
DNA Methylation Patterns in Relation to Acute Severity and Duration of Anxiety and Depression

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
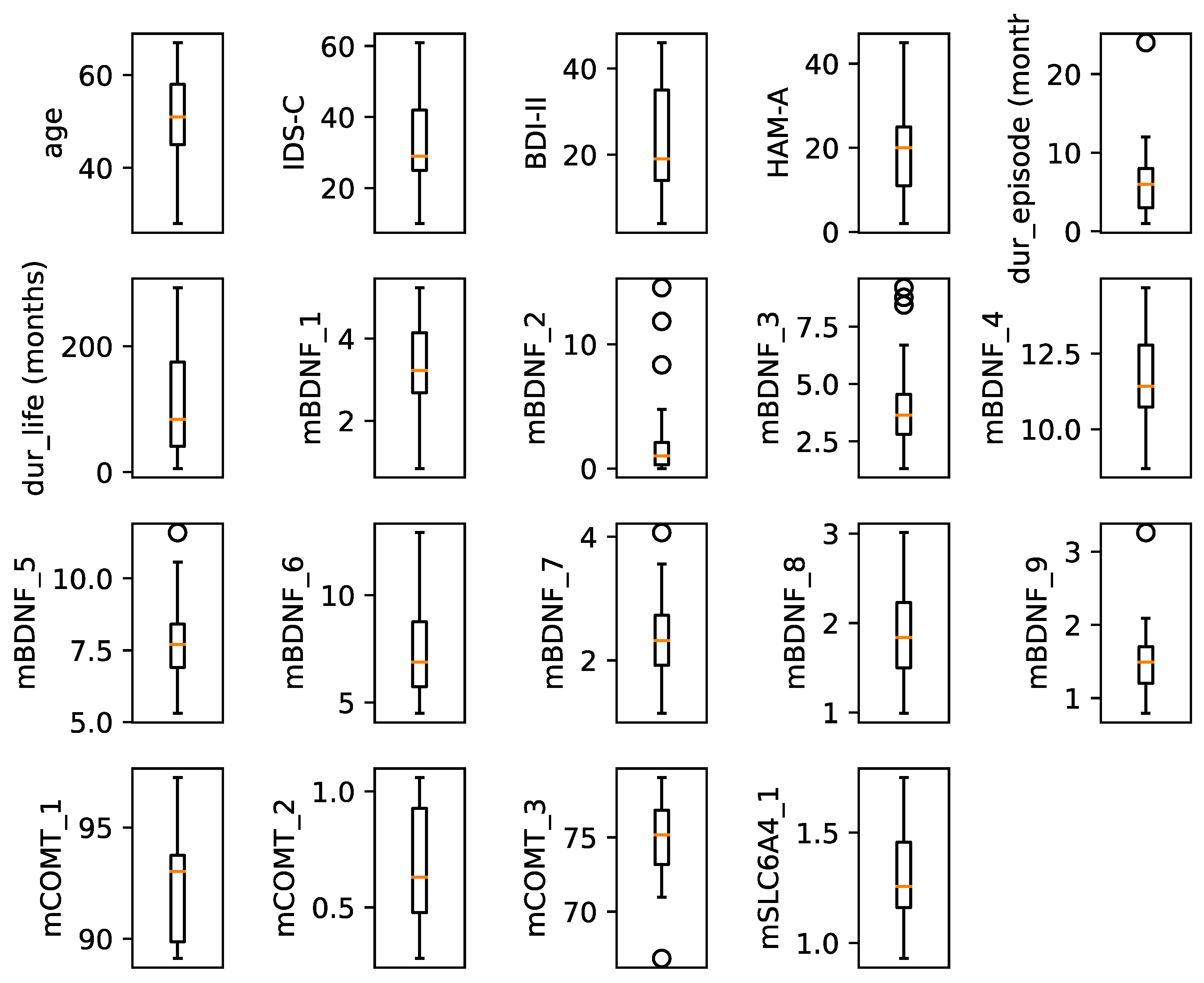
2.1. Participants
2.2. Study Design
2.3. DNA Methylation Analysis
2.3.1. Selection of Candidate Genes and Target Sequences
2.3.2. DNA Isolation and Bisulfite Conversion
2.3.3. Primer Design
2.3.4. Amplicon Generation and Sequencing
2.3.5. Library Preparation and NGS Sequencing
2.4. Bioinformatic and Statistical Analysis
3. Results
3.1. DNA Methylation Dynamics as a Flexible Change
3.2. DNA Methylation Dynamics as a Stable Change
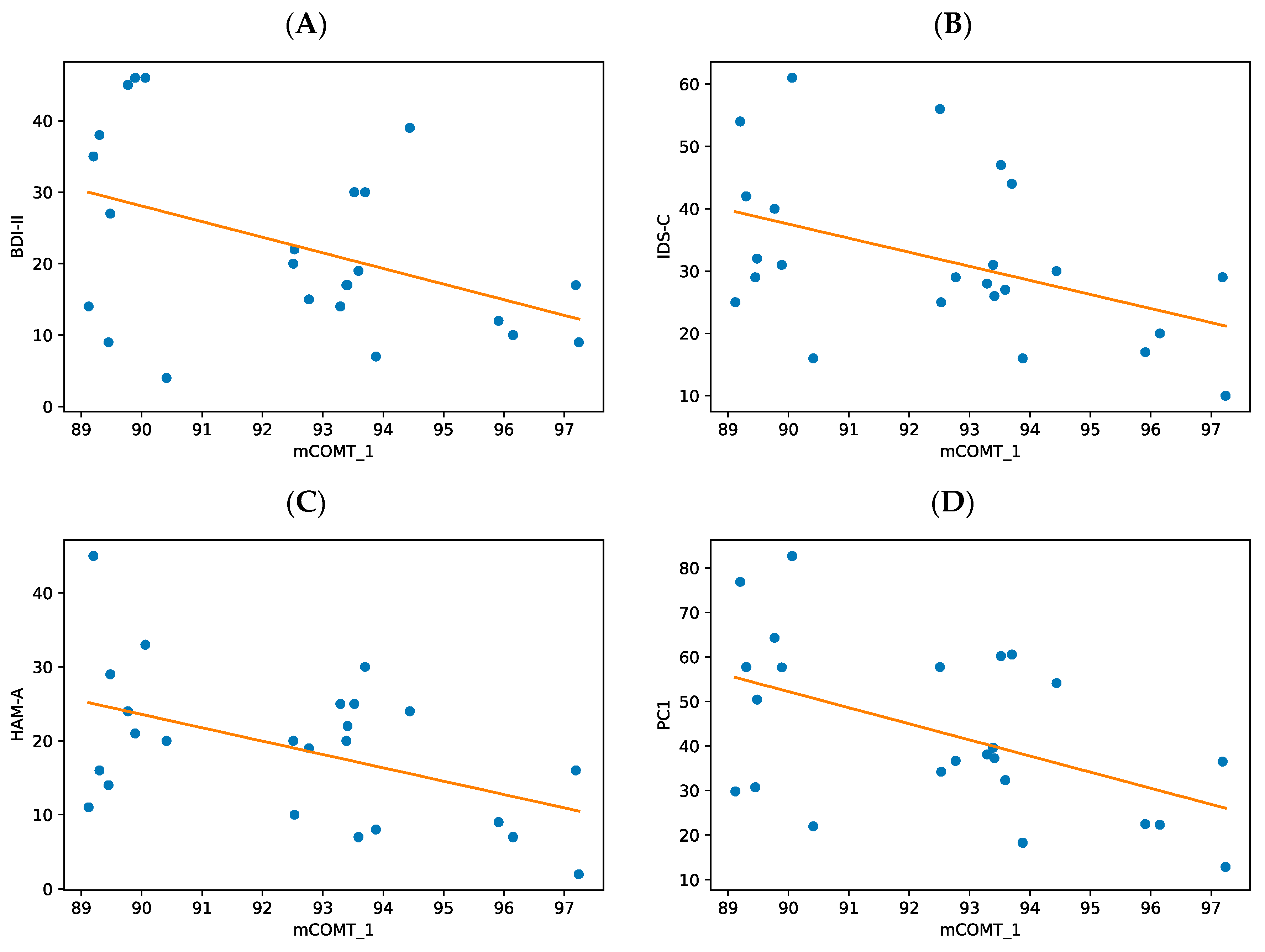
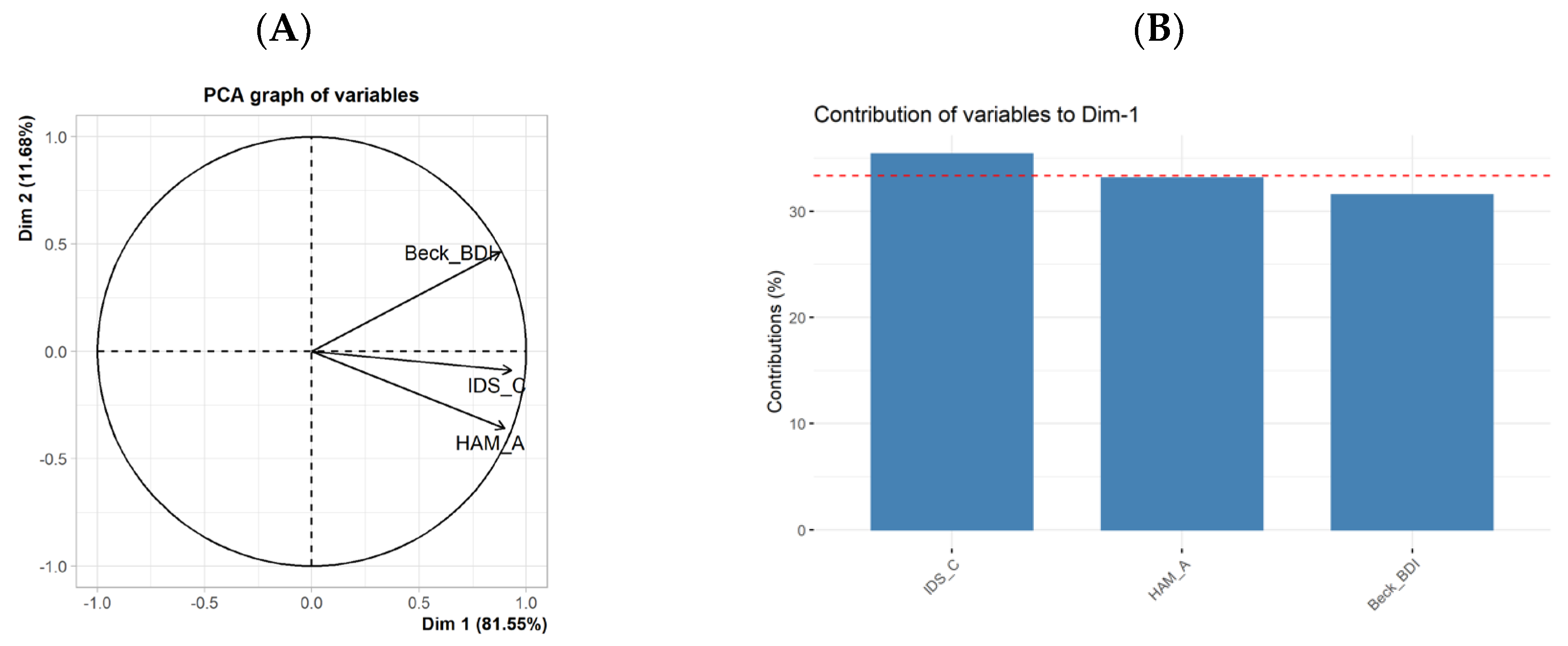
3.3. Differential Influences of BDI-II, HAM-A, and IDS-C on the DNA Methylation Levels
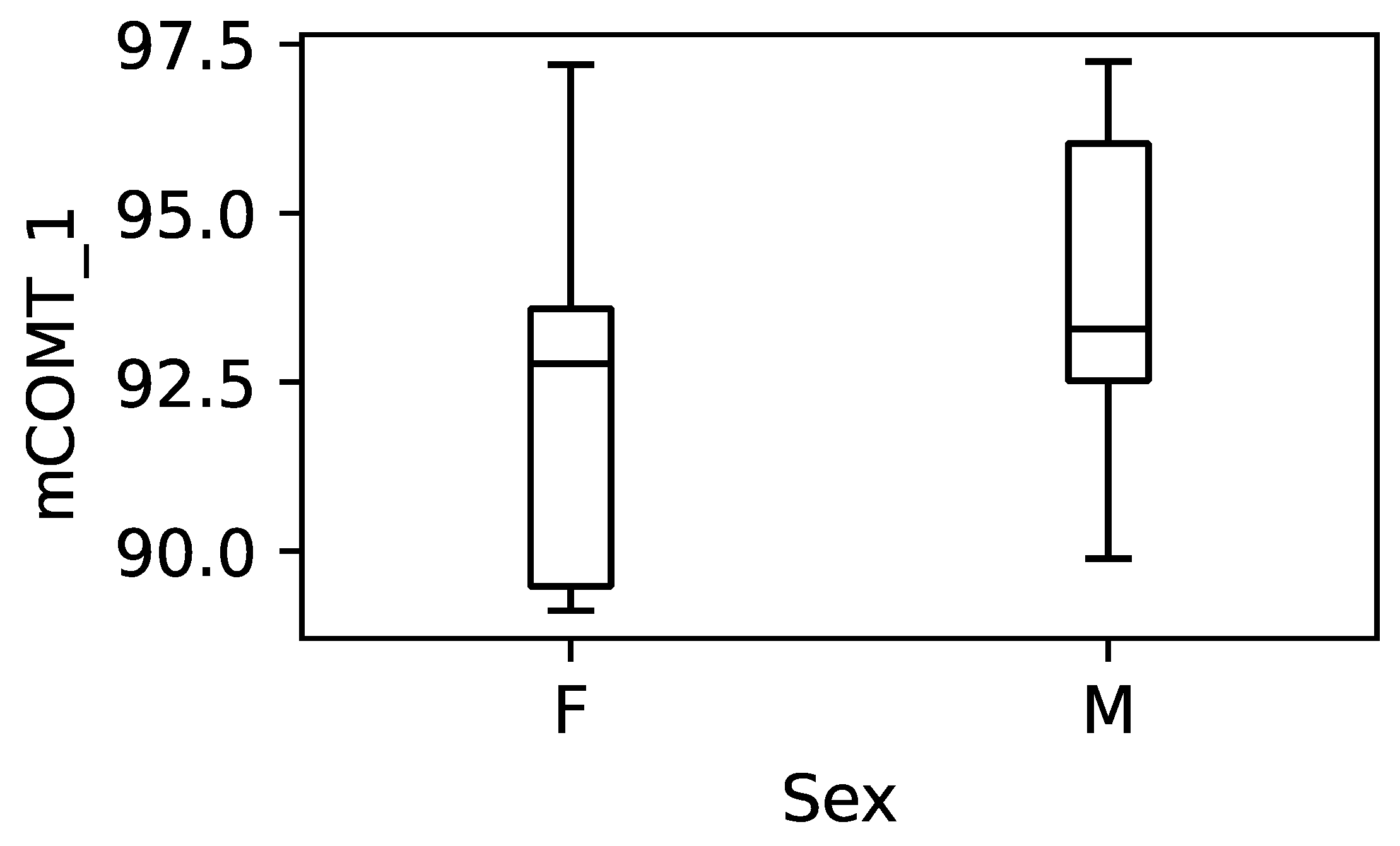
3.4. Effect of Gender on the DNA Methylation Levels
4. Discussion
4.1. DNA Methylation as a Flexible or Stable Epigenetic Change in Anxiety and Depression
4.2. DNA Methylation Levels Are Similarly Associated with Symptoms of Anxiety and Depression, but Not with Gender
4.3. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirschfeld, R.M.A. The Comorbidity of Major Depression and Anxiety Disorders: Recognition and Management in Primary Care. Prim. Care Companion J. Clin. Psychiatry 2001, 3, 244–254. [Google Scholar] [CrossRef]
- GBD 2019 Mental Disorders Collaborators. Global, Regional, and National Burden of 12 Mental Disorders in 204 Countries and Territories, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet Psychiatry 2022, 9, 137–150. [Google Scholar] [CrossRef]
- Burmeister, M.; McInnis, M.G.; Zöllner, S. Psychiatric Genetics: Progress amid Controversy. Nat. Rev. Genet. 2008, 9, 527–540. [Google Scholar] [CrossRef]
- Rusconi, F.; Battaglioli, E. Acute Stress-Induced Epigenetic Modulations and Their Potential Protective Role toward Depression. Front. Mol. Neurosci. 2018, 11, 184. [Google Scholar] [CrossRef]
- Sapolsky, R.M. Stress and the Brain: Individual Variability and the Inverted-U. Nat. Neurosci. 2015, 18, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, D.; Lightman, S.L.; Pariante, C.M. The HPA Axis in the Pathogenesis and Treatment of Depressive Disorders: Integrating Clinical and Molecular Findings. Psychopathol. Rev. 2016, 3, 64–76. [Google Scholar] [CrossRef]
- Hovens, J.G.F.M.; Giltay, E.J.; Spinhoven, P.; van Hemert, A.M.; Penninx, B.W.J.H. Impact of Childhood Life Events and Childhood Trauma on the Onset and Recurrence of Depressive and Anxiety Disorders. J. Clin. Psychiatry 2015, 76, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Lupien, S.J.; Juster, R.-P.; Raymond, C.; Marin, M.-F. The Effects of Chronic Stress on the Human Brain: From Neurotoxicity, to Vulnerability, to Opportunity. Front. Neuroendocrinol. 2018, 49, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Pan, F.; Tang, Y.; Huang, J.H. Editorial: Stress Induced Neural Changes in Emotional Disorders. Front. Psychiatry 2021, 12, 710691. [Google Scholar] [CrossRef]
