
Mouse CCL9 Chemokine Acts as Tumor Suppressor in a Murine Model of Colon Cancer
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. MTS Proliferation Assay
2.3. Migration Assay
2.4. Tumor Growth Assessment
2.5. Microarray
2.6. Statistical Analysis
3. Results
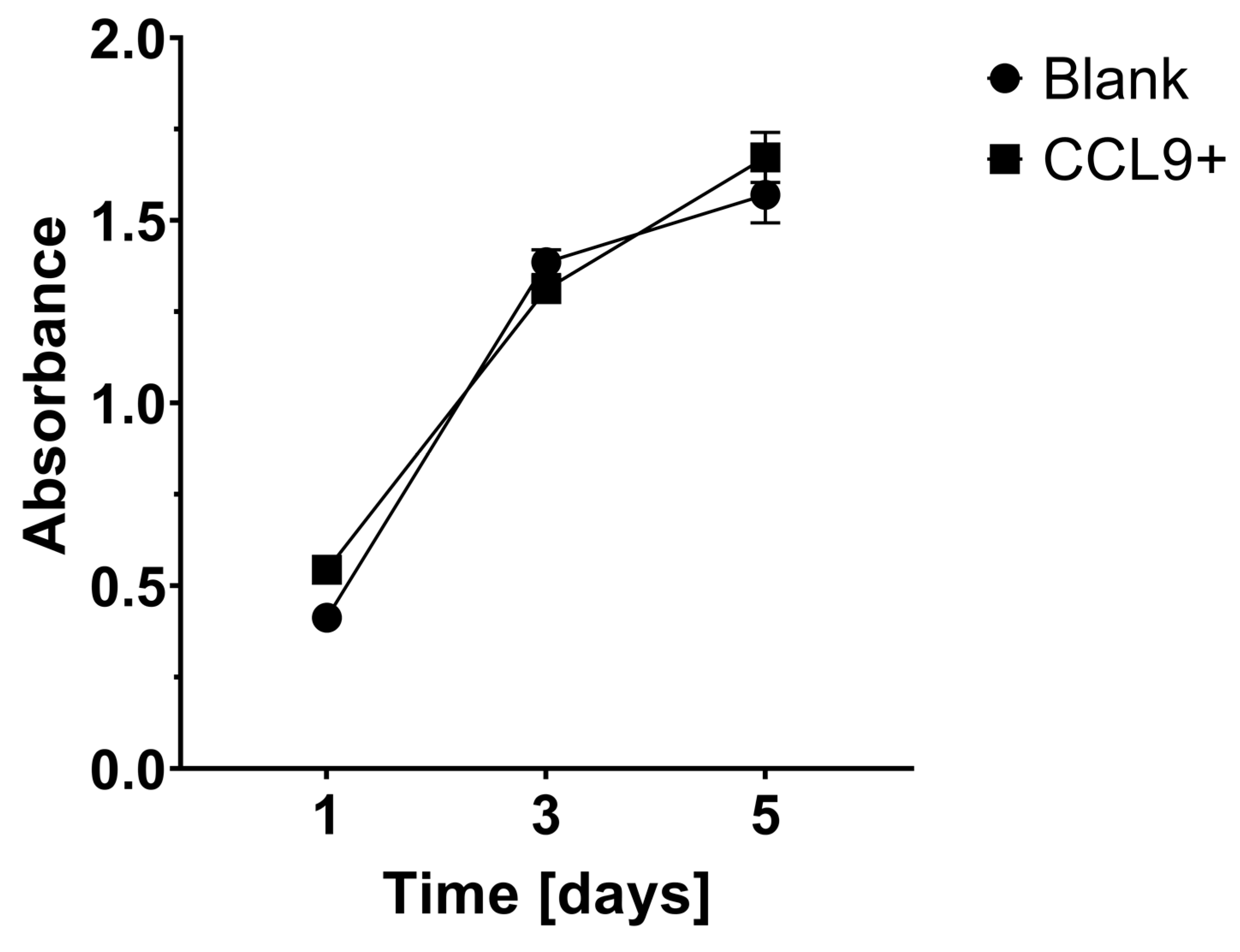
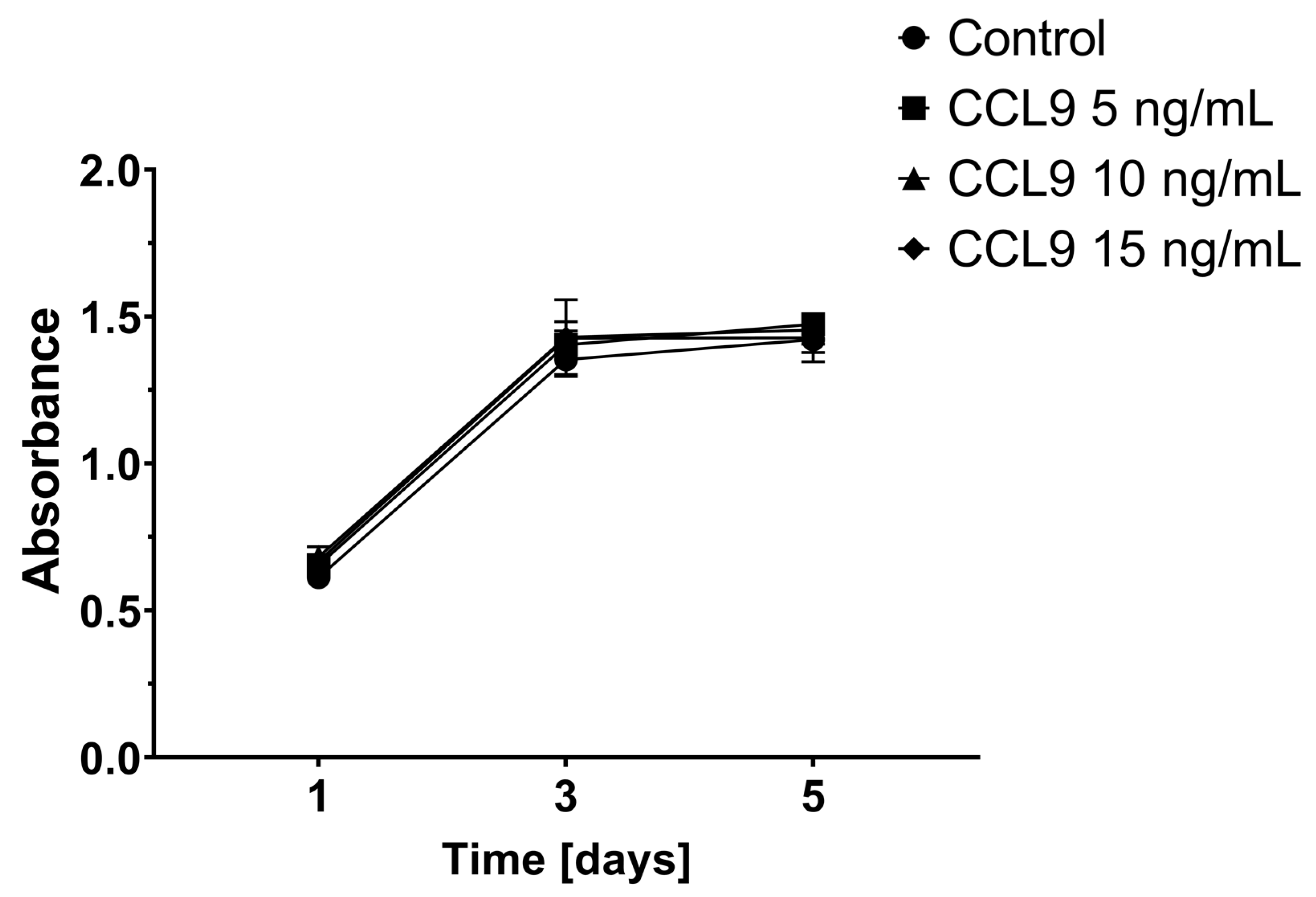
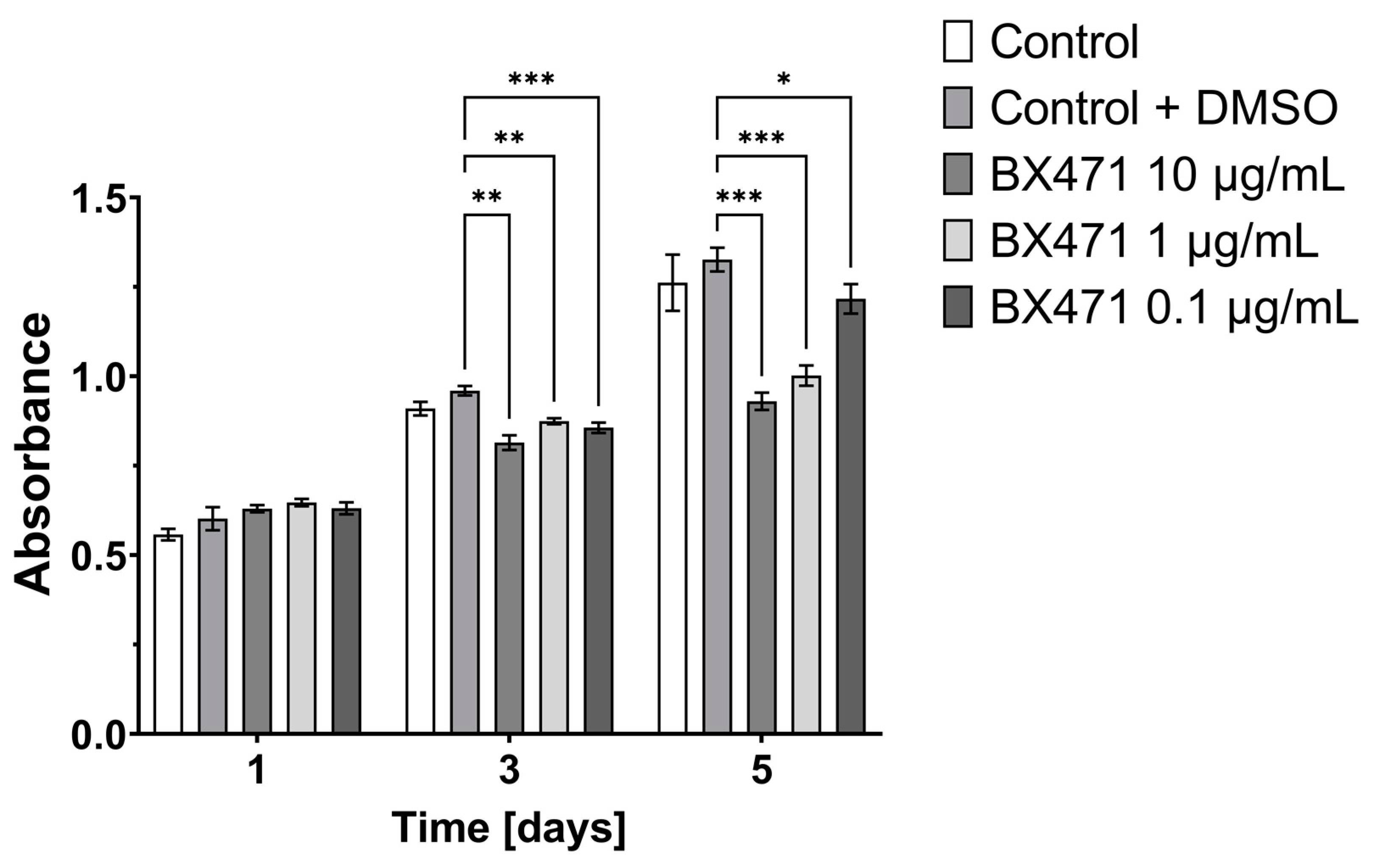
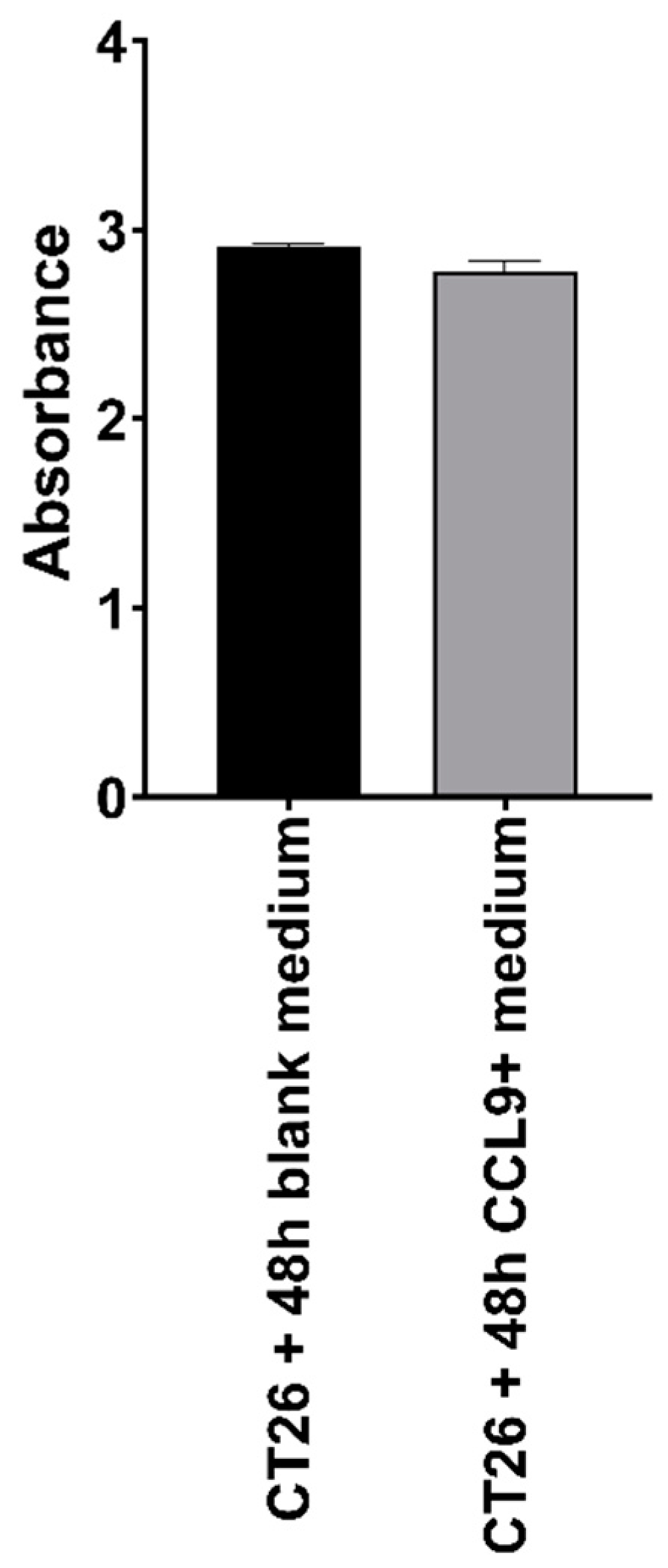
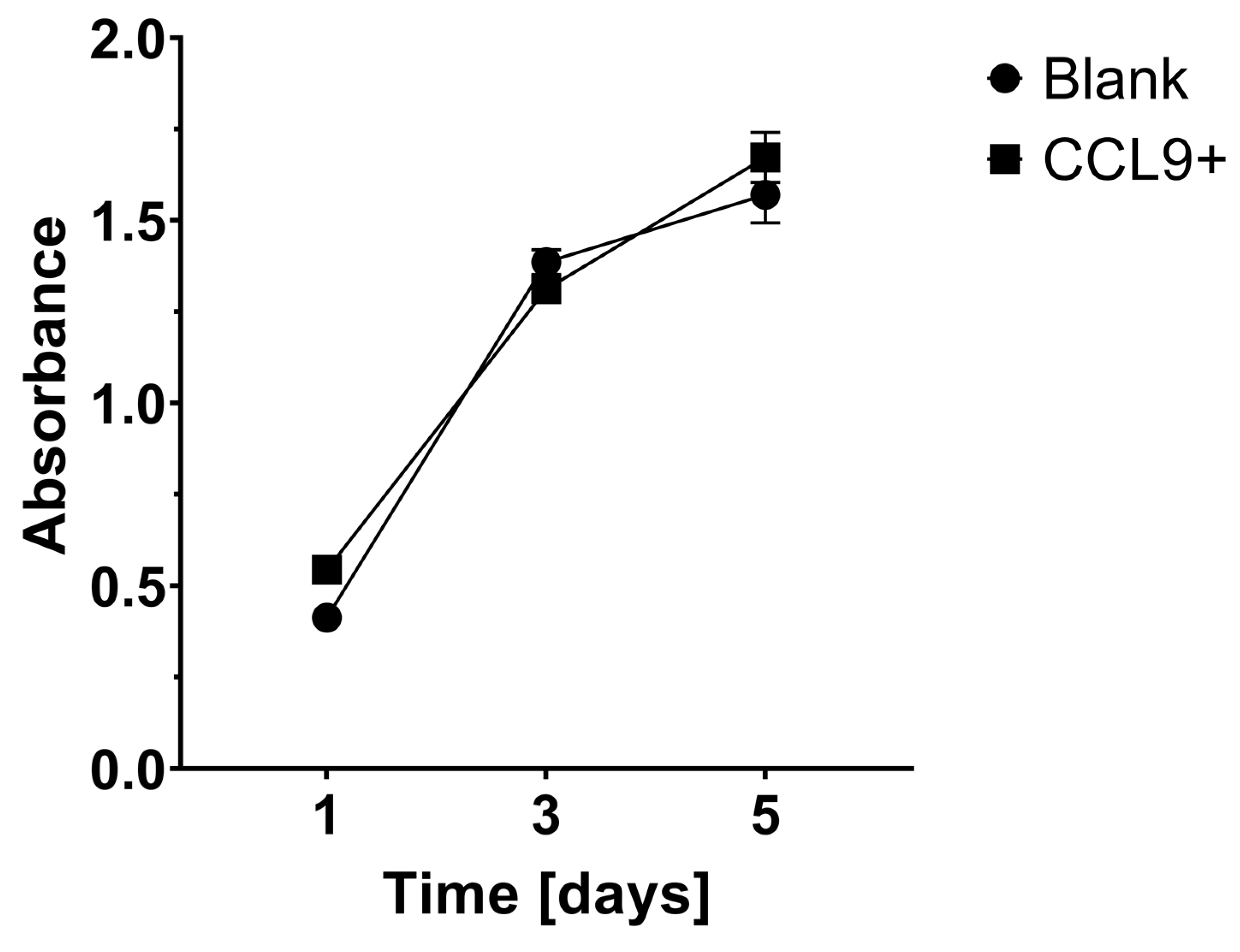
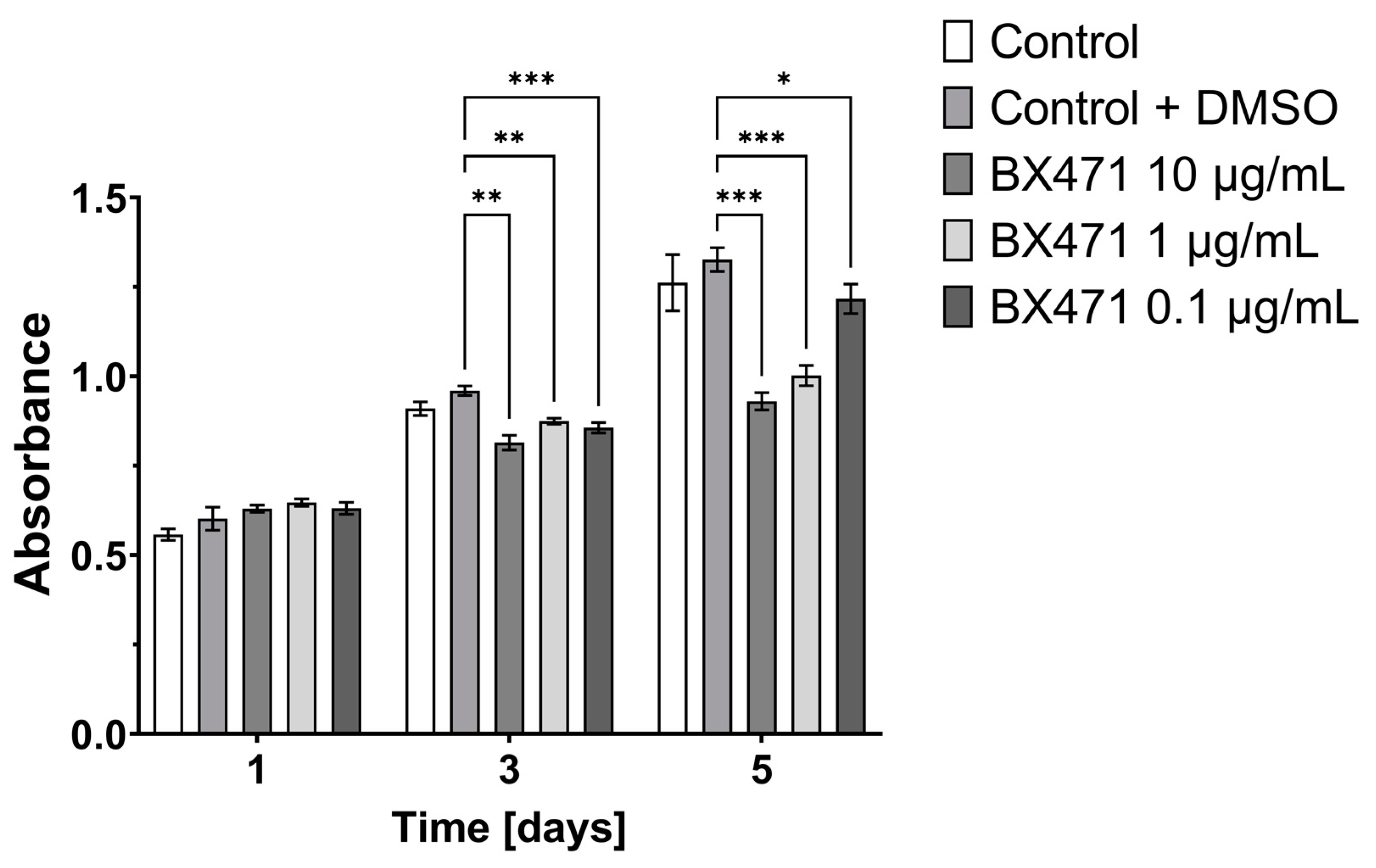
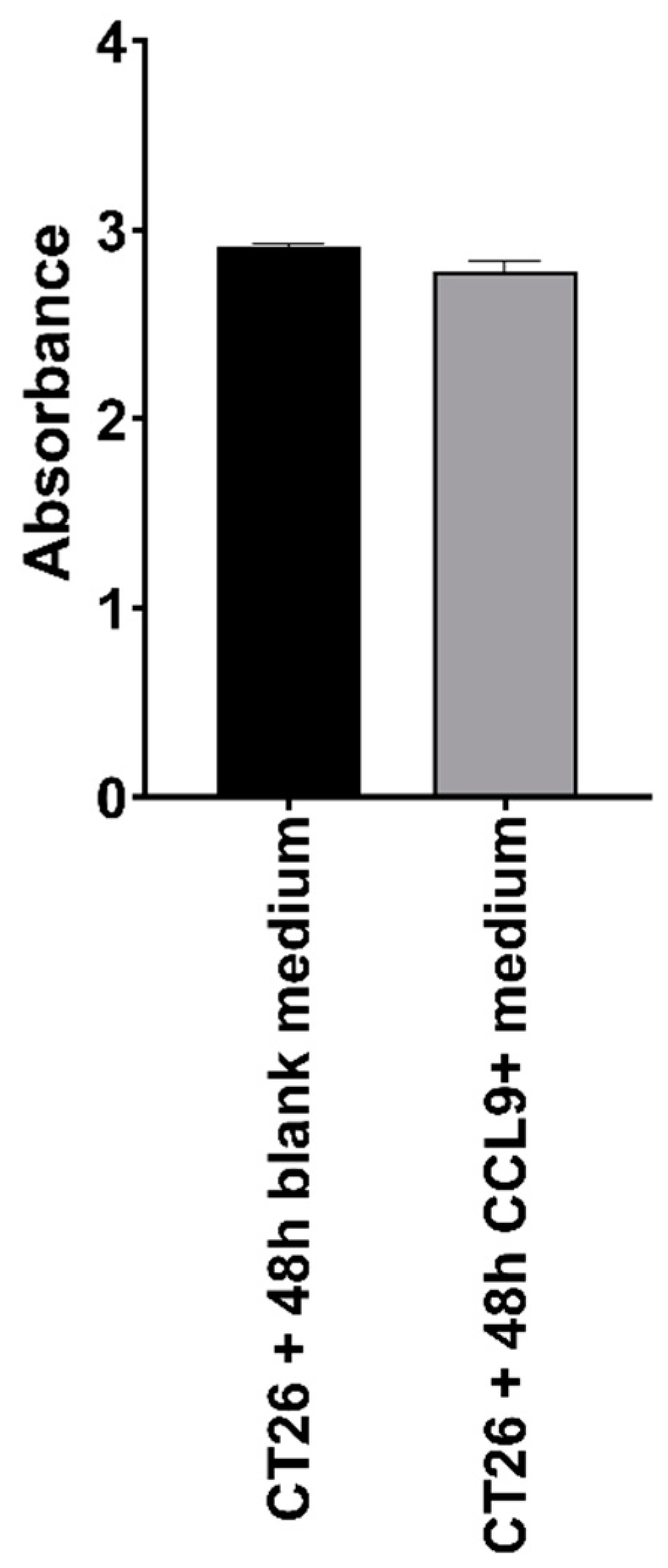
3.1. Effect of CCL9 or CCR1 Antagonist on CT26 Cell Proliferation In Vitro
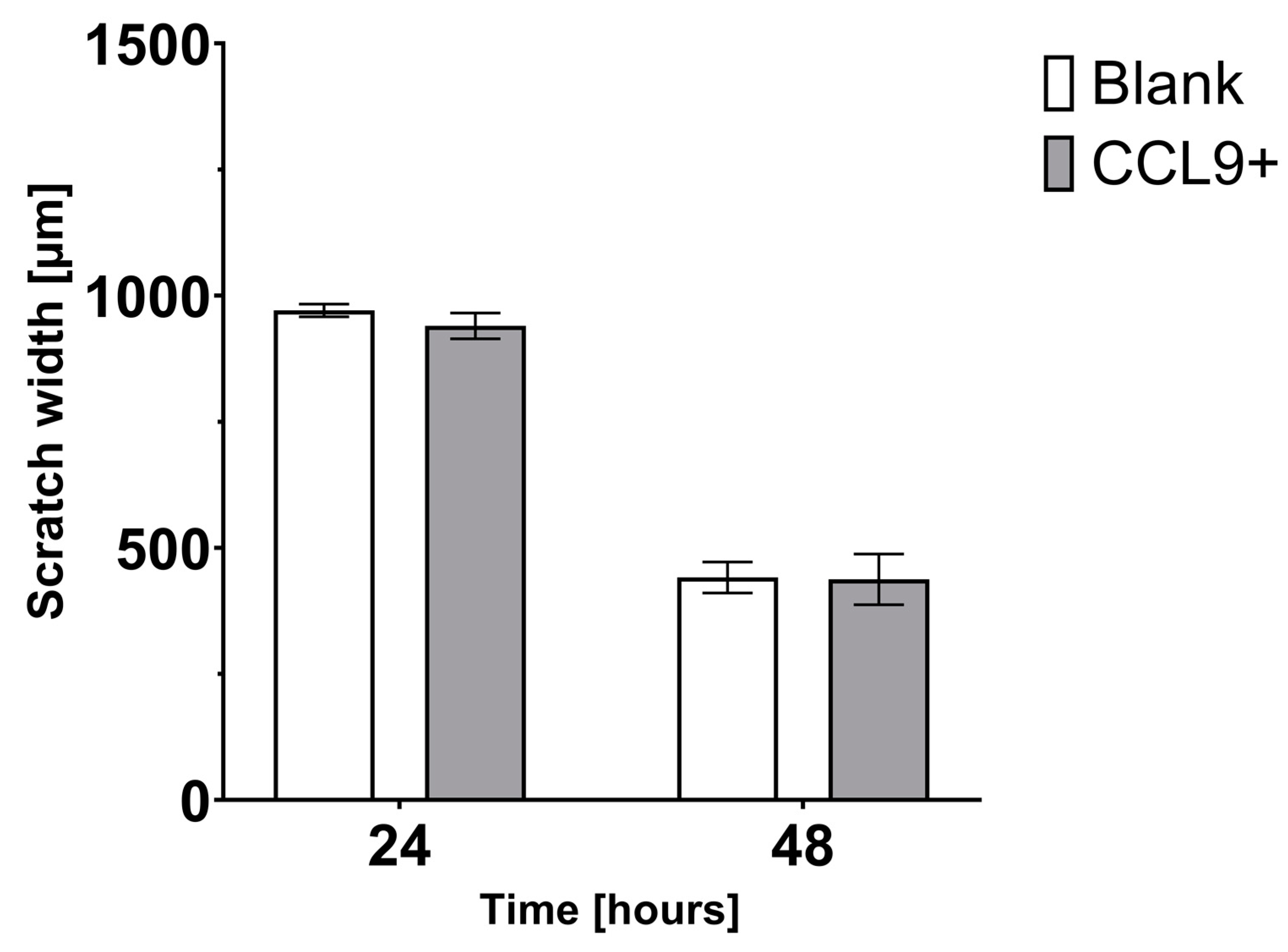
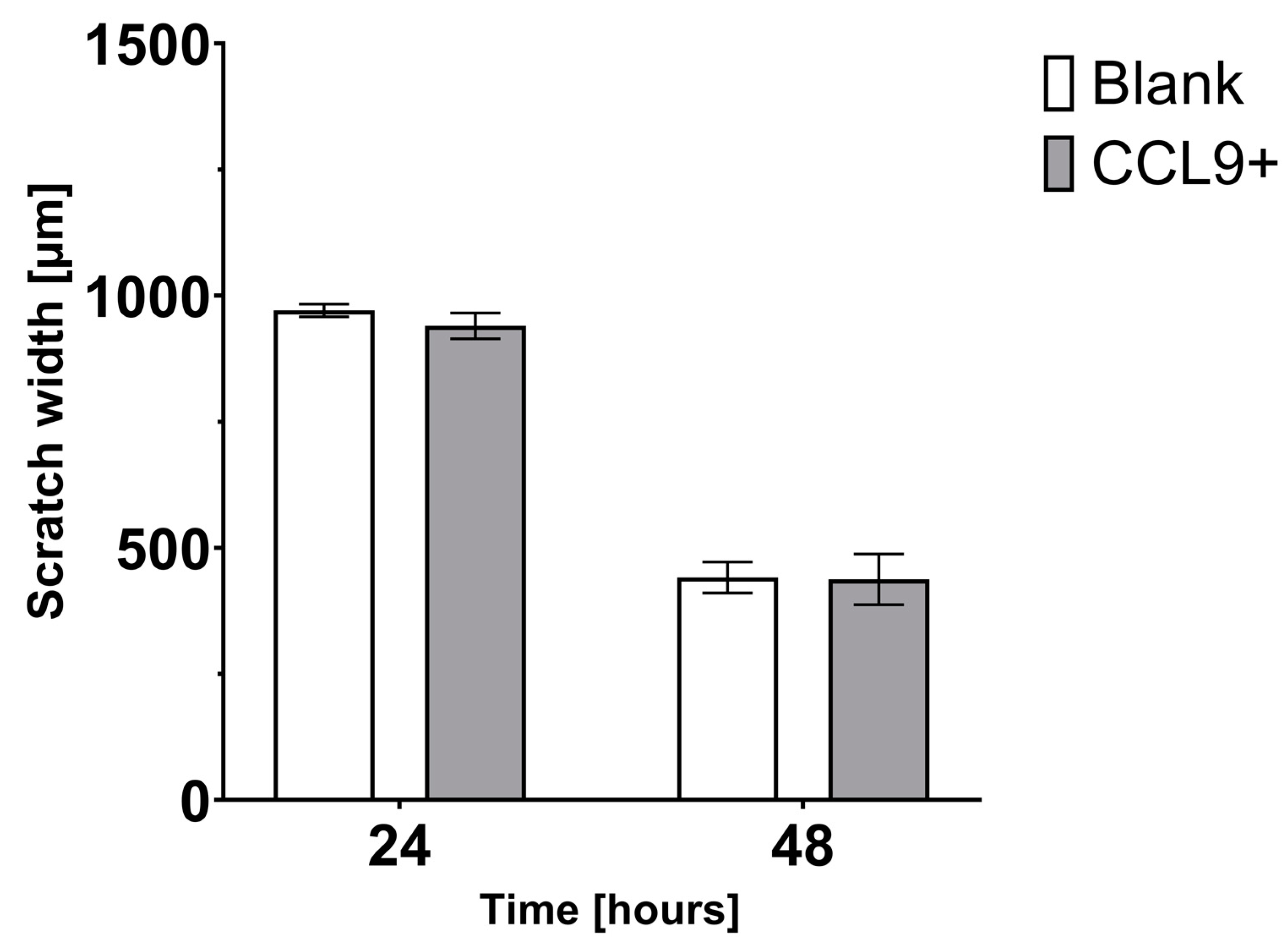
3.2. Effect of CCL9 on CT26 Cell Migration In Vitro
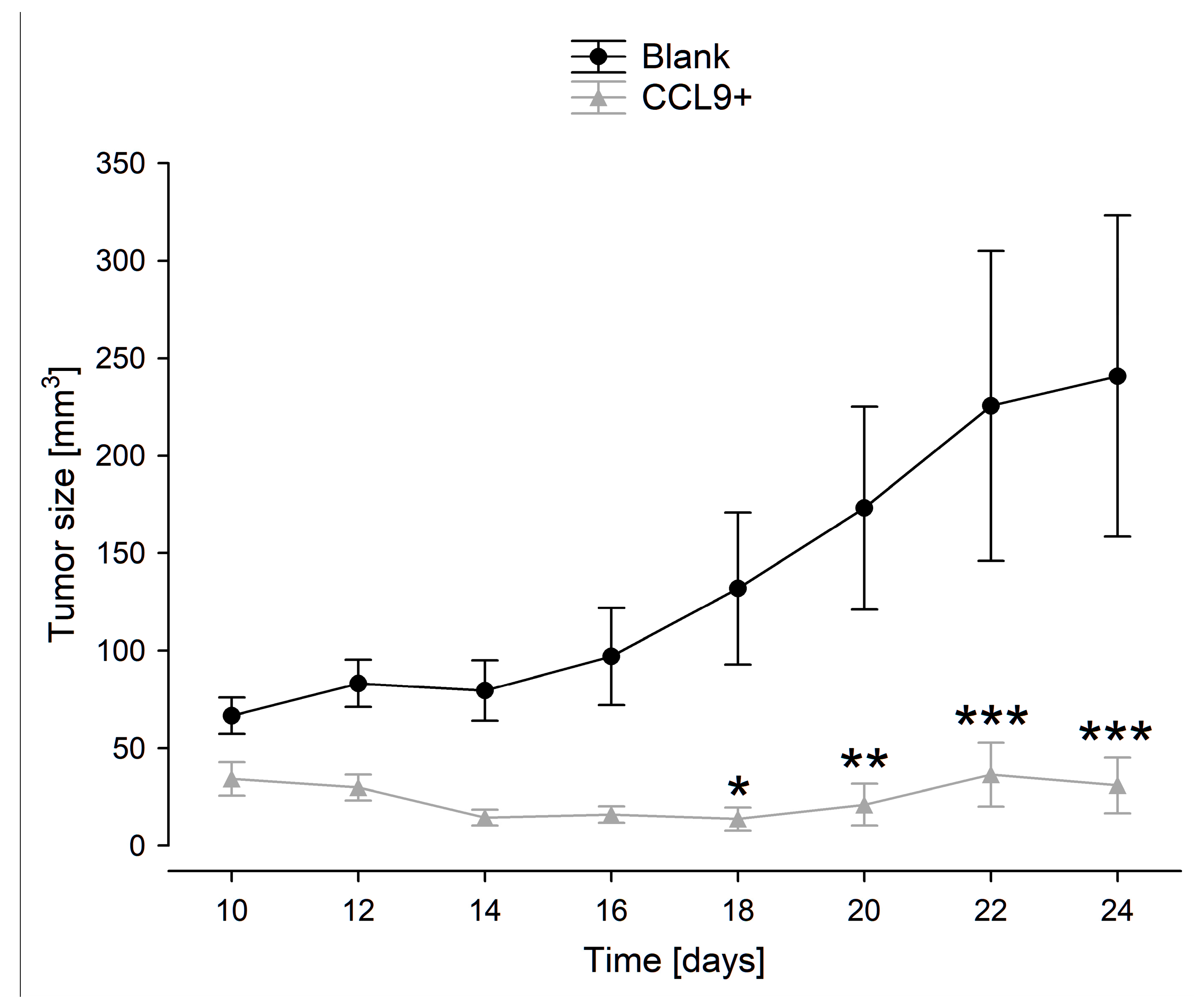
3.3. Tumor Growth in BALB/c Mice Inoculated with Blank Control or CCL9-Obverexpressing CT26 Cells
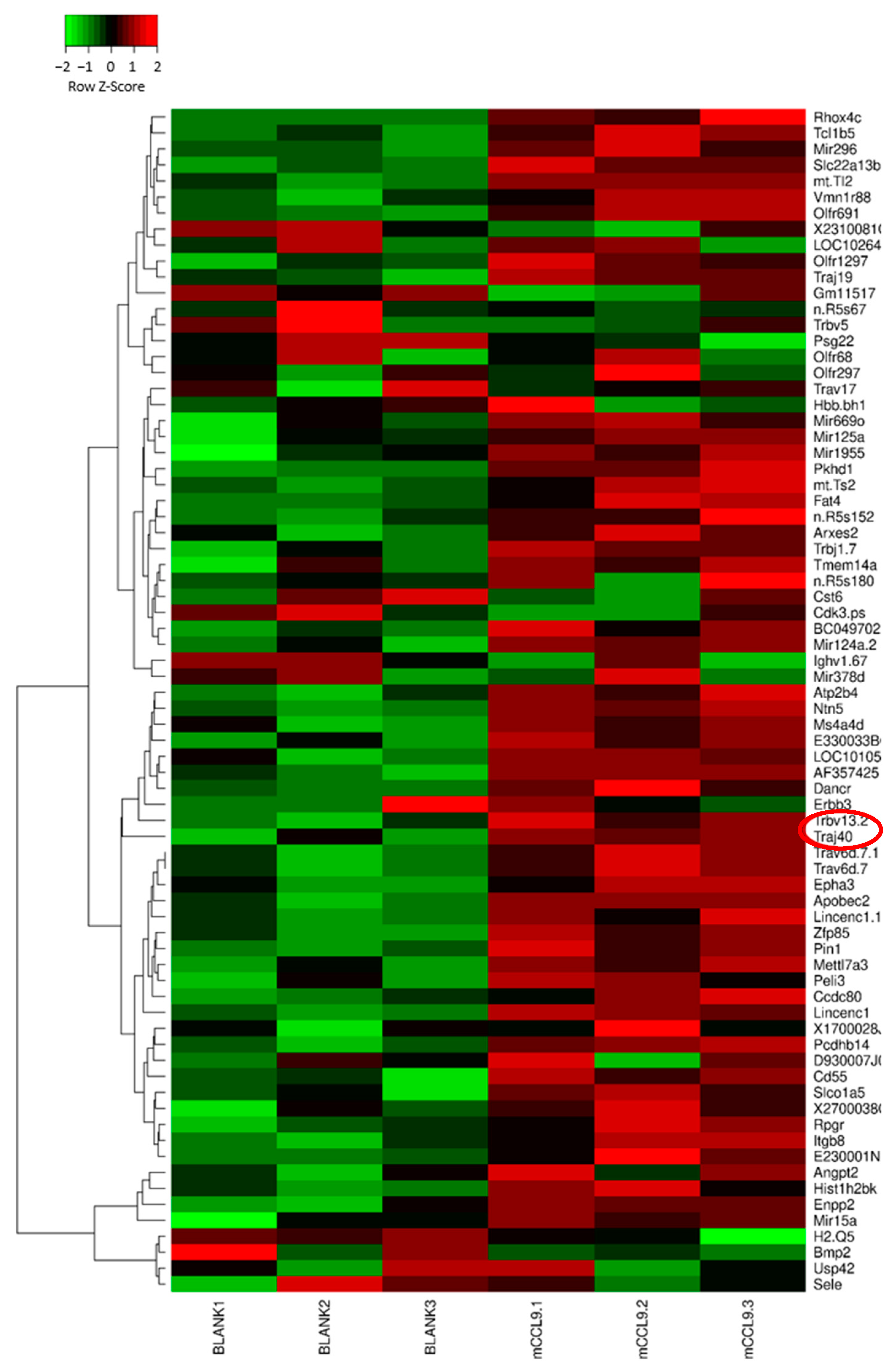
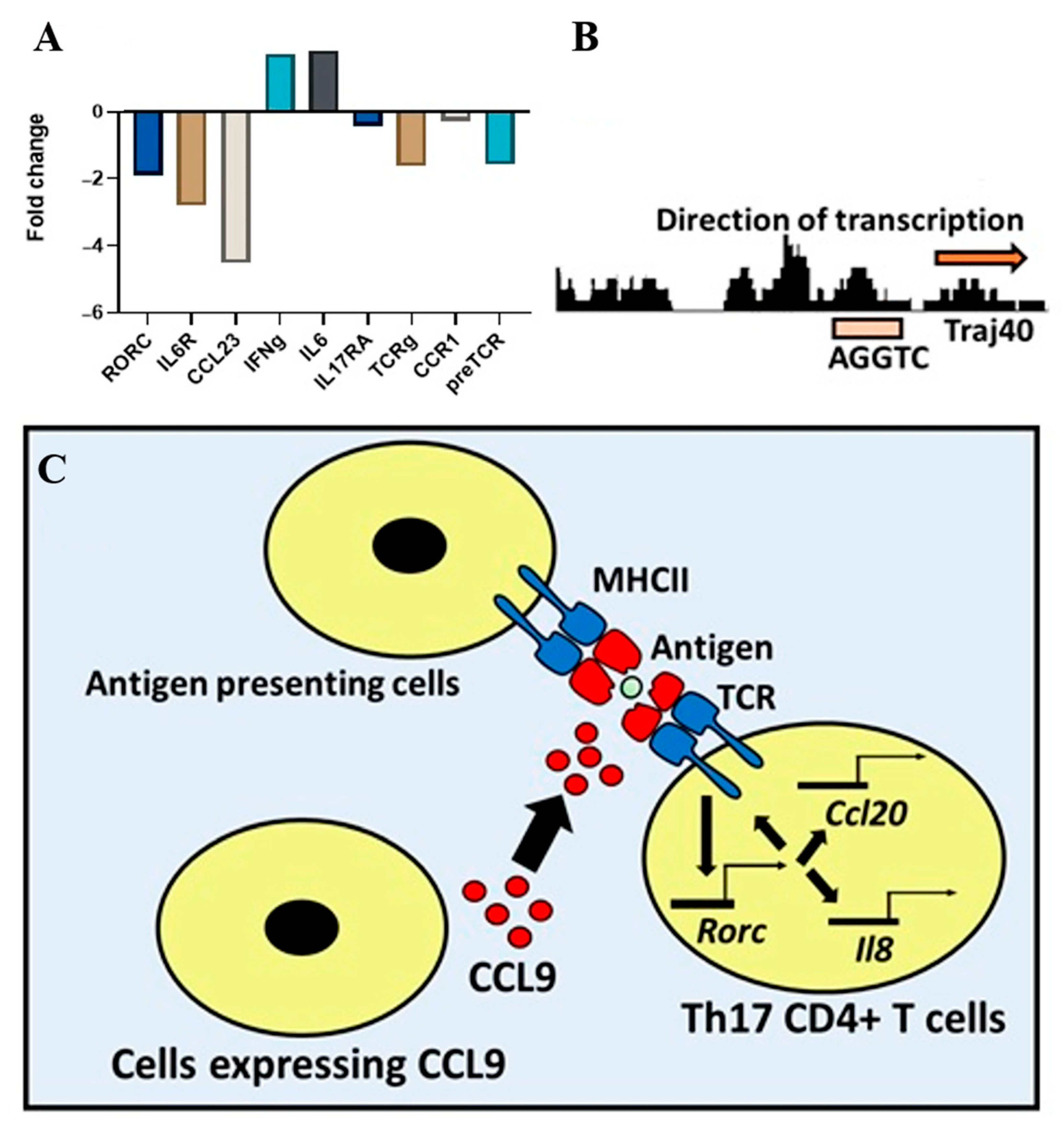
3.4. Microarray Results and Bioinformatics Analysis
4. Discussion
4.1. Perspectives for CCL9 Use in Anti-Cancer Therapy
4.2. Limitation of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in Colorectal Cancer: Rationale, Challenges and Potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Mickael, M.E.; Rajput, A.; Steyn, J.; Wiemerslage, L.; Bürglin, T. An Optimised Phylogenetic Method Sheds More Light on the Main Branching Events of Rhodopsin-like Superfamily. Comp. Biochem. Physiol. Part D Genom. Proteom. 2016, 20, 85–94. [Google Scholar] [CrossRef]
- Aman, A.; Piotrowski, T. Cell Migration during Morphogenesis. Dev. Biol. 2010, 341, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Middleton, J.; Patterson, A.M.; Gardner, L.; Schmutz, C.; Ashton, B.A. Leukocyte Extravasation: Chemokine Transport and Presentation by the Endothelium. Blood 2002, 100, 3853–3860. [Google Scholar] [CrossRef] [Green Version]
