
Genome-Wide Identification and Expression Analyses of the FAR1/FHY3 Gene Family Provide Insight into Inflorescence Development in Maize


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of ZmFAR1 Genes in Maize B73
2.2. Phylogenetic Analyses of ZmFAR1 Genes
2.3. Gene Structure and Conserved Motif Analysis of ZmFAR1 Genes
2.4. Chromosomal Location and Collinearity Analysis of ZmFAR1 Genes
2.5. Cis-Acting Elements and Interaction Network Analysis of ZmFAR1 Genes
2.6. Tissue-Specific Expression Analysis of ZmFAR1 Genes
2.7. Plant Materials and Stress Treatments
2.8. RNA Isolation and Quantitative qRT-PCR Analysis
2.9. Expression Quantitative Trait Loci (eQTL) Analysis
2.10. Statistical Analysis
3. Results
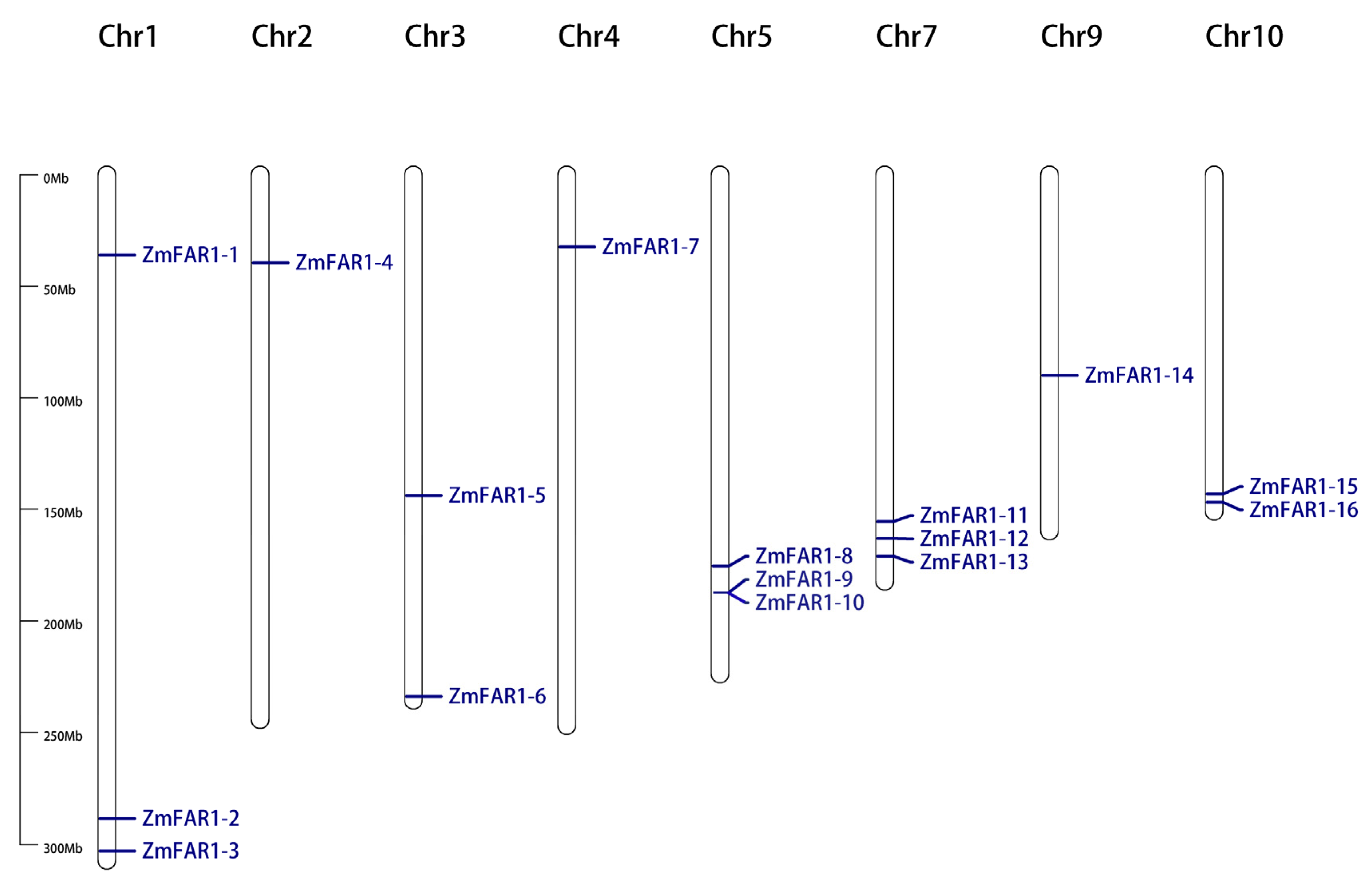
3.1. Identification and Chromosomal Location of ZmFAR1 Genes in Maize
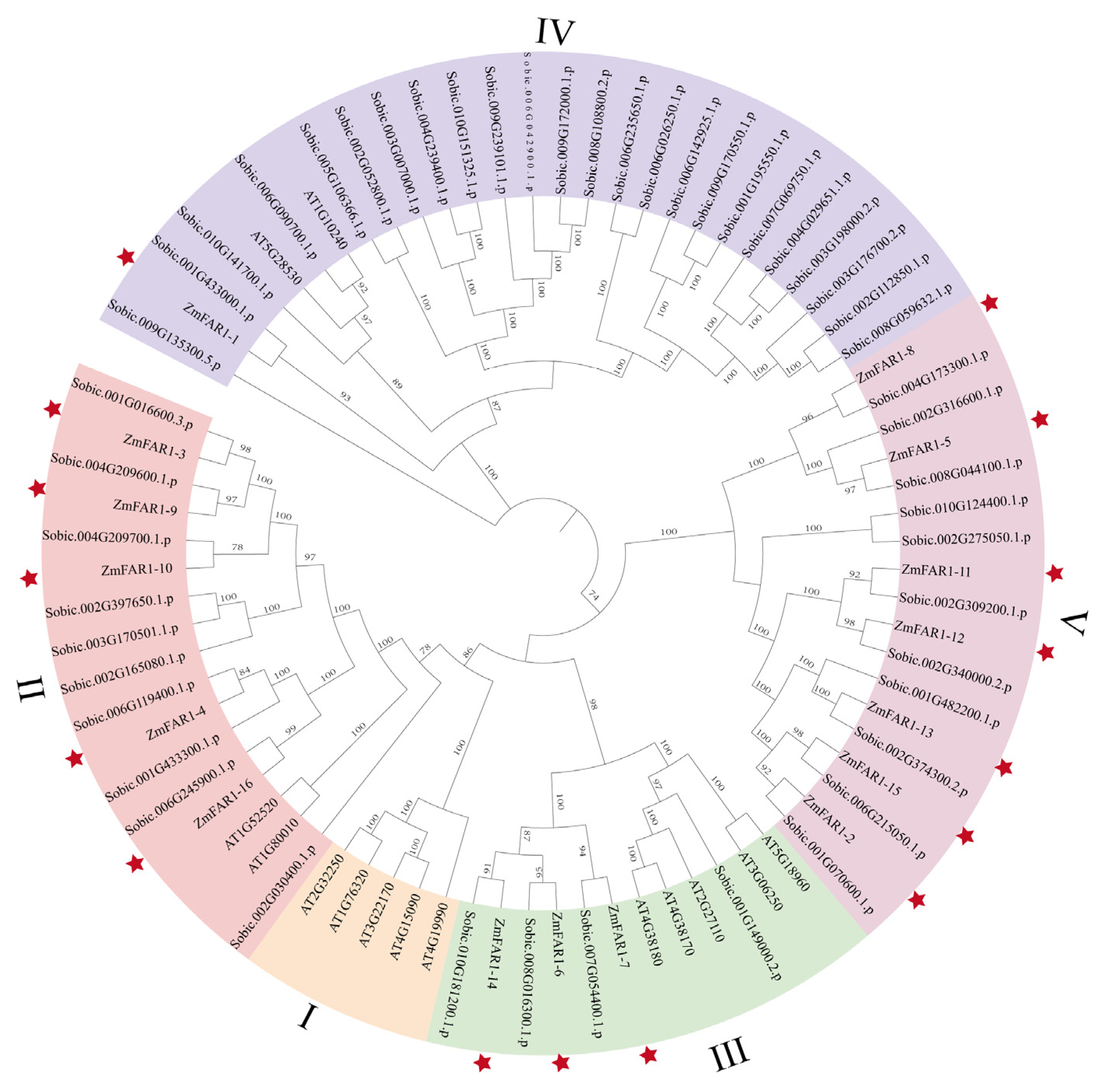
3.2. Phylogenetic Analysis of ZmFAR1 Genes
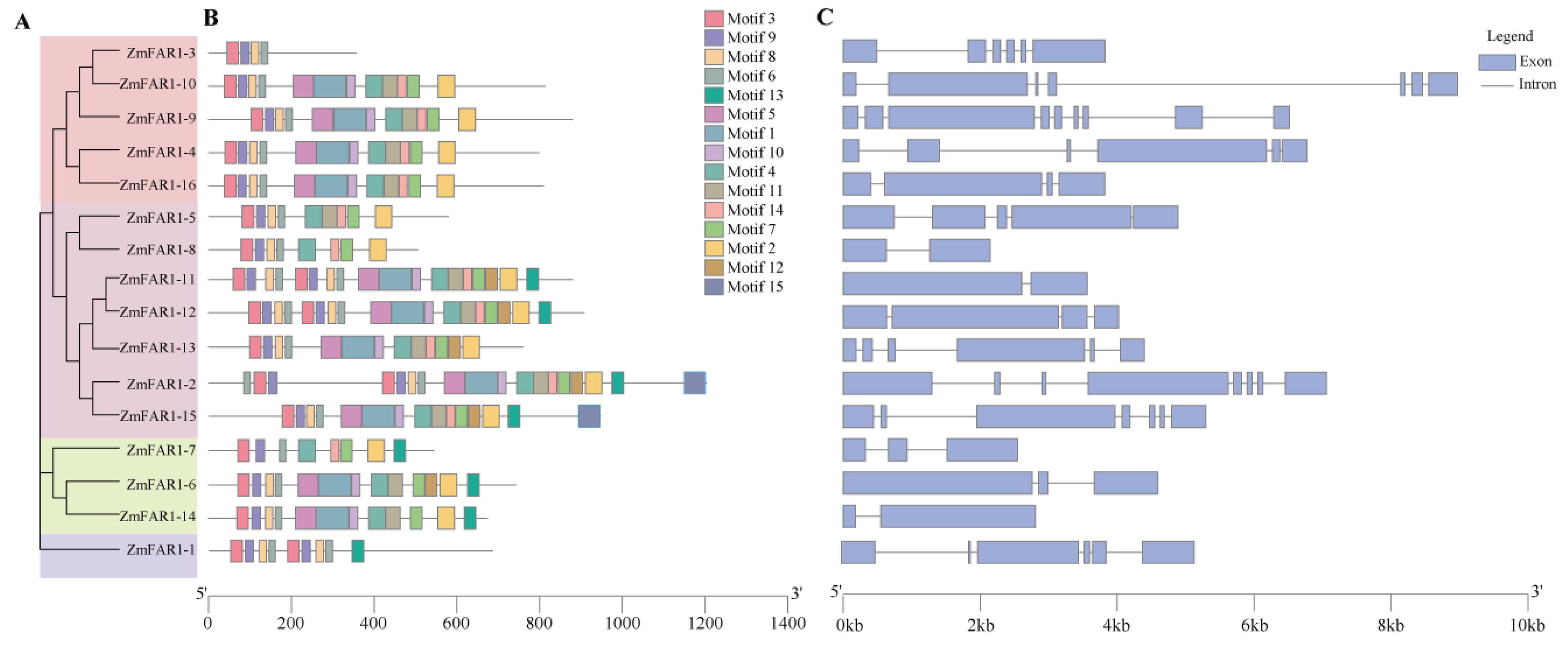
3.3. Conserved Structure and Motif Analyses of ZmFAR1 Genes
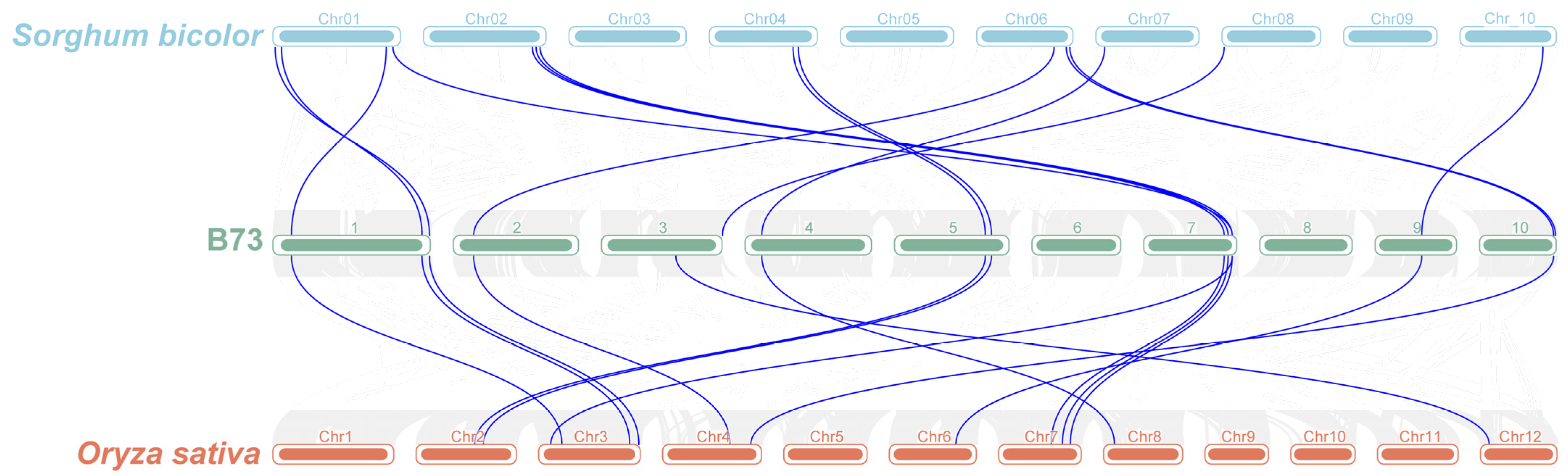
3.4. Collinearity Analysis of ZmFAR1 Genes
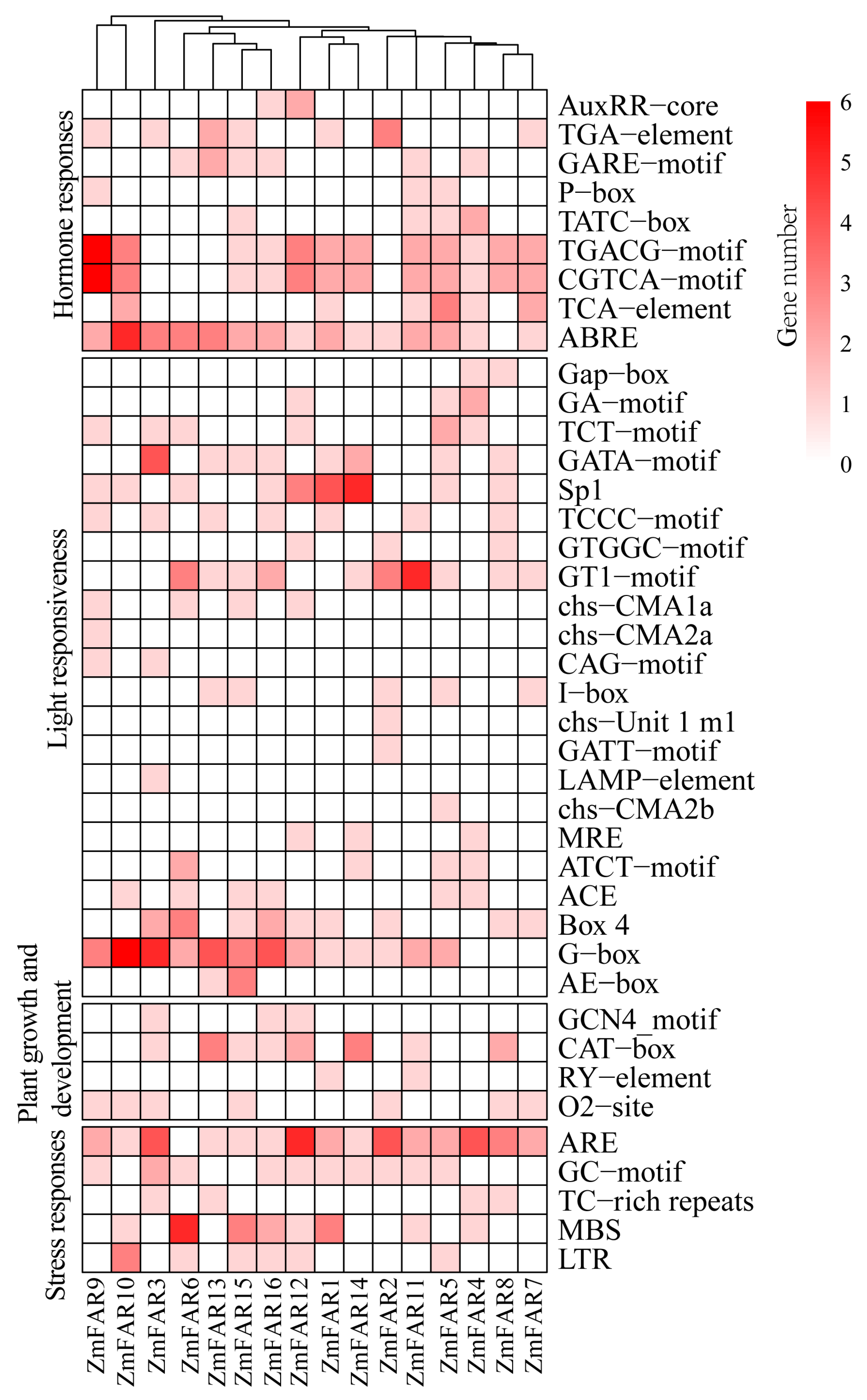
3.5. Cis-Acting Element Analysis of ZmFAR1 Genes
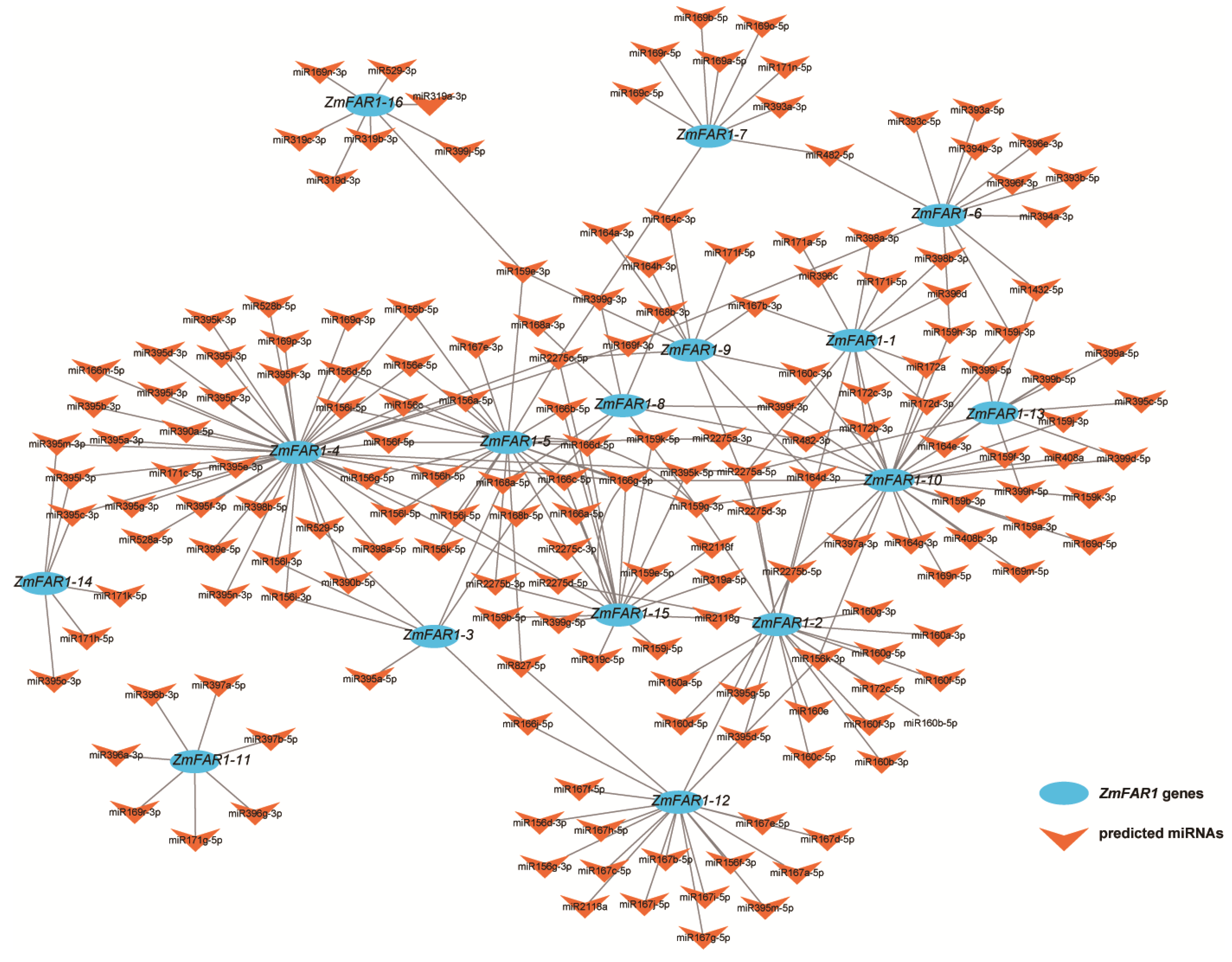
3.6. Target miRNA Prediction and Interactive Network Analysis of ZmFAR1 Genes
3.7. Expression Analysis of ZmFAR1 Genes
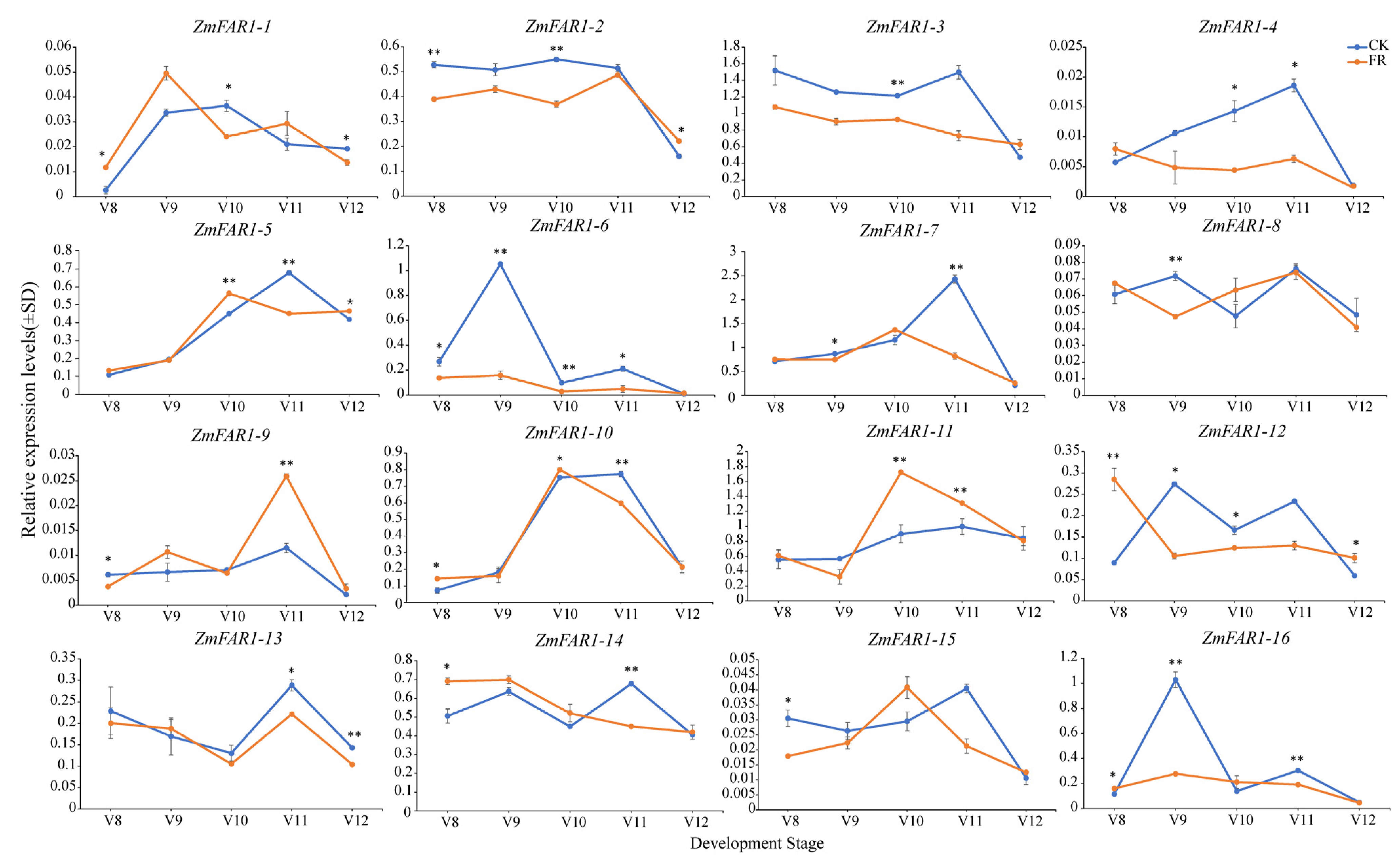
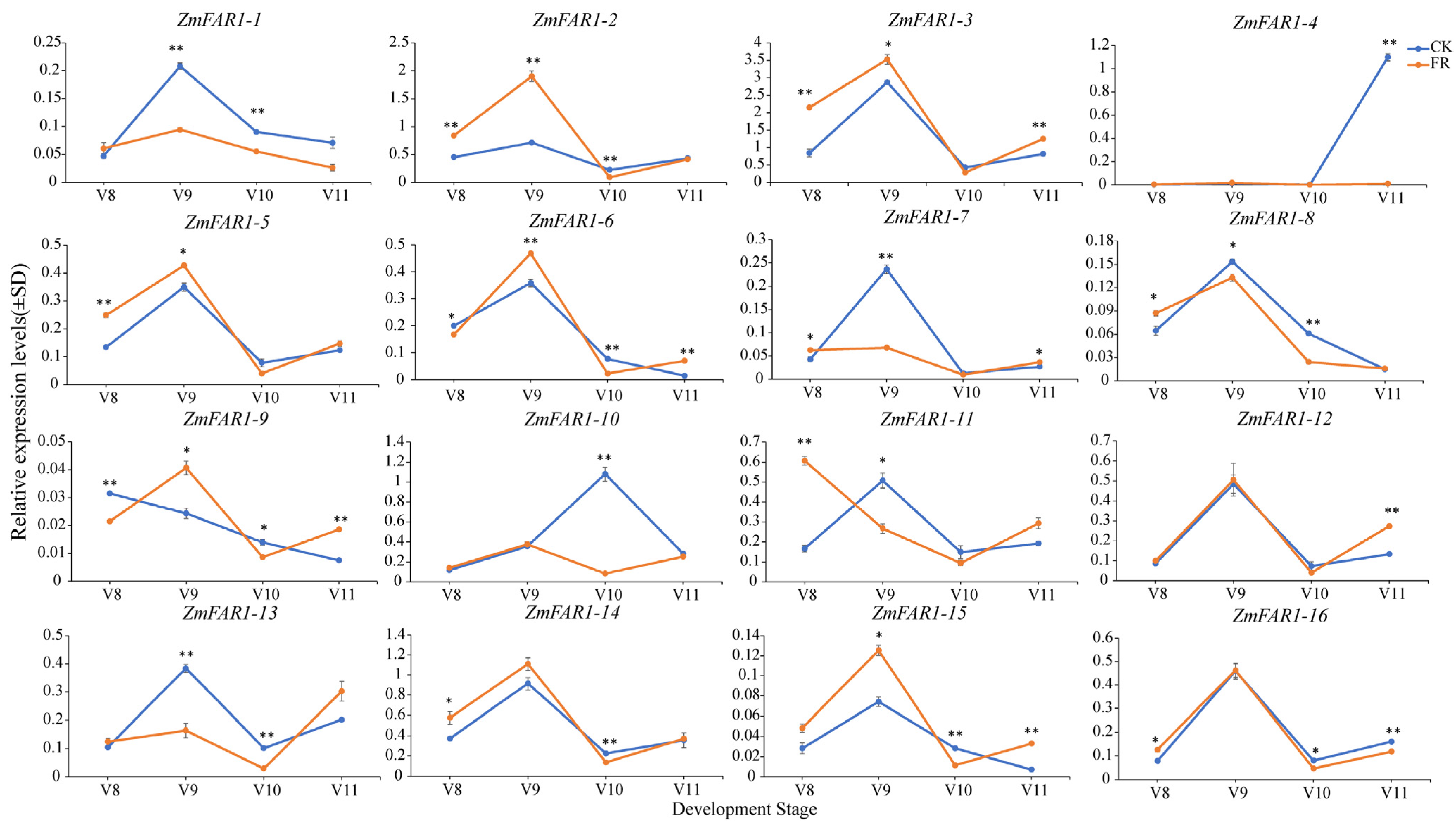
3.8. Expression Analysis of ZmFAR1 Genes under Far-Red Light
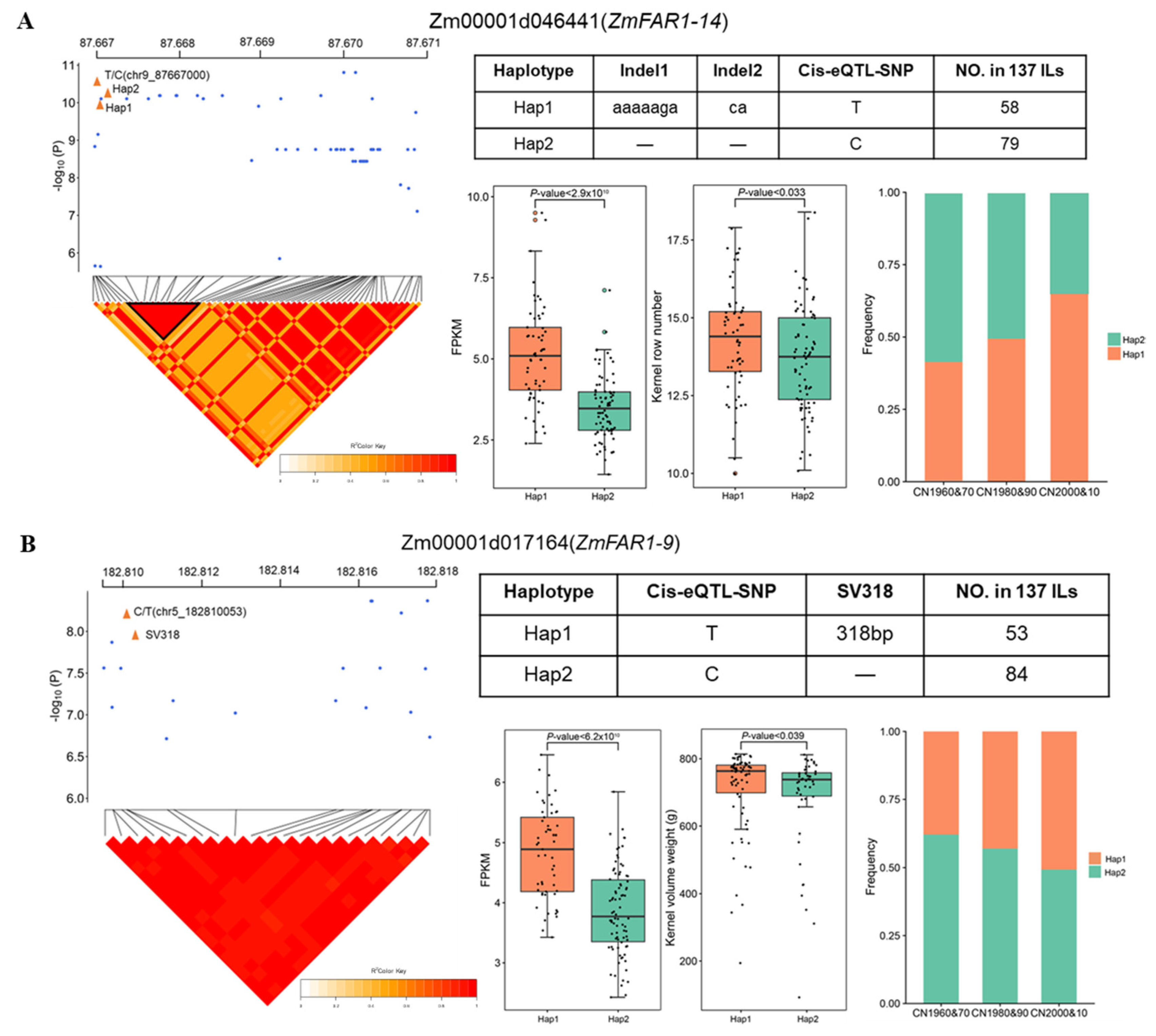
3.9. Cis-eQTL Analyses Identify the Causal Variations Associated with Gene Expression Variation and Agronomic Traits
4. Discussion
4.1. Characterization of the ZmFAR1 Family Members in Maize
4.2. Studies on the Structure, Evolutionary Characteristics, and Functions of Maize ZmFAR1 Genes