- Boyer, P. Do Anxiety and Depression Have a Common Pathophysiological Mechanism? Acta Psychiatr. Scand. Suppl. 2000, 102, 24–29. [Google Scholar] [CrossRef]
- Kalin, N.H. The Critical Relationship between Anxiety and Depression. AJP 2020, 177, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Ressler, K.J.; Nemeroff, C.B. Role of Serotonergic and Noradrenergic Systems in the Pathophysiology of Depression and Anxiety Disorders. Depress. Anxiety 2000, 12 (Suppl. S1), 2–19. [Google Scholar] [CrossRef] [PubMed]
- Tiller, J.W.G. Depression and Anxiety. Med. J. Aust. 2013, 199, S28–S31. [Google Scholar] [CrossRef] [PubMed]
- Mourtzi, N.; Sertedaki, A.; Charmandari, E. Glucocorticoid Signaling and Epigenetic Alterations in Stress-Related Disorders. Int. J. Mol. Sci. 2021, 22, 5964. [Google Scholar] [CrossRef] [PubMed]
- Zannas, A.S.; Chrousos, G.P. Epigenetic Programming by Stress and Glucocorticoids along the Human Lifespan. Mol. Psychiatry 2017, 22, 640–646. [Google Scholar] [CrossRef]
- Chmielewska, N.; Szyndler, J.; Maciejak, P.; Płaźnik, A. Epigenetic Mechanisms of Stress and Depression. Psychiatr. Pol. 2019, 53, 1413–1428. [Google Scholar] [CrossRef]
- Kuehner, J.N.; Yao, B. The Dynamic Partnership of Polycomb and Trithorax in Brain Development and Diseases. Epigenomes 2019, 3, 17–24. [Google Scholar] [CrossRef]
- Glajch, K.E.; Sadri-Vakili, G. Epigenetic Mechanisms Involved in Huntington’s Disease Pathogenesis. J. Huntington’s Dis. 2015, 4, 1–15. [Google Scholar] [CrossRef]
- Lu, T.; Aron, L.; Zullo, J.; Pan, Y.; Kim, H.; Chen, Y.; Yang, T.-H.; Kim, H.-M.; Drake, D.; Liu, X.S.; et al. REST and Stress Resistance in Ageing and Alzheimer’s Disease. Nature 2014, 507, 448–454. [Google Scholar] [CrossRef]
- Lin, E.; Tsai, S.-J. Epigenetics and Depression: An Update. Psychiatry Investig. 2019, 16, 654–661. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The Diverse Roles of DNA Methylation in Mammalian Development and Disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Fitz-James, M.H.; Cavalli, G. Molecular Mechanisms of Transgenerational Epigenetic Inheritance. Nat. Rev. Genet. 2022, 23, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Bose, R.; Spulber, S.; Kilian, P.; Heldring, N.; Lönnerberg, P.; Johnsson, A.; Conti, M.; Hermanson, O.; Ceccatelli, S. Tet3 Mediates Stable Glucocorticoid-Induced Alterations in DNA Methylation and Dnmt3a/Dkk1 Expression in Neural Progenitors. Cell Death Dis. 2015, 6, e1793. [Google Scholar] [CrossRef]
- Duan, Z.; Lu, J. DNA Methyltransferases in Depression: An Update. Front. Psychiatry 2020, 11, 538683. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Fries, G.R.; Zannas, A.S.; Hartmann, J.; Zschocke, J.; Hafner, K.; Carrillo-Roa, T.; Steinbacher, J.; Preißinger, S.N.; Hoeijmakers, L.; et al. Chaperoning Epigenetics: FKBP51 Decreases the Activity of DNMT1 and Mediates Epigenetic Effects of the Antidepressant Paroxetine. Sci. Signal. 2015, 8, ra119. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.; Keshavan, M. The Neurobiology of Depression: An Integrated View. Asian J. Psychiatr. 2017, 27, 101–111. [Google Scholar] [CrossRef]
