
Impact of Lysine Succinylation on the Biology of Fungi

Abstract
:1. Introduction
2. Post-Translational Modifications (PTMs)
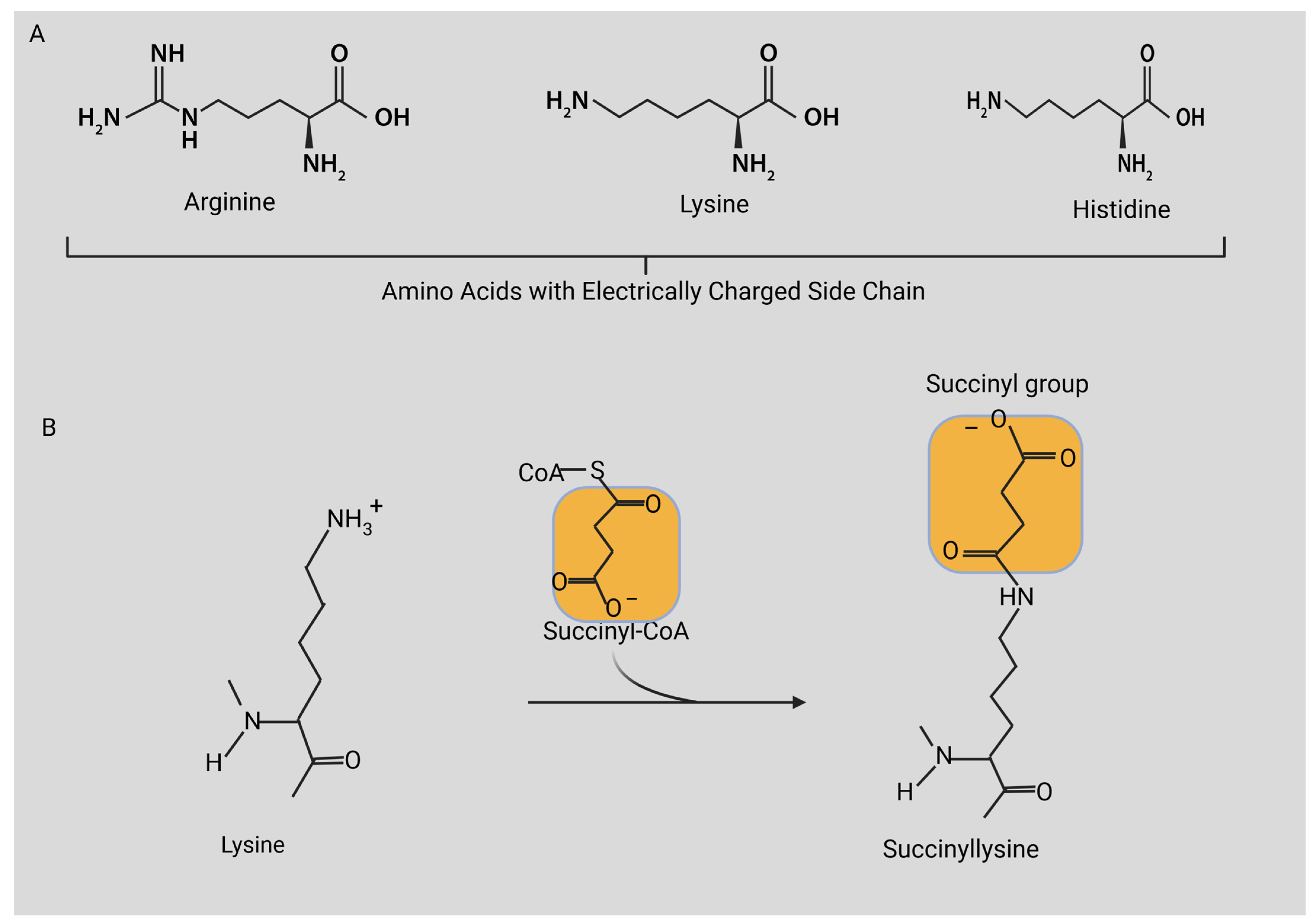
3. Protein Lysine Acylation

4. Discovery of Lysine Succinylation
5. Biochemical Process of Succinylation Modification
