%) | Pathologic N stage, n (%) | 0.036 | T4 | 0 (0%) | 2 (0.7%) | |||||||
| T2 | 47 (12.7%) | 48 (12.9%) | N0 | 126 (48.8%) | 116 (45%) | Pathologic N stage, n (%) | 0.011 | |||||||
| T3 | 33 (8.9%) | 47 (12.7%) | N1 | 4 (1.6%) | 12 (4.7%) | N0 | 29 (37.2%) | 21 (26.9%) | ||||||
| T4 | 8 (2.2%) | 5 (1.3%) | Pathologic M stage, n (%) | 0.771 | N1 | 5 (6.4%) | 19 (24.4%) | |||||||
| Pathologic N stage, n (%) | 0.659 | M0 | 214 (42.1%) | 215 (42.3%) | N2 | 2 (2.6%) | 2 (2.6%) | |||||||
| N0 | 124 (48.1%) | 130 (50.4%) | M1 | 38 (7.5%) | 41 (8.1%) | Pathologic M stage, n (%) | 1 | |||||||
| N1 | 1 (0.4%) | 3 (1.2%) | LUAD | n | 269 | 270 | M0 | 48 (46.2%) | 47 (45.2%) | |||||
| Pathologic M stage, n (%) | 0.625 | Pathologic T stage, n (%) | 0.554 | M1 | 4 (3.8%) | 5 (4.8%) | ||||||||
| M0 | 134 (49.3%) | 134 (49.3%) | T1 | 83 (15.5%) | 93 (17.4%) | LUSC | n | 251 | 251 | |||||
| M1 | 3 (1.1%) | 1 (0.4%) | T2 | 148 (27.6%) | 144 (26.9%) | Pathologic T stage, n (%) | 0.41 | |||||||
| MESO | n | 43 | 44 | T3 | 28 (5.2%) | 21 (3.9%) | T1 | 60 (12%) | 54 (10.8%) | |||||
| Pathologic T stage, n (%) | 0.464 | T4 | 8 (1.5%) | 11 (2.1%) | T2 | 142 (28.3%) | 152 (30.3%) | |||||||
| T1 | 7 (8.2%) | 7 (8.2%) | Pathologic N stage, n (%) | 0.193 | T3 | 40 (8%) | 31 (6.2%) | |||||||
| T2 | 12 (14.1%) | 14 (16.5%) | N0 | 184 (35.2%) | 166 (31.7%) | T4 | 9 (1.8%) | 14 (2.8%) | ||||||
| T3 | 14 (16.5%) | 18 (21.2%) | N1 | 43 (8.2%) | 54 (10.3%) | Pathologic N stage, n (%) | 0.635 | |||||||
| T4 | 9 (10.6%) | 4 (4.7%) | N2 | 30 (5.7%) | 44 (8.4%) | N0 | 160 (32.3%) | 160 (32.3%) | ||||||
| Pathologic N stage, n (%) | 0.063 | N3 | 1 (0.2%) | 1 (0.2%) | N1 | 61 (12.3%) | 70 (14.1%) | |||||||
| N0 | 18 (21.7%) | 26 (31.3%) | Pathologic M stage, n (%) | 0.571 | N2 | 23 (4.6%) | 17 (3.4%) | |||||||
| N1 | 4 (4.8%) | 6 (7.2%) | M0 | 182 (46.7%) | 183 (46.9%) | N3 | 3 (0.6%) | 2 (0.4%) | ||||||
| N2 | 17 (20.5%) | 9 (10.8%) | M1 | 11 (2.8%) | 14 (3.6%) | Pathologic M stage, n (%) | 0.473 | |||||||
| N3 | 3 (3.6%) | 0 (0%) | PAAD | n | 89 | 90 | M0 | 208 (49.6%) | 204 (48.7%) | |||||
| Pathologic M stage, n (%) | 1 | Pathologic T stage, n (%) | 0.515 | M1 | 5 (1.2%) | 2 (0.5%) | ||||||||
| M0 | 28 (46.7%) | 29 (48.3%) | T1 | 5 (2.8%) | 2 (1.1%) | PRAD | n | 250 | 251 | |||||
| M1 | 2 (3.3%) | 1 (1.7%) | T3 | 71 (40.1%) | 72 (40.7%) | Pathologic T stage, n (%) | 0.186 | |||||||
| READ | n | 83 | 83 | T4 | 2 (1.1%) | 1 (0.6%) | T2 | 87 (17.6%) | 102 (20.6%) | |||||