- Poltorak, A.N.; Bazzoni, F.; Smirnova, I.I.; Alejos, E.; Thompson, P.; Luheshi, G.; Rothwell, N.; Beutler, B. MIP-1 Gamma: Molecular Cloning, Expression, and Biological Activities of a Novel CC Chemokine That Is Constitutively Secreted in vivo. J. Inflamm. 1995, 45, 207–219. [Google Scholar] [PubMed]
- Kitamura, T.; Kometani, K.; Hashida, H.; Matsunaga, A.; Miyoshi, H.; Hosogi, H.; Aoki, M.; Oshima, M.; Hattori, M.; Takabayashi, A.; et al. SMAD4-Deficient Intestinal Tumors Recruit CCR1+ Myeloid Cells That Promote Invasion. Nat. Genet. 2007, 39, 467–475. [Google Scholar] [CrossRef]
- Nardi, V.; Naveiras, O.; Azam, M.; Daley, G.Q. ICSBP-Mediated Immune Protection against BCR-ABL–Induced Leukemia Requires the CCL6 and CCL9 Chemokines. Blood 2009, 113, 3813–3820. [Google Scholar] [CrossRef] [Green Version]
- Berahovich, R.D.; Miao, Z.; Wang, Y.; Premack, B.; Howard, M.C.; Schall, T.J. Proteolytic Activation of Alternative CCR1 Ligands in Inflammation. J. Immunol. 2005, 174, 7341–7351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Li, F.; Zhang, C.; Li, N.; Huang, H.; Shao, Z.; Zhang, M.; Zhan, X.; He, Y.; Ju, Z.; et al. Eosinophil-Derived Chemokine (HCCL15/23, MCCL6) Interacts with CCR1 to Promote Eosinophilic Airway Inflammation. Signal Transduct. Target. Ther. 2021, 6, 91. [Google Scholar] [CrossRef]