4.3. Functional Variations and Expression Patterns of ZmFAR1 Genes in Maize Ear Development and Phenotypic Variation
4.4. ZmFAR1 Genes’ Role in Maize Yield and Trait Optimization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Casal, J.J. Photoreceptor Signaling Networks in Plant Responses to Shade. Annu. Rev. Plant Biol. 2013, 64, 403. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, H.; Rappaport, F.; Boussac, A.; Messinger, J. Substrate–water exchange in photosystem II is arrested before dioxygen formation. Nat. Commun. 2014, 5, 4305. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Siddiqui, H.; Teng, Y.; Lin, R.; Wan, X.-Y.; Li, J.; Lau, O.-S.; Ouyang, X.; Dai, M.; Wan, J. Coordinated transcriptional regulation underlying the circadian clock in Arabidopsis. Nat. Cell Biol. 2011, 13, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H. Multifaceted roles of FHY3 and FAR1 in light signaling and beyond. Trends Plant Sci. 2015, 20, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Wang, H. Arabidopsis FHY3/FAR1 gene family and distinct roles of its members in light control of Arabidopsis development. Plant Physiol. 2004, 136, 4010–4022. [Google Scholar] [CrossRef]
- Ouyang, X.; Li, J.; Li, G.; Li, B.; Chen, B.; Shen, H.; Huang, X.; Mo, X.; Wan, X.; Lin, R. Genome-wide binding site analysis of FAR-RED ELONGATED HYPOCOTYL3 reveals its novel function in Arabidopsis development. Plant Cell 2011, 23, 2514–2535. [Google Scholar] [CrossRef]
- Ma, L.; Li, G. FAR1-related sequence (FRS) and FRS-related factor (FRF) family proteins in Arabidopsis growth and development. Front. Plant. Sci. 2018, 9, 692. [Google Scholar] [CrossRef]
- Du, J.; Zhang, L.; Ge, X.; Xiang, X.; Cao, D.; Yang, H.; Hu, J. Genome-wide identification and characterization of the FAR1/FHY3 family in Populus trichocarpa Torr. & Gray and expression analysis in light response. Forests 2021, 12, 1385. [Google Scholar]