5.1. Non-Enzymatic Lysine Succinylation by Succinyl-CoA
5.2. Enzymatic Lysine Succinylation
5.3. Enzymatic Desuccinylation
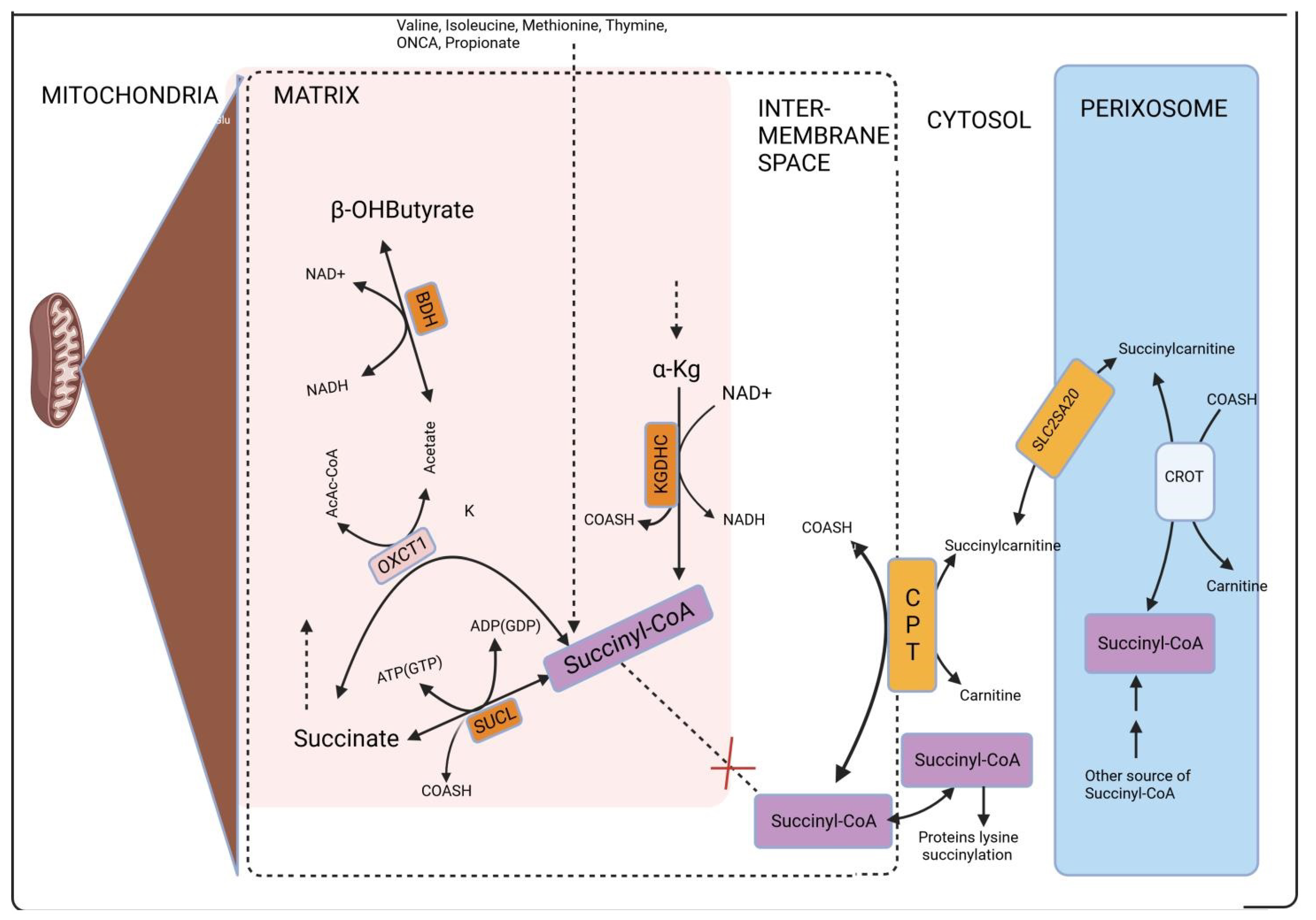
6. Mitochondria and Cytosol Localization of Lysine Succinylation
- i.