- Bouckaert, F.; Dols, A.; Emsell, L.; De Winter, F.-L.; Vansteelandt, K.; Claes, L.; Sunaert, S.; Stek, M.; Sienaert, P.; Vandenbulcke, M. Relationship between Hippocampal Volume, Serum BDNF, and Depression Severity Following Electroconvulsive Therapy in Late-Life Depression. Neuropsychopharmacology 2016, 41, 2741–2748. [Google Scholar] [CrossRef]
- Fossati, P.; Radtchenko, A.; Boyer, P. Neuroplasticity: From MRI to Depressive Symptoms. Eur. Neuropsychopharmacol. 2004, 14 (Suppl. S5), S503–S510. [Google Scholar] [CrossRef]
- Gatt, J.M.; Burton, K.L.O.; Williams, L.M.; Schofield, P.R. Specific and Common Genes Implicated across Major Mental Disorders: A Review of Meta-Analysis Studies. J. Psychiatr. Res. 2015, 60, 1–13. [Google Scholar] [CrossRef]
- Kiyohara, C.; Yoshimasu, K. Association between Major Depressive Disorder and a Functional Polymorphism of the 5-Hydroxytryptamine (Serotonin) Transporter Gene: A Meta-Analysis. Psychiatr. Genet. 2010, 20, 49–58. [Google Scholar] [CrossRef]
- López-León, S.; Janssens, A.C.J.W.; González-Zuloeta Ladd, A.M.; Del-Favero, J.; Claes, S.J.; Oostra, B.A.; van Duijn, C.M. Meta-Analyses of Genetic Studies on Major Depressive Disorder. Mol. Psychiatry 2008, 13, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Bakusic, J.; Schaufeli, W.; Claes, S.; Godderis, L. Stress, Burnout and Depression: A Systematic Review on DNA Methylation Mechanisms. J. Psychosom. Res. 2017, 92, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Klengel, T.; Pape, J.; Binder, E.B.; Mehta, D. The Role of DNA Methylation in Stress-Related Psychiatric Disorders. Neuropharmacology 2014, 80, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Rosenblat, J.D.; Brietzke, E.; Pan, Z.; Lee, Y.; Cao, B.; Zuckerman, H.; Kalantarova, A.; McIntyre, R.S. Stress, Epigenetics and Depression: A Systematic Review. Neurosci. Biobehav. Rev. 2019, 102, 139–152. [Google Scholar] [CrossRef]
- Smith, K.E.; Pollak, S.D. Early Life Stress and Development: Potential Mechanisms for Adverse Outcomes. J. Neurodevelop. Disord. 2020, 12, 34. [Google Scholar] [CrossRef]
- Jawahar, M.C.; Murgatroyd, C.; Harrison, E.L.; Baune, B.T. Epigenetic Alterations Following Early Postnatal Stress: A Review on Novel Aetiological Mechanisms of Common Psychiatric Disorders. Clin. Epigenet. 2015, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.-J.; Kim, J.-M.; Lee, J.-Y.; Kim, S.-Y.; Bae, K.-Y.; Kim, S.-W.; Shin, I.-S.; Kim, H.-R.; Shin, M.-G.; Yoon, J.-S. BDNF Promoter Methylation and Suicidal Behavior in Depressive Patients. J. Affect. Disord. 2013, 151, 679–685. [Google Scholar] [CrossRef]
- Kang, H.-J.; Kim, J.-M.; Stewart, R.; Kim, S.-Y.; Bae, K.-Y.; Kim, S.-W.; Shin, I.-S.; Shin, M.-G.; Yoon, J.-S. Association of SLC6A4 Methylation with Early Adversity, Characteristics and Outcomes in Depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 44, 23–28. [Google Scholar] [CrossRef]