- Liu, L.; Li, B.; Liu, X. FAR-RED ELONGATED HYPOCOTYL3 promotes floral meristem determinacy in Arabidopsis. Plant. Signal. Behav. 2016, 11, e1238545. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, M.; Li, G.; Yuan, L.; Xie, Y.; Wei, H.; Ma, X.; Li, Q.; Devlin, P.F.; Xu, X. Transcription factors FHY3 and FAR1 regulate light-induced CIRCADIAN CLOCK ASSOCIATED1 gene expression in Arabidopsis. Plant Cell 2020, 32, 1464–1478. [Google Scholar] [CrossRef]
- Ma, L.; Xue, N.; Fu, X.; Zhang, H.; Li, G. Arabidopsis thaliana FAR-RED ELONGATED HYPOCOTYLS3 (FHY3) and FAR-RED-IMPAIRED RESPONSE1 (FAR1) modulate starch synthesis in response to light and sugar. New Phytol. 2017, 213, 1682–1696. [Google Scholar] [CrossRef] [PubMed]
- Rea, A.C. Avoiding shade to grow taller but not always stronger: Phytochrome–jasmonic acid interplay. Am. Soc. Plant Biol. 2019, 31, 1941–1942. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, H.; Ma, M.; Li, Q.; Kong, D.; Sun, J.; Ma, X.; Wang, B.; Chen, C.; Xie, Y. Arabidopsis FHY3 and FAR1 regulate the balance between growth and defense responses under shade conditions. Plant Cell 2019, 31, 2089–2106. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, Y.; Ma, M.; Zhou, Q.; Zhao, Y.; Zhao, B.; Wang, B.; Wei, H.; Wang, H. Arabidopsis FHY3 and FAR1 integrate light and strigolactone signaling to regulate branching. Nat. Commun. 2020, 11, 1955. [Google Scholar] [CrossRef]
- Tang, W.; Ji, Q.; Huang, Y.; Jiang, Z.; Bao, M.; Lin, W.R. FAR-RED ELONGATED HYPOCOTYL3 and FAR-RED IMPAIRED RESPONSE1 transcription factors integrate light and abscisic acid signaling in Arabidopsis. Plant Physiol. 