- The conversion of succinylcarnitine to carnitine and succinyl-CoA is facilitated by carnitine palmitoyltransferase [130]. This enzyme is present in three isoforms, each with a tissue-specific distribution: isoform A is found in the liver, isoform B is located in the muscle, and isoform C is situated in the brain. All three isoforms are situated in the outer mitochondrial membrane [131,132], with isoform C also being present in the endoplasmic reticulum [133]. CoA can be found in the cytosol through various pathways, whereas succinyl carnitine can be transported from peroxisomes via the carrier protein SLC25A20. Initially identified as a “mitochondrial carnitine/acylcarnitine carrier protein”, it has since been recognized to also exist in peroxisomes. In peroxisomes, succinylcarnitine synthesis occurs through the following process: carnitine and succinyl-CoA are converted into succinylcarnitine and CoA with the involvement of carnitine O-octanoyltransferase (CROT), as discussed in more detail in [57]. In the context of human peroxisomes, the necessary carnitine might be brought in through one of the organic cation transporters (OCTs). In rodents, the OCTN3 (organic cation transporter 3) carnitine transporter (SLC22A21) is present [134,135]. Nevertheless, there is some debate regarding whether OCTN3 is consistently localized in peroxisomes [136].
- ii.
- The transportation of succinyl-CoA from the mitochondria to the cytosol relies on a model assumption presented in [137]. This assumption was part of an effort to create a comprehensive and high-quality genome-scale metabolic reconstruction by Thiele and Palsson. However, it has not been experimentally confirmed.
- iii.
- Adenosine triphosphate (ATP) + Coenzyme A (CoA) + succinate combine to form Adenosine monophosphate (AMP) + Inorganic pyrophosphate (PPi) + succinyl-CoA. This transformation is potentially facilitated by a microsomal dicarboxylyl-CoA synthetase (E.C. 6.2.1.23). In the rat liver and kidney, but not in the muscle tissue, dicarboxylic acids (succinate) can be converted into their CoA esters by this enzyme [138,139]. Nevertheless, there is a suggestion that this enzyme might not be effective on short-chain dicarboxylic acids like succinate. This is because enzyme activity tends to approach zero as the carbon chain length of the dicarboxylic acid decreases, and it is already minimal with a chain length of C = 5 [138] (Figure 4).