- Matosin, N.; Cruceanu, C.; Binder, E.B. Preclinical and Clinical Evidence of DNA Methylation Changes in Response to Trauma and Chronic Stress. Chronic Stress 2017, 1, 247054701771076. [Google Scholar] [CrossRef]
- Provenzi, L.; Giorda, R.; Beri, S.; Montirosso, R. SLC6A4 Methylation as an Epigenetic Marker of Life Adversity Exposures in Humans: A Systematic Review of Literature. Neurosci. Biobehav. Rev. 2016, 71, 7–20. [Google Scholar] [CrossRef]
- Bock, J.; Wainstock, T.; Braun, K.; Segal, M. Stress In Utero: Prenatal Programming of Brain Plasticity and Cognition. Biol. Psychiatry 2015, 78, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lei, L.; De Rooij, S.R.; King, S.; Matthews, S.G.; Metz, G.A.S.; Roseboom, T.J.; Szyf, M. Prenatal Stress and Epigenetics. Neurosci. Biobehav. Rev. 2020, 117, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Amare, A.T.; Schubert, K.O.; Baune, B.T. Pharmacogenomics in the Treatment of Mood Disorders: Strategies and Opportunities for Personalized Psychiatry. EPMA J. 2017, 8, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Brunoni, A.R.; Carracedo, A.; Amigo, O.M.; Pellicer, A.L.; Talib, L.; Carvalho, A.F.; Lotufo, P.A.; Benseñor, I.M.; Gattaz, W.; Cappi, C. Association of BDNF, HTR2A, TPH1, SLC6A4, and COMT Polymorphisms with TDCS and Escitalopram Efficacy: Ancillary Analysis of a Double-Blind, Placebo-Controlled Trial. Braz. J. Psychiatry 2020, 42, 128–135. [Google Scholar] [CrossRef]
- Rasmi, Y.; Shokati, A.; Hassan, A.; Aziz, S.G.-G.; Bastani, S.; Jalali, L.; Moradi, F.; Alipour, S. The Role of DNA Methylation in Progression of Neurological Disorders and Neurodegenerative Diseases as Well as the Prospect of Using DNA Methylation Inhibitors as Therapeutic Agents for Such Disorders. IBRO Neurosci. Rep. 2023, 14, 28–37. [Google Scholar] [CrossRef]
- Andreescu, C.; Lenze, E.J.; Dew, M.A.; Begley, A.E.; Mulsant, B.H.; Dombrovski, A.Y.; Pollock, B.G.; Stack, J.; Miller, M.D.; Reynolds, C.F. Effect of Comorbid Anxiety on Treatment Response and Relapse Risk in Late-Life Depression: Controlled Study. Br. J. Psychiatry 2007, 190, 344–349. [Google Scholar] [CrossRef]
- Greenlee, A.; Karp, J.F.; Dew, M.A.; Houck, P.; Andreescu, C.; Reynolds, C.F. Anxiety Impairs Depression Remission in Partial Responders during Extended Treatment in Late-Life. Depress. Anxiety 2010, 27, 451–456. [Google Scholar] [CrossRef]
- Mulsant, B.H.; Reynolds, C.F.; Shear, M.K.; Sweet, R.A.; Miller, M. Comorbid Anxiety Disorders in Late-Life Depression. Anxiety 1996, 2, 242–247. [Google Scholar] [CrossRef]
- Beck, A.T.; Steer, R.A.; Carbin, M.G. Psychometric Properties of the Beck Depression Inventory: Twenty-Five Years of Evaluation. Clin. Psychol. Rev. 1988, 8, 77–100. [Google Scholar] [CrossRef]