2013, 163, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Ma, M.; Liu, Y.; Wang, B.; Wei, H.; Kong, D.; Wang, H. Arabidopsis FHY3 and FAR1 function in age gating of leaf senescence. Front. Plant. Sci. 2021, 12, 770060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhu, W.; He, Y.; Fan, J.; Shi, J.; Fu, R.; Hu, J.; Li, L.; Zhang, D.; Liang, W. THERMOSENSITIVE BARREN PANICLE (TAP) is required for rice panicle and spikelet development at high ambient temperature. New Phytol. 2023, 237, 855–869. [Google Scholar] [CrossRef]
- Lu, Q.; Liu, H.; Hong, Y.; Liang, X.; Li, S.; Liu, H.; Li, H.; Wang, R.; Deng, Q.; Jiang, H. Genome-Wide Identification and Expression of FAR1 Gene Family Provide Insight Into Pod Development in Peanut (Arachis hypogaea). Front. Plant. Sci. 2022, 13, 893278. [Google Scholar] [CrossRef]
- Zhong, M.-C.; Jiang, X.-D.; Cui, W.-H.; Hu, J.-Y. Expansion and expression diversity of FAR1/FRS-like genes provides insights into flowering time regulation in roses. Plant Divers 2021, 43, 173–179. [Google Scholar] [CrossRef]
- Ruberti, I.; Sessa, G.; Ciolfi, A.; Possenti, M.; Carabelli, M.; Morelli, G. Plant adaptation to dynamically changing environment: The shade avoidance response. Biotechnol. Adv. 2012, 30, 1047–1058. [Google Scholar] [CrossRef]
- Roig-Villanova, I.; Martínez-García, J.F. Plant responses to vegetation proximity: A whole life avoiding shade. Front. Plant. Sci. 2016, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Lancaster, J.; Buhler, J.; Harris, B.; Chamberlain, R.D. Mercury BLASTP: Accelerating protein sequence alignment. ACM Trans. Reconfigurable Technol. Syst. 2008, 1, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Yupeng, W.; Haibao, T.; Debarry, J.D.; Xu, T.; Jingping, L.; Xiyin, W.; Tae-Ho, L.; Huizhe, J.; Barry, M.; Hui, G. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant Graphics for Data Analysis; Taylor & Francis: Oxford, UK, 2019. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Y.; Song, G.; Yang, D.; Xia, Z.; Sun, C.; Zhao, Y.; Hou, M.; Zhang, M.; Qi, Z. Gene expression and expression quantitative trait loci analyses uncover natural variations underlying the improvement of important agronomic traits during modern maize breeding. Plant J. Cell Mol. Biol. 2023, 115, 772–787. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Ritter, A.; Iñigo, S.; Fernández-Calvo, P.; Heyndrickx, K.S.; Dhondt, S.; Shi, H.; De Milde, L.; Vanden Bossche, R.; De Clercq, R.; Eeckhout, D. The transcriptional repressor complex FRS7-FRS12 regulates flowering time and growth in Arabidopsis. Nat. Commun. 2017, 8, 15235. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, P.; Li, Y.; Liu, Y.; Yang, N.; Yu, J.; Ma, X.; Sun, S.; Xia, R.; Liu, X. An overlooked paleotetraploidization in Cucurbitaceae. Mol. Biol. Evol. 2018, 35, 16–26. [Google Scholar] [CrossRef]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant. Sci. 2014, 217, 109–119. [Google Scholar] [CrossRef]
- Kong, D.; Li, C.; Xue, W.; Wei, H.; Ding, H.; Hu, G.; Zhang, X.; Zhang, G.; Zou, T.; Xian, Y. UB2/UB3/TSH4-anchored transcriptional networks regulate early maize inflorescence development in response to simulated shade. Plant Cell 2023, 35, 717–737. [Google Scholar] [CrossRef]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.-S. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef]
- Sun, S.; Zhou, Y.; Chen, J.; Shi, J.; Zhao, H.; Zhao, H.; Song, W.; Zhang, M.; Cui, Y.; Dong, X. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 2018, 50, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hou, M.; Shi, J.; Ku, L.; Song, W.; Li, C.; Ning, Q.; Li, X.; Li, C.; Zhao, B. De novo genome assembly and analyses of 12 founder inbred lines provide insights into maize heterosis. Nat. Genet. 