7. Lysine Succinylation in Fungi
8. Tools Used to Discover Lysine Succinylated Proteins and Sites
8.1. Conventional Techniques Employed in the Identification of Lysine Succinylation
8.2. Computational Tools to Predict Lysine Succinylation Sites
9. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hyde, K.D.; Al-Hatmi, A.M.; Andersen, B.; Boekhout, T.; Buzina, W.; Dawson, T.L.; Eastwood, D.C.; Jones, E.G.; de Hoog, S.; Kang, Y. The world’s ten most feared fungi. Fungal Divers. 2018, 93, 161–194. [Google Scholar] [CrossRef]
- Tumukunde, E.; Xie, R.; Wang, S. Updates on the Functions and Molecular Mechanisms of the Genes Involved in Aspergillus flavus Development and Biosynthesis of Aflatoxins. J. Fungi 2021, 7, 666. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.S.; Mannino, D.M.; Eaton, S.; Moss, M. The Epidemiology of Sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 2003, 348, 1546–1554. [Google Scholar] [CrossRef]
- Kronstad, J.W.; Attarian, R.; Cadieux, B.; Choi, J.; D’Souza, C.A.; Griffiths, E.J.; Geddes, J.M.H.; Hu, G.; Jung, W.H.; Kretschmer, M.; et al. Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat. Rev. Microbiol. 2011, 9, 193–203. [Google Scholar] [CrossRef]
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017, 16, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Geddes-McAlister, J.; Shapiro, R.S. New pathogens, new tricks: Emerging, drug-resistant fungal pathogens and future prospects for antifungal therapeutics. Ann. N. Y. Acad. Sci. 2019, 1435, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, N.P. Antifungal resistance: Current trends and future strategies to combat. Infect. Drug Resist. 2017, 10, 249–259. [Google Scholar] [CrossRef]
- Bermas, A.; Geddes-McAlister, J. Combatting the evolution of antifungal resistance in Cryptococcus neoformans. Mol. Microbiol. 2020, 114, 721–734. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Diekema, D.J. Rare and Emerging Opportunistic Fungal Pathogens: Concern for Resistance beyond Candida albicans and Aspergillus fumigatus. J. Clin. Microbiol. 2004, 42, 4419–4431. [Google Scholar] [CrossRef]
- Janbon, G.; Quintin, J.; Lanternier, F.; D’enfert, C. Studying fungal pathogens of humans and fungal infections: Fungal diversity and diversity of approaches. Microbes Infect. 2019, 21, 237–245. [Google Scholar] [CrossRef]
- Xu, H.; Chen, X.; Xu, X.; Shi, R.; Suo, S.; Cheng, K.; Zheng, Z.; Wang, M.; Wang, L.; Zhao, Y.; et al. Lysine Acetylation and Succinylation in HeLa Cells and their Essential Roles in Response to UV-induced Stress. Sci. Rep. 2016, 6, 30212. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Edit. 2005, 44, 7342–7372. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.-X.; Smet-Nocca, C.; Liokatis, S.; Thongwichian, R.; Kosten, J.; Yoon, M.-K.; Kriwacki, R.W.; Landrieu, I.; Lippens, G.; Selenko, P. Cell signaling, post-translational protein modifications and NMR spectroscopy. J. Biomol. NMR 2012, 54, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Leach, M.D.; Brown, A.J.P. Posttranslational Modifications of Proteins in the Pathobiology of Medically Relevant Fungi. Eukaryot. Cell 2012, 11, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Salomon, D.; Orth, K. What Pathogens Have Taught Us About Posttranslational Modifications. Cell Host Microbe 2013, 14, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Ball, B.; Bermas, A.; Carruthers-Lay, D.; Geddes-McAlister, J. Mass Spectrometry-Based Proteomics of Fungal Pathogenesis, Host–Fungal Interactions, and Antifungal Development. J. Fungi 2019, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, A.; Coish, J.M.; Yeung, J.; Muselius, B.; Gadjeva, M.; MacNeil, A.J.; Geddes-McAlister, J. Decoding communication patterns of the innate immune system by quantitative proteomics. J. Leukoc. Biol. 2019, 106, 1221–1232. [Google Scholar] [CrossRef]