- Trivedi, M.H.; Rush, A.J.; Ibrahim, H.M.; Carmody, T.J.; Biggs, M.M.; Suppes, T.; Crismon, M.L.; Shores-Wilson, K.; Toprac, M.G.; Dennehy, E.B.; et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in Public Sector Patients with Mood Disorders: A Psychometric Evaluation. Psychol. Med. 2004, 34, 73–82. [Google Scholar] [CrossRef]
- Bruss, G.S.; Gruenberg, A.M.; Goldstein, R.D.; Barber, J.P. Hamilton Anxiety Rating Scale Interview Guide: Joint Interview and Test-Retest Methods for Interrater Reliability. Psychiatry Res. 1994, 53, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Meng, L.; Pei, F.; Zheng, Y.; Leng, J. A Review of DNA Methylation in Depression. J. Clin. Neurosci. 2017, 43, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Na, K.-S.; Won, E.; Kang, J.; Kim, A.; Choi, S.; Tae, W.-S.; Kim, Y.-K.; Lee, M.-S.; Joe, S.-H.; Ham, B.-J. Differential Effect of COMT Gene Methylation on the Prefrontal Connectivity in Subjects with Depression versus Healthy Subjects. Neuropharmacology 2018, 137, 59–70. [Google Scholar] [CrossRef]
- Bishop, J.R.; Lee, A.M.; Mills, L.J.; Thuras, P.D.; Eum, S.; Clancy, D.; Erbes, C.R.; Polusny, M.A.; Lamberty, G.J.; Lim, K.O. Methylation of FKBP5 and SLC6A4 in Relation to Treatment Response to Mindfulness Based Stress Reduction for Posttraumatic Stress Disorder. Front. Psychiatry 2018, 9, 418. [Google Scholar] [CrossRef]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-Specific PCR: A Novel PCR Assay for Methylation Status of CpG Islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef]
- Wani, K.; Aldape, K.D. PCR Techniques in Characterizing DNA Methylation. In Clinical Applications of PCR; Luthra, R., Singh, R.R., Patel, K.P., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1392, pp. 177–186. ISBN 978-1-4939-3358-7. [Google Scholar]
- ThermoFisher Methyl Primer Express v1.0. Available online: https://resource.thermofisher.com/page/WE28396_2/ (accessed on 24 November 2021).
- Kouter, K.; Nikolac Perkovic, M.; Nedic Erjavec, G.; Milos, T.; Tudor, L.; Uzun, S.; Mimica, N.; Pivac, N.; Videtic Paska, A. Difference in Methylation and Expression of Brain-Derived Neurotrophic Factor in Alzheimer’s Disease and Mild Cognitive Impairment. Biomedicines 2023, 11, 235. [Google Scholar] [CrossRef]
- Institute of Enzymology BiSearch v2.63. Available online: http://bisearch.enzim.hu/ (accessed on 24 November 2021).
- Integrated DNA Technologies IDT. Oligo Analyzer. Available online: https://eu.idtdna.com/pages/tools/oligoanalyzer (accessed on 24 November 2021).
- Illumina PCR Amplicon, PCR Clean-Up, and Index PCR: 16S Metagenomic Sequencing Library Preparation. Available online: https://www.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 22 February 2023).
- Andrews, S. Fastqc: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 March 2022).
- Krueger, F. Trimgalore. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 20 March 2022).
- Krueger, F.; Andrews, S.R. Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Altuna Akalin MethylKit. Available online: https://bioconductor.org/packages/methylKit (accessed on 22 February 2023).