- Yan, H.H.; Jiang, J.; Pang, Y.; Achyut, B.R.; Lizardo, M.; Liang, X.; Hunter, K.; Khanna, C.; Hollander, C.; Yang, L. CCL9 Induced by TGFβ Signaling in Myeloid Cells Enhances Tumor Cell Survival in the Premetastatic Organ. Cancer Res. 2015, 75, 5283–5298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iotti, G.; Ferrari-Amorotti, G.; Rosafio, C.; Corradini, F.; Lidonnici, M.R.; Ronchetti, M.; Bardini, M.; Zhang, Y.; Martinez, R.; Blasi, F.; et al. Expression of CCL9/MIP-1γ Is Repressed by BCR/ABL and Its Restoration Suppresses in Vivo Leukemogenesis of 32D-BCR/ABL Cells. Oncogene 2007, 26, 3482–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.M.; Back, T.C.; Scarzello, A.J.; Subleski, J.J.; Hall, V.L.; Stauffer, J.K.; Chen, X.; Micic, D.; Alderson, K.; Murphy, W.J.; et al. Successful Immunotherapy with IL-2/Anti-CD40 Induces the Chemokine-Mediated Mitigation of an Immunosuppressive Tumor Microenvironment. Proc. Natl. Acad. Sci. USA 2009, 106, 19455–19460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, V.; Tallapragada, S.; Schaar, B.; Kamat, K.; Chanana, A.M.; Zhang, Y.; Patel, S.; Parkash, V.; Rinker-Schaeffer, C.; Folkins, A.K.; et al. Omental Macrophages Secrete Chemokine Ligands That Promote Ovarian Cancer Colonization of the Omentum via CCR1. Commun. Biol. 2020, 3, 524. [Google Scholar] [CrossRef]
- Steinbach, D. Identification of a Set of Seven Genes for the Monitoring of Minimal Residual Disease in Pediatric Acute Myeloid Leukemia. Clin. Cancer Res. 2006, 12, 2434–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, M.H.; Murray, G.I.; Stewart, K.N.; Norrie, G.; Mayer, C.; Hold, G.L.; Thomson, J.; Fyfe, N.; Hope, M.; Mowat, N.A.G.; et al. The Inflammatory Microenvironment in Colorectal Neoplasia. PLoS ONE 2011, 6, e15366. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Morishita, A.; Tani, J.; Sakamoto, T.; Fujita, K.; Katsura, A.; Tatsuta, M.; Nomura, T.; Yoneyama, H.; Iwama, H.; et al. Expression Profiles of 507 Proteins from a Biotin Label-Based Antibody Array in Human Colorectal Cancer. Oncol. Rep. 2014, 31, 1277–1281. [Google Scholar] [CrossRef]