2023, 55, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Deng, X.W. Arabidopsis FHY3 defines a key phytochrome A signaling component directly interacting with its homologous partner FAR1. EMBO J. 2002, 21, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; An, C.; Zhao, Y.; Xiao, Y.; Bao, L.; Gong, C.; Gao, Y. Genome-wide identification and characterization of the CsFHY3/FAR1 gene family and expression analysis under biotic and abiotic stresses in tea plants (Camellia sinensis). Plants 2021, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.M.; Mamidala, P.; Podile, A.R. Regulation of Polygalacturonase-inhibitory proteins in plants is highly dependent on stress and light responsive elements. Plant Omics 2009, 2, 238–249. [Google Scholar]
- Baek, K.; Lee, Y.; Nam, O.; Park, S.; Sim, S.J.; Jin, E. Introducing Dunaliella LIP promoter containing light-inducible motifs improves transgenic expression in Chlamydomonas reinhardtii. Biotechnol. J. 2016, 11, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ren, C.; Zou, L.; Wang, Y.; Li, S.; Liang, Z. Characterization of the GATA gene family in Vitis vinifera: Genome-wide analysis, expression profiles, and involvement in light and phytohormone response. Genome 2018, 61, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jafari, F.; Wang, H. Integration of light and hormone signaling pathways in the regulation of plant shade avoidance syndrome. Abiotech 2021, 2, 131–145. [Google Scholar] [CrossRef]
- Rouster, J.; Leah, R.; Mundy, J.; Cameron-Mills, V. Identification of a methyl jasmonate-responsive region in the promoter of a lipoxygenase 1 gene expressed in barley grain. Plant J. 1997, 11, 513–523. [Google Scholar] [CrossRef]
- Chu, Z.; Wang, X.; Li, Y.; Yu, H.; Li, J.; Lu, Y.; Li, H.; Ouyang, B. Genomic organization, phylogenetic and expression analysis of the B-BOX gene family in tomato. Front. Plant. Sci. 2016, 7, 1552. [Google Scholar] [CrossRef]