- Park, Y.; Cavalcante, R. MethylSig. Available online: https://bioconductor.org/packages/methylSig (accessed on 22 February 2023).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 21 February 2023).
- Horvath, S. DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Ryan, C.P. “Epigenetic Clocks”: Theory and Applications in Human Biology. Am. J. Hum. Biol. 2021, 33, e23488. [Google Scholar] [CrossRef]
- Allaire, J. RStudio: Integrated Development Environment for R; RStudio: Boston, MA, USA, 2012; Volume 770, pp. 165–171. [Google Scholar]
- Kundakovic, M.; Rocks, D. Sex Hormone Fluctuation and Increased Female Risk for Depression and Anxiety Disorders: From Clinical Evidence to Molecular Mechanisms. Front. Neuroendocrinol. 2022, 66, 101010. [Google Scholar] [CrossRef]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA Methylation Analysis in Nonalcoholic Fatty Liver Disease Suggests Distinct Disease-Specific and Remodeling Signatures after Bariatric Surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef]
- Dugué, P.-A.; Jung, C.-H.; Joo, J.E.; Wang, X.; Wong, E.M.; Makalic, E.; Schmidt, D.F.; Baglietto, L.; Severi, G.; Southey, M.C.; et al. Smoking and Blood DNA Methylation: An Epigenome-Wide Association Study and Assessment of Reversibility. Epigenetics 2020, 15, 358–368. [Google Scholar] [CrossRef]
- Izquierdo, A.G.; Crujeiras, A.B. Obesity-Related Epigenetic Changes After Bariatric Surgery. Front. Endocrinol. 2019, 10, 232. [Google Scholar] [CrossRef]
- Tsaprouni, L.G.; Yang, T.-P.; Bell, J.; Dick, K.J.; Kanoni, S.; Nisbet, J.; Viñuela, A.; Grundberg, E.; Nelson, C.P.; Meduri, E.; et al. Cigarette Smoking Reduces DNA Methylation Levels at Multiple Genomic Loci but the Effect Is Partially Reversible upon Cessation. Epigenetics 2014, 9, 1382–1396. [Google Scholar] [CrossRef] [PubMed]
- Babb, J.A.; Carini, L.M.; Spears, S.L.; Nephew, B.C. Transgenerational Effects of Social Stress on Social Behavior, Corticosterone, Oxytocin, and Prolactin in Rats. Horm. Behav. 2014, 65, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, A.; Mansuy, I.M. Inter- and Transgenerational Inheritance of Behavioral Phenotypes. Curr. Opin. Behav. Sci. 2019, 25, 96–101. [Google Scholar] [CrossRef]
- Sales, A.J.; Guimarães, F.S.; Joca, S.R.L. DNA Methylation in Stress and Depression: From Biomarker to Therapeutics. Acta Neuropsychiatr. 2021, 33, 217–241. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; D’Arcy, C.; Li, X.; Zhang, T.; Joober, R.; Meng, X. What Do DNA Methylation Studies Tell Us about Depression? A Systematic Review. Transl. Psychiatry 2019, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Brody, G.H.; Miller, G.E.; Yu, T.; Beach, S.R.H.; Chen, E. Supportive Family Environments Ameliorate the Link Between Racial Discrimination and Epigenetic Aging: A Replication Across Two Longitudinal Cohorts. Psychol. Sci. 2016, 27, 530–541. [Google Scholar] [CrossRef]
- Shields, A.E.; Wise, L.A.; Ruiz-Narvaez, E.A.; Seddighzadeh, B.; Byun, H.-M.; Cozier, Y.C.; Rosenberg, L.; Palmer, J.R.; Baccarelli, A.A. Childhood Abuse, Promoter Methylation of Leukocyte NR3C1 and the Potential Modifying Effect of Emotional Support. Epigenomics 2016, 8, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA Methylation Prevents Spurious Transcription Initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Morinobu, S.; Fuchikami, M.; Segawa, M.; Yokomaku, K.; Kataoka, T.; Okamoto, Y.; Yamawaki, S.; Inoue, T.; Kusumi, I.; et al. The Potential of SLC6A4 Gene Methylation Analysis for the Diagnosis and Treatment of Major Depression. J. Psychiatr. Res. 2014, 53, 47–53. [Google Scholar] [CrossRef]
- Peng, H.; Zhu, Y.; Strachan, E.; Fowler, E.; Bacus, T.; Roy-Byrne, P.; Goldberg, J.; Vaccarino, V.; Zhao, J. Childhood Trauma, DNA Methylation of Stress-Related Genes, and Depression: Findings From Two Monozygotic Twin Studies. Psychosom. Med. 2018, 80, 599–608. [Google Scholar] [CrossRef]
- Zhao, J.; Goldberg, J.; Bremner, J.D.; Vaccarino, V. Association Between Promoter Methylation of Serotonin Transporter Gene and Depressive Symptoms: A Monozygotic Twin Study. Psychosom. Med. 2013, 75, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Stewart, R.; Kang, H.J.; Kim, S.W.; Shin, I.S.; Kim, H.R.; Shin, M.G.; Kim, J.T.; Park, M.S.; Cho, K.H.; et al. A Longitudinal Study of SLC6A4 DNA Promoter Methylation and Poststroke Depression. J. Psychiatr. Res. 2013, 47, 1222–1227. [Google Scholar] [CrossRef]
- Iga, J.; Watanabe, S.; Numata, S.; Umehara, H.; Nishi, A.; Kinoshita, M.; Inoshita, M.; Shimodera, S.; Fujita, H.; Ohmori, T. Association Study of Polymorphism in the Serotonin Transporter Gene Promoter, Methylation Profiles, and Expression in Patients with Major Depressive Disorder: 5HTT Gene Expression and Methylation Profiles in Depression. Hum. Psychopharmacol. Clin. Exp. 2016, 31, 193–199. [Google Scholar] [CrossRef]