- Ball, B.; Langille, M.; Geddes-McAlister, J. Fun(gi)omics: Advanced and Diverse Technologies to Explore Emerging Fungal Pathogens and Define Mechanisms of Antifungal Resistance. MBio 2020, 11, 10–1128. [Google Scholar] [CrossRef]
- Witze, E.S.; Old, W.M.; Resing, K.A.; Ahn, N.G. Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 2007, 4, 798–806. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Peterson, S.E.; Loring, J.F. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014, 24, 143–160. [Google Scholar] [CrossRef]
- Humphrey, S.J.; James, D.E.; Mann, M. Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends Endocrinol. Metab. 2015, 26, 676–687. [Google Scholar] [CrossRef]
- Hitosugi, T.; Chen, J. Post-translational modifications and the Warburg effect. Oncogene 2014, 33, 4279–4285. [Google Scholar] [CrossRef]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef]
- Yang, Y.; Gibson, G.E. Succinylation Links Metabolism to Protein Functions. Neurochem. Res. 2019, 44, 2346–2359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-Mediated Lysine Desuccinylation Impacts Diverse Metabolic Pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.B.; Mylonakis, E. Our Paths Might Cross: The Role of the Fungal Cell Wall Integrity Pathway in Stress Response and Cross Talk with Other Stress Response Pathways. Eukaryot. Cell 2009, 8, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Colak, G.; Xie, Z.; Zhu, A.Y.; Dai, L.; Lu, Z.; Zhang, Y.; Wan, X.; Chen, Y.; Cha, Y.H.; Lin, H.; et al. Identification of Lysine Succinylation Substrates and the Succinylation Regulatory Enzyme CobB in Escherichia coli. Mol. Cell. Proteom. 2013, 12, 3509–3520. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Ferreira, G.C. Enzyme complexes important for the glutamate–glutamine cycle. In Glutamate/GABA-Glutamine Cycle—Amino Acid Neurotransmitter Homeostasis; Springer: Cham, Switzerland, 2016; pp. 59–98. [Google Scholar]
- Nijhawan, A.; Jain, M.; Tyagi, A.K.; Khurana, J.P. Genomic Survey and Gene Expression Analysis of the Basic Leucine Zipper Transcription Factor Family in Rice. Plant Physiol. 2008, 146, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.L.; Lehninger, A.L.; Cox, M.M. Lehninger Principles of Biochemistry; Macmillan: New York, NY, USA, 2008. [Google Scholar]
- Peng, C.; Lu, Z.; Xie, Z.; Cheng, Z.; Chen, Y.; Tan, M.; Luo, H.; Zhang, Y.; He, W.; Yang, K.; et al. The First Identification of Lysine Malonylation Substrates and Its Regulatory Enzyme. Mol. Cell. Proteom. 2011, 10, M111.012658. [Google Scholar] [CrossRef] [PubMed]
- Hopke, A.; Brown, A.J.P.; Hall, R.A.; Wheeler, R.T. Dynamic Fungal Cell Wall Architecture in Stress Adaptation and Immune Evasion. Trends Microbiol. 2018, 26, 284–295. [Google Scholar] [CrossRef]
- Sanz, A.B.; García, R.; Rodríguez-Peña, J.M.; Arroyo, J. The CWI Pathway: Regulation of the Transcriptional Adaptive Response to Cell Wall Stress in Yeast. J. Fungi 2017, 4, 1. [Google Scholar] [CrossRef]