| Pathologic T stage, n (%) | 0.162 | T2 | 10 (5.6%) | 14 (7.9%) | T3 | 157 (31.8%) | 137 (27.7%) | |||||||
| T1 | 2 (1.2%) | 7 (4.3%) | Pathologic N stage, n (%) | 0.887 | T4 | 4 (0.8%) | 7 (1.4%) | |||||||
| T3 | 62 (37.8%) | 51 (31.1%) | N0 | 24 (13.8%) | 26 (14.9%) | Pathologic N stage, n (%) | 0.804 | |||||||
| T4 | 7 (4.3%) | 7 (4.3%) | N1 | 61 (35.1%) | 63 (36.2%) | N0 | 173 (40.4%) | 175 (40.9%) | ||||||
| T2 | 11 (6.7%) | 17 (10.4%) | Pathologic M stage, n (%) | 1 | N1 | 41 (9.6%) | 39 (9.1%) | |||||||
| Pathologic N stage, n (%) | 0.963 | M0 | 34 (40%) | 46 (54.1%) | STAD | n | 187 | 188 | ||||||
| N0 | 43 (26.5%) | 41 (25.3%) | M1 | 2 (2.4%) | 3 (3.5%) | Pathologic T stage, n (%) | 0.416 | |||||||
| N1 | 22 (13.6%) | 23 (14.2%) | SKCM | n | 236 | 236 | T1 | 11 (3%) | 8 (2.2%) | |||||
| N2 | 17 (10.5%) | 16 (9.9%) | Pathologic T stage, n (%) | 0.848 | T3 | 87 (23.7%) | 81 (22.1%) | |||||||
| Pathologic M stage, n (%) | 0.794 | T1 | 23 (6.3%) | 19 (5.2%) | T4 | 43 (11.7%) | 57 (15.5%) | |||||||
| M0 | 62 (41.6%) | 64 (43%) | T2 | 39 (10.7%) | 40 (11%) | T2 | 42 (11.4%) | 38 (10.4%) | ||||||
| M1 | 12 (8.1%) | 11 (7.4%) | T3 | 43 (11.8%) | 48 (13.2%) | Pathologic N stage, n (%) | 0.303 | |||||||
| TGCT | n | 69 | 70 | T4 | 79 (21.6%) | 74 (20.3%) | N0 | 59 (16.5%) | 52 (14.6%) | |||||
| Pathologic T stage, n (%) | 0.682 | Pathologic N stage, n (%) | 0.24 | N1 | 41 (11.5%) | 56 (15.7%) | ||||||||
| T1 | 39 (28.3%) | 41 (29.7%) | N0 | 108 (26%) | 128 (30.8%) | N2 | 37 (10.4%) | 38 (10.6%) | ||||||
| T2 | 27 (19.6%) | 25 (18.1%) | N1 | 42 (10.1%) | 32 (7.7%) | N3 | 41 (11.5%) | 33 (9.2%) | ||||||
| T3 | 2 (1.4%) | 4 (2.9%) | N2 | 25 (6%) | 24 (5.8%) | Pathologic M stage, n (%) | 0.056 | |||||||
| Pathologic N stage, n (%) | 0.601 | N3 | 32 (7.7%) | 24 (5.8%) | M0 | 159 (44.8%) | 171 (48.2%) | |||||||
| N0 | 16 (25%) | 35 (54.7%) | Pathologic M stage, n (%) | 0.568 | M1 | 17 (4.8%) | 8 (2.3%) | |||||||
| N1 | 5 (7.8%) | 6 (9.4%) | M0 | 209 (47.1%) | 210 (47.3%) | UVM | n | 40 | 40 | |||||
| N2 | 1 (1.6%) | 1 (1.6%) | M1 | 11 (2.5%) | 14 (3.2%) | Pathologic M stage, n (%) | 0.835 | |||||||
| Pathologic M stage, n (%) | 0.153 | THCA | n | 256 | 256 | M0 | 25 (32.1%) | 26 (33.3%) | ||||||
| M0 | 59 (47.6%) | 61 (49.2%) | Pathologic T stage, n (%) | 0.043 | M1 | 2 (2.6%) | 2 (2.6%) | |||||||
| M1 | 0 (0%) | 4 (3.2%) | T1 | 68 (13.3%) | 75 (14.7%) | |||||||||
| T2 | 73 (14.3%) | 96 (18.8%) | ||||||||||||
| T3 | 100 (19.6%) | 75 (14.7%) | ||||||||||||