- Liu, L.Z.; Zhang, Z.; Zheng, B.H.; Shi, Y.; Duan, M.; Ma, L.J.; Wang, Z.C.; Dong, L.Q.; Dong, P.P.; Shi, J.Y.; et al. CCL15 Recruits Suppressive Monocytes to Facilitate Immune Escape and Disease Progression in Hepatocellular Carcinoma. Hepatology 2019, 69, 143–159. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wu, J.; Zhang, W.; Zhang, N.; Guo, H. Identification of Serum CCL15 in Hepatocellular Carcinoma. Br. J. Cancer 2013, 108, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Kurzejamska, E.; Sacharczuk, M.; Landázuri, N.; Kovtonyuk, O.; Łazarczyk, M.; Ananthaseshan, S.; Gaciong, Z.; Religa, P. Effect of Chemokine (C-C Motif) Ligand 7 (CCL7) and Its Receptor (CCR2) Expression on Colorectal Cancer Behaviors. Int. J. Mol. Sci. 2019, 20, 686. [Google Scholar] [CrossRef] [Green Version]
- Ciofani, M.; Madar, A.; Galan, C.; Sellars, M.; MacE, K.; Pauli, F.; Agarwal, A.; Huang, W.; Parkurst, C.N.; Muratet, M.; et al. A Validated Regulatory Network for Th17 Cell Specification. Cell 2012, 151, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Mickael, M.E.; Bhaumik, S.; Basu, R. Retinoid-Related Orphan Receptor RORγt in CD4+ T-Cell–Mediated Intestinal Homeostasis and Inflammation. Am. J. Pathol. 2020, 190, 1984–1999. [Google Scholar] [CrossRef] [PubMed]
- Mickael, M.E.; Bhaumik, S.; Chakraborti, A.; Umfress, A.A.; van Groen, T.; Macaluso, M.; Totenhagen, J.; Sorace, A.G.; Bibb, J.A.; Standaert, D.G.; et al. RORγt-Expressing Pathogenic CD4+ T Cells Cause Brain Inflammation during Chronic Colitis. J. Immunol. 2022, 208, 2054–2066. [Google Scholar] [CrossRef]