- Wang, X.; Guo, C.; Peng, J.; Li, C.; Wan, F.; Zhang, S.; Zhou, Y.; Yan, Y.; Qi, L.; Sun, K. ABRE-BINDING FACTORS play a role in the feedback regulation of ABA signaling by mediating rapid ABA induction of ABA co-receptor genes. New Phytol. 2019, 221, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Chen, C.; Pokhrel, S.; Ma, W.; Huang, K.; Patel, P.; Wang, F.; Xu, J.; Liu, Z.; Li, J. 24-nt reproductive phasiRNAs are broadly present in angiosperms. Nat. Commun. 2019, 10, 627. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Zhang, H.; Arikit, S.; Huang, K.; Nan, G.-L.; Walbot, V.; Meyers, B.C. Spatiotemporally dynamic, cell-type–dependent premeiotic and meiotic phasiRNAs in maize anthers. Proc. Natl. Acad. Sci. USA 2015, 112, 3146–3151. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Guo, Z.; Li, L. Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev. Biol. 2013, 380, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tao, P.; Xu, F.; He, P.; Wang, J. Function of Soybean miR159 Family Members in Plant Responses to Low Phosphorus, High Salinity, and Abscisic Acid Treatment. Agronomy 2023, 13, 1798. [Google Scholar] [CrossRef]
- Chen, J.-F.; Zhao, Z.-X.; Li, Y.; Li, T.-T.; Zhu, Y.; Yang, X.-M.; Zhou, S.-X.; Wang, H.; Zhao, J.-Q.; Pu, M. Fine-tuning roles of Osa-miR159a in rice immunity against Magnaporthe oryzae and development. Rice 2021, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, S.; Wu, W.; Li, L.; Jiang, T.; Zheng, B. Clearance of maternal barriers by paternal miR159 to initiate endosperm nuclear division in Arabidopsis. Nat. Commun. 2018, 9, 5011. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Q.; Wu, Y.; Lemmon, Z.H.; Xu, G.; Huang, C.; Liang, Y.; Xu, D.; Li, D.; Doebley, J.F. Genome-wide analysis of transcriptional variability in a large maize-teosinte population. Mol. Plant 2018, 11, 443–459. [Google Scholar] [CrossRef]