- Bahn, Y.-S. Master and Commander in Fungal Pathogens: The Two-Component System and the HOG Signaling Pathway. Eukaryot. Cell 2008, 7, 2017–2036. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Mehrabi, R.; Xu, J.-R. Mitogen-Activated Protein Kinase Pathways and Fungal Pathogenesis. Eukaryot. Cell 2007, 6, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Román, E.; Arana, D.M.; Nombela, C.; Alonso-Monge, R.; Pla, J. MAP kinase pathways as regulators of fungal virulence. Trends Microbiol. 2007, 15, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Juvvadi, P.R.; Lee, S.C.; Heitman, J.; Steinbach, W.J. Calcineurin in fungal virulence and drug resistance: Prospects for harnessing targeted inhibition of calcineurin for an antifungal therapeutic approach. Virulence 2017, 8, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Mattos, E.C.; Silva, L.P.; Valero, C.; de Castro, P.A.; dos Reis, T.F.; Ribeiro, L.F.C.; Marten, M.R.; Silva-Rocha, R.; Westmann, C.; Silva, C.H.T.d.P.d.; et al. The Aspergillus fumigatus Phosphoproteome Reveals Roles of High-Osmolarity Glycerol Mitogen-Activated Protein Kinases in Promoting Cell Wall Damage and Caspofungin Tolerance. MBio 2020, 11, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-J.; Hou, Y.-H.; Chen, Y.-L. The histone acetyltransferase GcnE regulates conidiation and biofilm formation in Aspergillus fumigatus. Med. Mycol. 2020, 58, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Shivarathri, R.; Tscherner, M.; Zwolanek, F.; Singh, N.K.; Chauhan, N.; Kuchler, K. The Fungal Histone Acetyl Transferase Gcn5 Controls Virulence of the Human Pathogen Candida albicans through Multiple Pathways. Sci. Rep. 2019, 9, 9445. [Google Scholar] [CrossRef]
- Bates, S.; Hughes, H.B.; Munro, C.A.; Thomas, W.P.; MacCallum, D.M.; Bertram, G.; Atrih, A.; Ferguson, M.A.; Brown, A.J.; Odds, F.C.; et al. Outer Chain N-Glycans Are Required for Cell Wall Integrity and Virulence of Candida albicans. J. Biol. Chem. 2006, 281, 90–98. [Google Scholar] [CrossRef]
- Gentzsch, M.; Tanner, W. The PMT gene family: Protein O-glycosylation in Saccharomyces cerevisiae is vital. EMBO J. 1996, 15, 5752–5759. [Google Scholar] [CrossRef] [PubMed]
- Prill, S.K.H.; Klinkert, B.; Timpel, C.; Gale, C.A.; Schröppel, K.; Ernst, J.F. Pmt family of candida albicans: Five protein mannosyltransferase isoforms affect growth, morphogenesis and antifungal resistance. Mol. Microbiol. 2005, 55, 546–560. [Google Scholar] [CrossRef] [PubMed]
- Rouabhia, M.; Schaller, M.; Corbucci, C.; Vecchiarelli, A.; Prill, S.K.-H.; Giasson, L.; Ernst, J.F. Virulence of the Fungal Pathogen Candida albicans Requires the Five Isoforms of Protein Mannosyltransferases. Infect. Immun. 2005, 73, 4571–4580. [Google Scholar] [CrossRef] [PubMed]
- Olson, G.M.; Fox, D.S.; Wang, P.; Alspaugh, J.A.; Buchanan, K.L. Role of Protein O-Mannosyltransferase Pmt4 in the Morphogenesis and Virulence of Cryptococcus neoformans. Eukaryot. Cell 2007, 6, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Willger, S.D.; Ernst, J.F.; Alspaugh, J.A.; Lengeler, K.B. Characterization of the PMT Gene Family in Cryptococcus neoformans. PLoS ONE 2009, 4, e6321. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, T.; Yang, J.; Chen, L.; Liu, B.; Wei, C.; Wang, L.; Jin, Q. The first succinylome profile of Trichophyton rubrum reveals lysine succinylation on proteins involved in various key cellular processes. BMC Genom. 2017, 18, 577. [Google Scholar] [CrossRef] [PubMed]
- Fachin, A.L.; Ferreira-Nozawa, M.S.; Maccheroni, W., Jr.; Martinez-Rossi, N.M. Role of the abc transporter trumdr2 in terbinafine, 4-nitroquinoline n-oxide and ethidium bromide susceptibility in trichophyton rubrum. J. Med. Microbiol. 2006, 55, 1093–1099. [Google Scholar] [CrossRef]
- Sanglard, D.; Ischer, F.; Monod, M.; Bille, J. Cloning of Candida albicans genes conferring resistance to azole antifungal agents: Characterization of CDR2, a new multidrug ABC transporter gene. Microbiology 1997, 143, 405–416. [Google Scholar] [CrossRef]