- Bhaumik, S.; Mickael, M.E.; Moran, M.; Spell, M.; Basu, R. RORγt Promotes Foxp3 Expression by Antagonizing the Effector Program in Colonic Regulatory T Cells. J. Immunol. 2021, 207, 2027–2038. [Google Scholar] [CrossRef]
- Choy, E.; Rose-John, S. Interleukin-6 as a Multifunctional Regulator: Inflammation, Immune Response, and Fibrosis. J. Scleroderma Relat. Disord. 2017, 2, S1–S5. [Google Scholar] [CrossRef] [Green Version]
- Torre, D.; Lachmann, A.; Ma’ayan, A. BioJupies: Automated Generation of Interactive Notebooks for RNA-Seq Data Analysis in the Cloud. Cell Syst. 2018, 7, 556–561.e3. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hays, E.; Rama, M.; Bonavida, B. Cell-mediated immune resistance in cancer. Cancer Drug Resist 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amicarella, F.; Muraro, M.G.; Hirt, C.; Cremonesi, E.; Padovan, E.; Mele, V.; Governa, V.; Han, J.; Huber, X.; Droeser, R.A.; et al. Dual Role of Tumour-Infiltrating T Helper 17 Cells in Human Colorectal Cancer. Gut 2017, 66, 692–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubick, N.; Flournoy, P.C.H.; Enciu, A.-M.; Manda, G.; Mickael, M.-E. Drugs Modulating CD4+ T Cells Blood–Brain Barrier Interaction in Alzheimer’s Disease. Pharmaceutics 2020, 12, 880. [Google Scholar] [CrossRef]
- Kamali, A.N.; Noorbakhsh, S.M.; Hamedifar, H.; Jadidi-Niaragh, F.; Yazdani, R.; Bautista, J.M.; Azizi, G. A role for Th1-like Th17 cells in the pathogenesis of inflammatory and autoimmune disorders. Mol. Immunol. 2018, 105, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Crome, S.Q.; MacDonald, K.G.; Dai, E.L.; Mager, D.L.; Levings, M.K. Human Th1 and Th17 Cells Exhibit Epigenetic Stability at Signature Cytokine and Transcription Factor Loci. J. Immunol. 2011, 187, 5615–5626. [Google Scholar] [CrossRef] [Green Version]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef]