- Lam, D.; Ancelin, M.L.; Ritchie, K.; Freak-Poli, R.; Saffery, R.; Ryan, J. Genotype-Dependent Associations between Serotonin Transporter Gene (SLC6A4) DNA Methylation and Late-Life Depression. BMC Psychiatry 2018, 18, 282. [Google Scholar] [CrossRef]
- Kang, H.; Kim, K.; Kim, J.; Kim, S.; Park, M.; Kim, H.; Shin, M.; Cho, K.; Kim, J. A Longitudinal Study of the Associations of BDNF Genotype and Methylation with Poststroke Anxiety. Int. J. Geriatr. Psychiatry 2019, 34, 1706–1714. [Google Scholar] [CrossRef]
- Kang, H.J.; Kim, J.M.; Kim, S.Y.; Kim, S.W.; Shin, I.S.; Kim, H.R.; Park, M.H.; Shin, M.G.; Yoon, J.H.; Yoon, J.S. A Longitudinal Study of BDNF Promoter Methylation and Depression in Breast Cancer. Psychiatry Investig. 2015, 12, 523–531. [Google Scholar] [CrossRef]
- Song, Y.; Miyaki, K.; Suzuki, T.; Sasaki, Y.; Tsutsumi, A.; Kawakami, N.; Shimazu, A.; Takahashi, M.; Inoue, A.; Kan, C.; et al. Altered DNA Methylation Status of Human Brain Derived Neurotrophis Factor Gene Could Be Useful as Biomarker of Depression. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165B, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Nour El Huda, A.R.; Norsidah, K.Z.; Nabil Fikri, M.R.; Hanisah, M.N.; Kartini, A.; Norlelawati, A.T. DNA Methylation of Membrane-Bound Catechol-O-Methyltransferase in Malaysian Schizophrenia Patients: DNA Methylation, COMT, and Schizophrenia. Psychiatry Clin. Neurosci. 2018, 72, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleky, H.M.; Cheng, K.-H.; Faraone, S.V.; Wilcox, M.; Glatt, S.J.; Gao, F.; Smith, C.L.; Shafa, R.; Aeali, B.; Carnevale, J.; et al. Hypomethylation of MB-COMT Promoter Is a Major Risk Factor for Schizophrenia and Bipolar Disorder. Hum. Mol. Genet. 2006, 15, 3132–3145. [Google Scholar] [CrossRef]
- Nohesara, S.; Ghadirivasfi, M.; Mostafavi, S.; Eskandari, M.-R.; Ahmadkhaniha, H.; Thiagalingam, S.; Abdolmaleky, H.M. DNA Hypomethylation of MB-COMT Promoter in the DNA Derived from Saliva in Schizophrenia and Bipolar Disorder. J. Psychiatr. Res. 2011, 45, 1432–1438. [Google Scholar] [CrossRef]
- Walton, E.; Liu, J.; Hass, J.; White, T.; Scholz, M.; Roessner, V.; Gollub, R.; Calhoun, V.D.; Ehrlich, S. MB-COMT Promoter DNA Methylation Is Associated with Working-Memory Processing in Schizophrenia Patients and Healthy Controls. Epigenetics 2014, 9, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Shirvani-Farsani, Z.; Maloum, Z.; Bagheri-Hosseinabadi, Z.; Vilor-Tejedor, N.; Sadeghi, I. DNA Methylation Signature as a Biomarker of Major Neuropsychiatric Disorders. J. Psychiatr. Res. 2021, 141, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Polli, A.; Hendrix, J.; Ickmans, K.; Bakusic, J.; Ghosh, M.; Monteyne, D.; Velkeniers, B.; Bekaert, B.; Nijs, J.; Godderis, L. Genetic and Epigenetic Regulation of Catechol-O-Methyltransferase in Relation to Inflammation in Chronic Fatigue Syndrome and Fibromyalgia. J. Transl. Med. 2022, 20, 487. [Google Scholar] [CrossRef]
- Ursini, G.; Bollati, V.; Fazio, L.; Porcelli, A.; Iacovelli, L.; Catalani, A.; Sinibaldi, L.; Gelao, B.; Romano, R.; Rampino, A.; et al. Stress-Related Methylation of the Catechol-O-Methyltransferase Val158 Allele Predicts Human Prefrontal Cognition and Activity. J. Neurosci. 2011, 31, 6692–6698. [Google Scholar] [CrossRef]
- Wiegand, A.; Blickle, A.; Brückmann, C.; Weller, S.; Nieratschker, V.; Plewnia, C. Dynamic DNA Methylation Changes in the COMT Gene Promoter Region in Response to Mental Stress and Its Modulation by Transcranial Direct Current Stimulation. Biomolecules 2021, 11, 1726. [Google Scholar] [CrossRef]
- Lee, R.S.; Tamashiro, K.L.K.; Yang, X.; Purcell, R.H.; Huo, Y.; Rongione, M.; Potash, J.B.; Wand, G.S. A Measure of Glucocorticoid Load Provided by DNA Methylation of Fkbp5 in Mice. Psychopharmacology 2011, 218, 303–312. [Google Scholar] [CrossRef]
- Homan, P.; Neumeister, A.; Nugent, A.C.; Charney, D.S.; Drevets, W.C.; Hasler, G. Serotonin versus Catecholamine Deficiency: Behavioral and Neural Effects of Experimental Depletion in Remitted Depression. Transl. Psychiatry 2015, 5, e532. [Google Scholar] [CrossRef] [PubMed]
- Antypa, N.; Drago, A.; Serretti, A. The Role of COMT Gene Variants in Depression: Bridging Neuropsychological, Behavioral and Clinical Phenotypes. Neurosci. Biobehav. Rev. 2013, 37, 1597–1610. [Google Scholar] [CrossRef] [PubMed]
- Burani, K.; Nelson, B.D. Gender Differences in Anxiety: The Mediating Role of Sensitivity to Unpredictable Threat. Int. J. Psychophysiol. 2020, 153, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.S.; Mezulis, A.H. Gender Differences in Depression: Biological, Affective, Cognitive, and Sociocultural Factors. Harv. Rev. Psychiatry 2020, 28, 4–13. [Google Scholar] [CrossRef]
- Kovács, T.; Szabó-Meleg, E.; Ábrahám, I.M. Estradiol-Induced Epigenetically Mediated Mechanisms and Regulation of Gene Expression. Int. J. Mol. Sci. 2020, 21, 3177. [Google Scholar] [CrossRef]
- Webb, L.M.; Phillips, K.E.; Ho, M.C.; Veldic, M.; Blacker, C.J. The Relationship between DNA Methylation and Antidepressant Medications: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 826. [Google Scholar] [CrossRef]