| T4 | 14 (2.7%) | 9 (1.8%) | ||||||||||||
| Pathologic N stage, n (%) | 0.077 | |||||||||||||
| N0 | 104 (22.5%) | 125 (27.1%) | ||||||||||||
| N1 | 125 (27.1%) | 108 (23.4%) | ||||||||||||
| Pathologic M stage, n (%) | 0.214 | |||||||||||||
| M0 | 140 (47.5%) | 146 (49.5%) | ||||||||||||
| M1 | 2 (0.7%) | 7 (2.4%) | ||||||||||||
| Tumor | Characteristics | Total (N) | OR (95% CI) | p-Value | Tumor | Characteristics | Total (N) | OR (95% CI) | p-Value | Tumor | Characteristics | Total (N) | OR (95% CI) | p-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | Pathologic T stage (T3&T4 vs. T1&T2) | 77 | 2.300 (0.865–6.116) | 0.095 | BLCA | Pathologic T stage (T3&T4 vs. T1&T2) | 378 | 0.768 (0.499–1.182) | 0.23 | BRCA | Pathologic T stage (T2&T3&T4 vs. T1) | 1084 | 0.986 (0.750–1.295) | 0.918 |
| Pathologic N stage (N1 vs. N0) | 77 | 1.179 (0.291–4.770) | 0.818 | Pathologic N stage (N1&N2&N3 vs. N0) | 368 | 0.671 (0.436–1.032) | 0.069 | Pathologic N stage (N1&N2&N3 vs. N0) | 1068 | 1.170 (0.920–1.488) | 0.199 | |||
| CESC | Pathologic T stage (T2&T3&T4 vs. T1) | 243 | 0.945 (0.568–1.573) | 0.829 | Pathologic M stage (M1 vs. M0) | 212 | 3.798 (0.800–18.022) | 0.093 | Pathologic M stage (M1 vs. M0) | 925 | 1.477 (0.598–3.647) | 0.398 | ||
| Pathologic N stage (N1 vs. N0) | 195 | 0.838 (0.457–1.537) | 0.568 | CHOL | Pathologic T stage (T2&T3&T4 vs. T1) | 35 | 4.800 (1.147–20.085) | 0.032 | COAD | Pathologic T stage (T3&T4 vs. T1&T2) | 477 | 0.804 (0.512–1.265) | 0.346 | |
| Pathologic M stage (M1&MX vs. M0) | 256 | 1.023 (0.625–1.674) | 0.928 | Pathologic N stage (N1 vs. N0) | 30 | 5.091 (0.496–52.285) | 0.171 | Pathologic N stage (N1&N2 vs. N0) | 478 | 0.757 (0.525–1.092) | 0.136 | |||
| ESCA | Pathologic T stage (T3&T4 vs. T1&T2) | 145 | 1.435 (0.743–2.772) | 0.282 | Pathologic M stage (M1 vs. M0) | 32 | 1.393 (0.200–9.711) | 0.738 | Pathologic M stage (M1 vs. M0) | 415 | 0.969 (0.572–1.640) | 0.905 | ||
| Pathologic N stage (N1&N2&N3 vs. N0) | 144 | 0.632 (0.326–1.223) | 0.173 | HNSC | Pathologic T stage (T3&T4 vs. T1&T2) | 448 | 0.818 (0.560–1.194) | 0.297 | KICH | Pathologic T stage (T2&T3&T4 vs. T1) | 65 | 0.583 (0.200–1.699) | 0.323 | |
| KIRC | Pathologic T stage (T2&T3&T4 vs. T1) | 541 | 0.716 (0.511–1.005) | 0.053 | Pathologic N stage (N1&N2&N3 vs. N0) | 411 | 1.241 (0.838–1.840) | 0.282 | Pathologic N stage (N1&N2 vs. N0) | 44 | 2.156 (0.323–14.410) | 0.428 | ||