- Andrade, A.C.; Van Nistelrooy, J.G.M.; Peery, R.B.; Skatrud, P.L.; De Waard, M.A. The role of ABC transporters from Aspergillus nidulans in protection against cytotoxic agents and in antibiotic production. Mol. Genet. Genom. 2000, 263, 966–977. [Google Scholar] [CrossRef]
- Smith, S.E.; Csank, C.; Reyes, G.; Ghannoum, M.A.; Berlin, V. Candida albicans rho1 is required for cell viability in vitro and in vivo. FEMS Yeast Res. 2002, 2, 103–111. [Google Scholar]
- Lam, W.C.; Gerik, K.J.; Lodge, J.K. Role of Cryptococcus neoformans Rho1 GTPases in the PKC1 Signaling Pathway in Response to Thermal Stress. Eukaryot. Cell 2013, 12, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Price, M.S.; Nichols, C.B.; Alspaugh, J.A. The Cryptococcus neoformans Rho-GDP Dissociation Inhibitor Mediates Intracellular Survival and Virulence. Infect. Immun. 2008, 76, 5729–5737. [Google Scholar] [CrossRef] [PubMed]
- Cowen, L.E.; Singh, S.D.; Köhler, J.R.; Collins, C.; Zaas, A.K.; Schell, W.A.; Aziz, H.; Mylonakis, E.; Perfect, J.R.; Whitesell, L.; et al. Harnessing Hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc. Natl. Acad. Sci. USA 2009, 106, 2818–2823. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Yang, M.; Yue, Y.; Ge, F.; Li, Y.; Guo, X.; Zhang, J.; Zhang, F.; Nie, X.; Wang, S. Lysine Succinylation Contributes to Aflatoxin Production and Pathogenicity in Aspergillus flavus. Mol. Cell. Proteom. 2018, 17, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Chornyi, S.; Ijlst, L.; van Roermund, C.W.T.; Wanders, R.J.A.; Waterham, H.R. Peroxisomal Metabolite and Cofactor Transport in Humans. Front. Cell Dev. Biol. 2021, 8, 1753. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, M.; Ge, F.; Jiang, B.; Hu, R.; Zhou, X.; Yang, Y.; Liu, M. Lysine Succinylation of VBS Contributes to Sclerotia Development and Aflatoxin Biosynthesis in Aspergillus flavus. Mol. Cell. Proteom. 2023, 22, 100490. [Google Scholar] [CrossRef] [PubMed]
- Hayaishi, O.; Veda, K. Poly (ADP-Ribose) and ADP-Ribosylation of Proteins. Annu. Rev. Biochem. 1977, 46, 95–116. [Google Scholar] [CrossRef]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine Glutarylation Is a Protein Posttranslational Modification Regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef]
- Chen, Y.; Sprung, R.; Tang, Y.; Ball, H.; Sangras, B.; Kim, S.C.; Falck, J.R.; Peng, J.; Gu, W.; Zhao, Y. Lysine Propionylation and Butyrylation Are Novel Post-translational Modifications in Histones. Mol. Cell. Proteom. 2007, 6, 812–819. [Google Scholar] [CrossRef]
- Dai, L.; Peng, C.; Montellier, E.; Lu, Z.; Chen, Y.; Ishii, H.; Debernardi, A.; Buchou, T.; Rousseaux, S.; Jin, F.; et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 2014, 10, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ning, Z.; Mayne, J.; Yang, Y.; Deeke, S.A.; Walker, K.; Farnsworth, C.L.; Stokes, M.P.; Couture, J.-F.; Mack, D.; et al. Widespread protein lysine acetylation in gut microbiome and its alterations in patients with Crohn’s disease. Nat. Commun. 2020, 11, 4120. [Google Scholar] [CrossRef]