- Liu, S.; Li, C.; Wang, H.; Wang, S.; Yang, S.; Liu, X.; Yan, J.; Li, B.; Beatty, M.; Zastrow-Hayes, G. Mapping regulatory variants controlling gene expression in drought response and tolerance in maize. Genome Biol. 2020, 21, 163. [Google Scholar] [CrossRef]
- Wu, X.; Feng, H.; Wu, D.; Yan, S.; Dai, M. Using high-throughput multiple optical phenotyping to decipher the genetic architecture of maize drought tolerance. Genome Biol. 2021, 22, 185. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, H.; Jing, D.; Liu, C.; Xie, X.; Zhang, L.; Chen, X.; Li, C. Genome-Wide Identification and Expression Analyses of the FAR1/FHY3 Gene Family Provide Insight into Inflorescence Development in Maize. Curr. Issues Mol. Biol. 2024, 46, 430-449. https://doi.org/10.3390/cimb46010027
Tang H, Jing D, Liu C, Xie X, Zhang L, Chen X, Li C. Genome-Wide Identification and Expression Analyses of the FAR1/FHY3 Gene Family Provide Insight into Inflorescence Development in Maize. Current Issues in Molecular Biology. 2024; 46(1):430-449. https://doi.org/10.3390/cimb46010027
Chicago/Turabian StyleTang, Huaijun, De Jing, Cheng Liu, Xiaoqing Xie, Lei Zhang, Xunji Chen, and Changyu Li. 2024. "Genome-Wide Identification and Expression Analyses of the FAR1/FHY3 Gene Family Provide Insight into Inflorescence Development in Maize" Current Issues in Molecular Biology 46, no. 1: 430-449. https://doi.org/10.3390/cimb46010027
APA StyleTang, H., Jing, D., Liu, C., Xie, X., Zhang, L., Chen, X., & Li, C. (2024). Genome-Wide Identification and Expression Analyses of the FAR1/FHY3 Gene Family Provide Insight into Inflorescence Development in Maize. Current Issues in Molecular Biology, 46(1), 430-449. https://doi.org/10.3390/cimb46010027