- Chen, K.; Liu, Q.; Tsang, L.L.; Ye, Q.; Chan, H.C.; Sun, Y.; Jiang, X. Human Mscs Promotes Colorectal Cancer Epithelial–Mesenchymal Transition and Progression via Ccl5/β-Catenin/Slug Pathway. Cell Death Dis. 2017, 8, e2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.Y.; Kim, S.H.; Cho, D.; Kim, T.S. Analysis of the Expression Profiles of Cytokines and Cytokine-Related Genes during the Progression of Breast Cancer Growth in Mice. Oncol. Rep. 2009, 22, 1141–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medler, T.R.; Coussens, L.M. Duality of the Immune Response in Cancer: Lessons Learned from Skin. J. Investig. Dermatol. 2014, 134, E23–E28. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, K.; Saikawa, Y.; Takeuchi, H.; Suda, K.; Fukuda, K.; Nakamura, R.; Takahashi, T.; Kawakubo, H.; Wada, N.; Miyasho, T.; et al. The Neutrophil Elastase Inhibitor Sivelestat Suppresses Accelerated Gastrointestinal Tumor Growth via Peritonitis after Cecal Ligation and Puncture. Anticancer Res. 2013, 33, 3653–3659. [Google Scholar]
- Selby, M.J.; Engelhardt, J.J.; Johnston, R.J.; Lu, L.-S.; Han, M.; Thudium, K.; Yao, D.; Quigley, M.; Valle, J.; Wang, C.; et al. Preclinical Development of Ipilimumab and Nivolumab Combination Immunotherapy: Mouse Tumor Models, In Vitro Functional Studies, and Cynomolgus Macaque Toxicology. PLoS ONE 2016, 11, e0161779. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Peng, D.; Lecanda, J.; Schmitz, V.; Barajas, M.; Qian, C.; Prieto, J. In Vivo Gene Transfer of CD40 Ligand into Colon Cancer Cells Induces Local Production of Cytokines and Chemokines, Tumor Eradication and Protective Antitumor Immunity. Gene Ther. 2000, 7, 1467–1476. [Google Scholar] [CrossRef] [Green Version]