- Menke, A.; Binder, E.B. Epigenetic Alterations in Depression and Antidepressant Treatment. Dialogues Clin. Neurosci. 2014, 16, 395–404. [Google Scholar] [CrossRef]
- Mohammadi, S.; Beh-Pajooh, A.; Ahmadimanesh, M.; Amini, M.; Ghazi-Khansari, M.; Moallem, S.A.; Hosseini, R.; Nourian, Y.H.; Ghahremani, M.H. Evaluation of DNA Methylation in BDNF, SLC6A4, NR3C1 and FKBP5 before and after Treatment with Selective Serotonin-Reuptake Inhibitor in Major Depressive Disorder. Epigenomics 2022, 14, 1269–1280. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amplicon | Position (Human Genome Build 19) and Length of the Target Sequence | Number of CpG Islands | Functional Significance |
|---|---|---|---|
| BDNF_1 | chr11:27744260–27744605 (−), 346 | 22 | The brain-derived neurotrophic factor regulates growth, differentiation, maintenance, death/survival, and plasticity of neurons [53] |
| BDNF_2 | chr11:27743702–27743960 (−), 259 | 10 | |
| BDNF_3 | chr11:27743454–27743762 (−), 309 | 20 | |
| BDNF_4 | chr11:27741988–27742250 (−), 263 | 13 | |
| BDNF_5 | chr11:27740916–27741131 (−), 216 | 16 | |
| BDNF_6 | chr11:27740607–27740901 (−), 295 | 30 | |
| BDNF_7 | chr11:27721638–27721854 (−), 217 | 19 | |
| BDNF_8 | chr11:27722466–27722696 (−), 231 | 13 | |
| BDNF_9 | chr11:27722209–27722487 (−), 279 | 23 | |
| COMT_1 | chr22:19951071–19951343, 273 | 14 | The catechol-O-methyl transferase (COMT) determines prefrontal dopaminergic availability [54] |
| COMT_2 | chr22:19929042–19929349, 308 | 36 | |
| COMT_3 | chr22:19950002–19950320, 319 | 13 | |
| SLC6A4_1 | chr17:28562753–28563050 (−), 298 | 29 | The serotonin transporter is associated with depression treatment outcomes [55] |
| SLC6A4_2 | chr17:28563277–28563552 (−), 276 | 7 |
| n | Flexible Model | Stable Model | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| BDI-II | HAM-A | IDS-C | Dur_Episode | Dur_Total | |||||||
| r | p | r | p | r | p | r | p | r | p | ||
| BDNF_1 | 24 | 0.185 | 0.386 | 0.271 | 0.2 | 0.203 | 0.342 | 0.134 | 0.53 | −0.250 | 0.238 |
| BDNF_2 | 21 | −0.329 | 0.145 | −0.23 | 0.316 | −0.219 | 0.341 | −0.221 | 0.377 | 0.081 | 0.748 |
| BDNF_3 | 24 | −0.138 | 0.52 | −0.233 | 0.273 | −0.236 | 0.267 | 0.168 | 0.466 | 0.150 | 0.515 |
| BDNF_4 | 25 | 0.13 | 0.534 | 0.117 | 0.578 | 0.049 | 0.818 | 0.201 | 0.336 | −0.192 | 0.356 |
| BDNF_5 | 25 | 0.038 | 0.856 | 0.165 | 0.431 | 0.042 | 0.844 | 0.241 | 0.255 | −0.041 | 0.847 |
| BDNF_6 | 24 | 0.068 | 0.753 | 0.025 | 0.909 | 0.125 | 0.559 | 0.062 | 0.774 | −0.102 | 0.634 |
| BDNF_7 | 25 | −0.229 | 0.27 | −0.038 | 0.856 | −0.188 | 0.367 | 0.091 | 0.674 | 0.235 | 0.269 |
| BDNF_8 | 25 | −0.067 | 0.752 | 0.146 | 0.485 | −0.221 | 0.288 | 0.002 | 0.993 | 0.221 | 0.288 |
| BDNF_9 | 25 | −0.222 | 0.285 | −0.24 | 0.248 | −0.245 | 0.239 | 0.264 | 0.212 | −0.036 | 0.868 |
| COMT_1 | 24 | −0.436 | 0.033 * | −0.48 | 0.018 * | −0.446 | 0.029 * | −0.100 | 0.643 | 0.335 | 0.110 |
| COMT_2 | 24 | −0.256 | 0.227 | 0.188 | 0.379 | −0.11 | 0.608 | 0.068 | 0.752 | −0.157 | 0.462 |
| COMT_3 | 25 | −0.023 | 0.914 | −0.07 | 0.739 | −0.085 | 0.685 | −0.307 | 0.114 | 0.145 | 0.498 |
| SLC6A4_1 | 24 | −0.144 | 0.503 | −0.064 | 0.766 | −0.256 | 0.228 | −0.004 | 0.986 | 0.119 | 0.579 |
| SLC6A4_2 | 25 | −0.007 | 0.973 | −0.084 | 0.691 | −0.125 | 0.552 | −0.241 | 0.246 | 0.154 | 0.463 |
| Age | 25 | 0.208 | 0.318 | 0.255 | 0.218 | 0.394 | 0.051 | 0.105 | 0.614 | 0.021 | 0.919 |
| BDI-II | IDS-C | HAM-A | |
|---|---|---|---|
| BDI-II | |||
| IDS-C | r(23) = 0.73, p < 0.001 | ||
| HAM-A | r(23) = 0.66, p < 0.001 | r(23) = 0.78, p < 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidovič, E.; Pelikan, S.; Atanasova, M.; Kouter, K.; Pileckyte, I.; Oblak, A.; Novak Šarotar, B.; Videtič Paska, A.; Bon, J. DNA Methylation Patterns in Relation to Acute Severity and Duration of Anxiety and Depression. Curr. Issues Mol. Biol. 2023, 45, 7286-7303. https://doi.org/10.3390/cimb45090461
Vidovič E, Pelikan S, Atanasova M, Kouter K, Pileckyte I, Oblak A, Novak Šarotar B, Videtič Paska A, Bon J. DNA Methylation Patterns in Relation to Acute Severity and Duration of Anxiety and Depression. Current Issues in Molecular Biology. 2023; 45(9):7286-7303. https://doi.org/10.3390/cimb45090461
Chicago/Turabian StyleVidovič, Eva, Sebastian Pelikan, Marija Atanasova, Katarina Kouter, Indre Pileckyte, Aleš Oblak, Brigita Novak Šarotar, Alja Videtič Paska, and Jurij Bon. 2023. "DNA Methylation Patterns in Relation to Acute Severity and Duration of Anxiety and Depression" Current Issues in Molecular Biology 45, no. 9: 7286-7303. https://doi.org/10.3390/cimb45090461
APA StyleVidovič, E., Pelikan, S., Atanasova, M., Kouter, K., Pileckyte, I., Oblak, A., Novak Šarotar, B., Videtič Paska, A., & Bon, J. (2023). DNA Methylation Patterns in Relation to Acute Severity and Duration of Anxiety and Depression. Current Issues in Molecular Biology, 45(9), 7286-7303. https://doi.org/10.3390/cimb45090461