| Pathologic N stage (N1 vs. N0) | 258 | 3.259 (1.022–10.388) | 0.046 | Pathologic M stage (M1 vs. M0) | 189 | 75169169.1684 (0.000–Inf) | 0.997 | Pathologic M stage (M1 vs. M0) | 36 | 1.267 (0.073–21.968) | 0.871 | |||
| Pathologic M stage (M1 vs. M0) | 508 | 1.074 (0.664–1.736) | 0.771 | KIRP | Pathologic T stage (T2&T3&T4 vs. T1) | 289 | 1.441 (0.881–2.358) | 0.146 | LIHC | Pathologic T stage (T2&T3&T4 vs. T1) | 371 | 1.310 (0.871–1.970) | 0.194 | |
| LUAD | Pathologic T stage (T2&T3&T4 vs. T1) | 536 | 0.854 (0.595–1.225) | 0.390 | Pathologic N stage (N1&N2 vs. N0) | 78 | 4.143 (1.489–11.527) | 0.006 | Pathologic N stage (N1 vs. N0) | 258 | 2.862 (0.294–27.877) | 0.365 | ||
| Pathologic N stage (N1&N2&N3 vs. N0) | 523 | 1.483 (1.027–2.141) | 0.035 | Pathologic M stage (M1 vs. M0) | 104 | 1.277 (0.323–5.049) | 0.728 | Pathologic M stage (M1 vs. M0) | 272 | 0.333 (0.034–3.245) | 0.344 | |||
| Pathologic M stage (M1 vs. M0) | 390 | 1.266 (0.560–2.862) | 0.571 | LUSC | Pathologic T stage (T2&T3&T4 vs. T1) | 502 | 1.146 (0.754–1.741) | 0.523 | MESO | Pathologic T stage (T3&T4 vs. T1&T2) | 85 | 0.865 (0.369–2.030) | 0.74 | |
| PAAD | Pathologic T stage (T3&T4 vs. T1&T2) | 177 | 0.937 (0.432–2.036) | 0.870 | Pathologic N stage (N1&N2&N3 vs. N0) | 496 | 1.023 (0.708–1.478) | 0.904 | Pathologic N stage (N1&N2&N3 vs. N0) | 83 | 0.433 (0.179–1.045) | 0.063 | ||
| Pathologic N stage (N1 vs. N0) | 174 | 0.953 (0.494–1.839) | 0.887 | Pathologic M stage (M1 vs. M0) | 419 | 0.408 (0.078–2.126) | 0.287 | Pathologic M stage (M1 vs. M0) | 60 | 0.483 (0.041–5.628) | 0.561 | |||
| Pathologic M stage (M1 vs. M0) | 85 | 1.109 (0.176–7.004) | 0.913 | PRAD | Pathologic T stage (T3&T4 vs. T2) | 494 | 0.763 (0.530–1.098) | 0.145 | READ | Pathologic T stage (T3&T4 vs. T1&T2) | 164 | 0.455 (0.213–0.973) | 0.042 | |
| SKCM | Pathologic T stage (T3&T4 vs. T1&T2) | 365 | 1.051 (0.680–1.625) | 0.824 | Pathologic N stage (N1 vs. N0) | 428 | 0.940 (0.578–1.529) | 0.804 | Pathologic N stage (N1&N2 vs. N0) | 162 | 1.049 (0.566–1.943) | 0.880 | ||
| Pathologic N stage (N1&N2&N3 vs. N0) | 415 | 0.682 (0.461–1.007) | 0.054 | STAD | Pathologic T stage (T3&T4 vs. T1&T2) | 367 | 1.223 (0.771–1.941) | 0.393 | Pathologic M stage (M1 vs. M0) | 149 | 0.888 (0.365–2.162) | 0.794 | ||
| Pathologic M stage (M1 vs. M0) | 444 | 1.267 (0.562–2.855) | 0.569 | Pathologic N stage (N1&N2&N3 vs. N0) | 357 | 1.211 (0.773–1.897) | 0.403 | UVM | Pathologic M stage (M1&MX vs. M0) | 78 | 0.769 (0.301–1.963) | 0.583 | ||