- Itatani, Y.; Kawada, K.; Fujishita, T.; Kakizaki, F.; Hirai, H.; Matsumoto, T.; Iwamoto, M.; Inamoto, S.; Hatano, E.; Hasegawa, S.; et al. Loss of SMAD4 from Colorectal Cancer Cells Promotes CCL15 Expression to Recruit CCR1+ Myeloid Cells and Facilitate Liver Metastasis. Gastroenterology 2013, 145, 1064–1075.e11. [Google Scholar] [CrossRef] [Green Version]
- Mohit, E.; Rafati, S. Chemokine-Based Immunotherapy: Delivery Systems and Combination Therapies. Immunotherapy 2012, 4, 807–840. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The Human Tumor Microbiome Is Composed of Tumor Type-Specific Intracellular Bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, M.; Le’Negrate, G.; Krajewska, M.; Reed, J.C. Salmonella Typhimurium Engineered to Produce CCL21 Inhibit Tumor Growth. Cancer Immunol. Immunother. 2009, 58, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Din, M.O.; Danino, T.; Prindle, A.; Skalak, M.; Selimkhanov, J.; Allen, K.; Julio, E.; Atolia, E.; Tsimring, L.S.; Bhatia, S.N.; et al. Synchronized Cycles of Bacterial Lysis for in Vivo Delivery. Nature 2016, 536, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Savage, T.M.; Vincent, R.L.; Rae, S.S.; Huang, L.H.; Ahn, A.; Pu, K.; Li, F.; Santos-Alexis, K.D.L.; Coker, C.; Danino, T.; et al. Chemokines expressed by engineered bacteria recruit and orchestrate antitumor immunity. Sci. Adv. 2023, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Balato, A.; Fishelevich, R.; Chapoval, A.; Mann, D.; Gaspari, A. Th17/Tc17 infiltration and associated cytokine gene expression in elicitation phase of allergic contact dermatitis. Br. J. Dermatol. 2009, 161, 1301–1306. [Google Scholar] [CrossRef] [Green Version]