| TGCT | Pathologic T stage (T2&T3 vs. T1) | 138 | 0.951 (0.484–1.870) | 0.885 | Pathologic M stage (M1 vs. M0) | 355 | 0.438 (0.184–1.042) | 0.062 | ||||||
| Pathologic N stage (N1&N2 vs. N0) | 64 | 0.533 (0.154–1.844) | 0.321 | THCA | Pathologic T stage (T3&T4 vs. T1&T2) | 510 | 0.608 (0.424–0.870) | 0.007 | ||||||
| Pathologic M stage (M1 vs. M0) | 124 | 56035110.1225 (0.000–Inf) | 0.994 | Pathologic N stage (N1 vs. N0) | 462 | 0.719 (0.499–1.037) | 0.077 | |||||||
| Pathologic M stage (M1 vs. M0) | 295 | 3.356 (0.685–16.433) | 0.135 |
| Name | P_6 | P_0 | P_4 | P_1 |
|---|---|---|---|---|
| drugScore | 0.785028 | 0.745974 | 0.723506 | 0.680187 |
| volume | 277.86 | 485.15 | 309.3 | 419.38 |
| surface | 312.4 | 685.27 | 243.85 | 613 |
| depth | 21.1 | 16.15 | 18.27 | 15.25 |
| surf/vol | 1.124307205 | 1.412490982 | 0.788393146 | 1.46168153 |
| ell c/a | 0.05 | 0.09 | 0.1 | 0.11 |
| ell b/a | 0.14 | 0.2 | 0.29 | 0.79 |
| siteAtms | 135 | 97 | 145 | 124 |
| accept | 34 | 25 | 42 | 33 |
| donor | 16 | 12 | 15 | 11 |
| hydrophobicity | 0.18 | 0.53 | 0.08 | 0.37 |
| Cs | 92 | 67 | 97 | 81 |
| Ns | 25 | 12 | 26 | 23 |
| Os | 16 | 17 | 17 | 19 |
| Ss | 2 | 1 | 5 | 1 |
| Xs | 0 | 0 | 0 | 0 |
| negAA | 0.04 | 0.09 | 0.03 | 0.07 |
| posAA | 0.24 | 0.18 | 0.25 | 0.26 |
| polarAA | 0.2 | 0.27 | 0.38 | 0.37 |
| apolarAA | 0.52 | 0.45 | 0.34 | 0.3 |
| ALA | 3 | 1 | 2 | 2 |
| ARG | 4 | 3 | 5 | 3 |
| ASN | 0 | 0 | 2 | 1 |
| ASP | 1 | 1 | 0 | 1 |
| CYS | 0 | 0 | 4 | 2 |
| GLN | 1 | 0 | 1 | 2 |
| GLU | 0 | 1 | 1 | 1 |
| GLY | 0 | 0 | 2 | 0 |
| HIS | 2 | 0 | 3 | 3 |
| ILE | 2 | 3 | 2 | 0 |
| LEU | 1 | 2 | 1 | 1 |
| LYS | 0 | 1 | 0 | 1 |
| MET | 2 | 1 | 2 | 2 |
| PHE | 0 | 0 | 0 | 0 |
| PRO | 2 | 1 | 1 | 0 |
| SER | 3 | 2 | 1 | 4 |
| THR | 0 | 3 | 0 | 1 |
| TRP | 0 | 0 | 0 | 0 |
| TYR | 1 | 1 | 2 | 0 |
| VAL | 3 | 2 | 3 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Du, J.; Cheng, X.; Wei, J. Integrative Analysis of the Role of TP53 in Human Pan-Cancer. Curr. Issues Mol. Biol. 2023, 45, 9606-9633. https://doi.org/10.3390/cimb45120601
Liu T, Du J, Cheng X, Wei J. Integrative Analysis of the Role of TP53 in Human Pan-Cancer. Current Issues in Molecular Biology. 2023; 45(12):9606-9633. https://doi.org/10.3390/cimb45120601
Chicago/Turabian StyleLiu, Tingting, Jin Du, Xiangshu Cheng, and Jianshe Wei. 2023. "Integrative Analysis of the Role of TP53 in Human Pan-Cancer" Current Issues in Molecular Biology 45, no. 12: 9606-9633. https://doi.org/10.3390/cimb45120601