- Adams, T.; Sakamoto, K.; Ahangari, F.; Munivar, A.; Kaminski, N. Single Cell RNA-Sequencing Reveals Distinct Effects of Inhibition of FENDRR, a Long Non-Coding RNA Implicated in Fibroblast to Myofibroblast Differentiation. Am. J. Respir. Crit. Care Med. 2017, 195, A4664. Available online: https://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2017.195.1_MeetingAbstracts.A4664 (accessed on 27 February 2023).




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łazarczyk, M.; Kurzejamska, E.; Mickael, M.-E.; Poznański, P.; Skiba, D.; Sacharczuk, M.; Gaciong, Z.; Religa, P. Mouse CCL9 Chemokine Acts as Tumor Suppressor in a Murine Model of Colon Cancer. Curr. Issues Mol. Biol. 2023, 45, 3446-3461. https://doi.org/10.3390/cimb45040226
Łazarczyk M, Kurzejamska E, Mickael M-E, Poznański P, Skiba D, Sacharczuk M, Gaciong Z, Religa P. Mouse CCL9 Chemokine Acts as Tumor Suppressor in a Murine Model of Colon Cancer. Current Issues in Molecular Biology. 2023; 45(4):3446-3461. https://doi.org/10.3390/cimb45040226
Chicago/Turabian StyleŁazarczyk, Marzena, Ewa Kurzejamska, Michel-Edwar Mickael, Piotr Poznański, Dominik Skiba, Mariusz Sacharczuk, Zbigniew Gaciong, and Piotr Religa. 2023. "Mouse CCL9 Chemokine Acts as Tumor Suppressor in a Murine Model of Colon Cancer" Current Issues in Molecular Biology 45, no. 4: 3446-3461. https://doi.org/10.3390/cimb45040226
APA StyleŁazarczyk, M., Kurzejamska, E., Mickael, M. -E., Poznański, P., Skiba, D., Sacharczuk, M., Gaciong, Z., & Religa, P. (2023). Mouse CCL9 Chemokine Acts as Tumor Suppressor in a Murine Model of Colon Cancer. Current Issues in Molecular Biology, 45(4), 3446-3461. https://doi.org/10.3390/cimb45040226