- Xie, Z.; Dai, J.; Dai, L.; Tan, M.; Cheng, Z.; Wu, Y.; Boeke, J.D.; Zhao, Y. Lysine Succinylation and Lysine Malonylation in Histones. Mol. Cell. Proteom. 2012, 11, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Schölz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine Succinylation Is a Frequently Occurring Modification in Prokaryotes and Eukaryotes and Extensively Overlaps with Acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Huang, J.Y.; Schwer, B.; Verdin, E. SIRT3 Regulates Mitochondrial Protein Acetylation and Intermediary Metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Colak, G.; Pougovkina, O.; Dai, L.; Tan, M.; Te Brinke, H.; Huang, H.; Cheng, Z.; Park, J.; Wan, X.; Liu, X. Proteomic and biochemical studies of lysine malonylation suggest its malonic aciduria-associated regulatory role in mitochondrial function and fatty acid oxidation. Mol. Cell Proteom. 2015, 14, 3056–3071. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Zhao, Y. Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Mol. Cell. Proteom. 2015, 14, 2308–2315. [Google Scholar] [CrossRef]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 Is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 Regulates the Mitochondrial Lysine Succinylome and Metabolic Networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, M.J.; Cha, S.H.; Sidhaye, A.; Chohnan, S.; Cline, G.; Shulman, G.I.; Lane, M.D. Regulation of hypothalamic malonyl-coa by central glucose and leptin. Proc. Natl. Acad. Sci. USA 2007, 104, 19285–19290. [Google Scholar] [CrossRef] [PubMed]
- Ottaway, J.; McClellan, J.; Saunderson, C. Succinic thiokinase and metabolic control. Int. J. Biochem. 1981, 13, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Trefely, S.; Lovell, C.D.; Snyder, N.W.; Wellen, K.E. Compartmentalised acyl-coa metabolism and roles in chromatin regulation. Mol. Metab. 2020, 38, 100941. [Google Scholar] [CrossRef] [PubMed]
- Simithy, J.; Sidoli, S.; Yuan, Z.-F.; Coradin, M.; Bhanu, N.V.; Marchione, D.M.; Klein, B.J.; Bazilevsky, G.A.; McCullough, C.E.; Magin, R.S.; et al. Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 2017, 8, 1141. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Sivanand, S.; Viney, I.; Wellen, K.E. Spatiotemporal control of acetyl-coa metabolism in chromatin regulation. Trends Biochem. Sci. 2018, 43, 61–74. [Google Scholar] [CrossRef]
- Parker, C.W.; Kern, M.; Eisen, H.N. Polyfunctional dinitrophenyl haptens as reagents for elicitation of immediate type allergic skin responses. J. Exp. Med. 1962, 115, 789–801. [Google Scholar] [CrossRef]
- Kidwai, S.A.; Ansari, A.A.; Salahuddin, A. Effect of succinylation (3-carboxypropionylation) on the conformation and immunological activity of ovalbumin. Biochem. J. 1976, 155, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.; Becher, D.; Büttner, K.; Biran, D.; Hecker, M.; Ron, E.Z. Probing the active site of homoserine trans-succinylase. FEBS Lett. 2004, 577, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; He, X.; Ye, D.; Lin, Y.; Yu, H.; Yao, C.; Huang, L.; Zhang, J.; Wang, F.; Xu, S.; et al. NADP+-IDH Mutations Promote Hypersuccinylation that Impairs Mitochondria Respiration and Induces Apoptosis Resistance. Mol. Cell 2015, 60, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Galam, L.; Failla, A.; Soundararajan, R.; Lockey, R.F.; Kolliputi, N. 4-Hydroxynonenal regulates mitochondrial function in human small airway epithelial cells. Oncotarget 2015, 6, 41508–41521. [Google Scholar] [CrossRef] [PubMed]
- Waxman, A.B.; Kolliputi, N. IL-6 Protects against Hyperoxia-Induced Mitochondrial Damage via Bcl-2–Induced Bak Interactions with Mitofusions. Am. J. Respir. Cell Mol. Biol. 2009, 41, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Kolliputi, N.; Waxman, A.B.; Narala, V.R.; Fukumoto, J.; Hernández-Cuervo, H.; Patil, S.S.; Krishnamurthy, S.; Breitzig, M.; Galam, L.; Soundararajan, R.; et al. IL-6 cytoprotection in hyperoxic acute lung injury occurs via PI3K/Akt-mediated Bax phosphorylation. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L6–L16. [Google Scholar] [CrossRef]
- Wagner, G.R.; Hirschey, M.D. Nonenzymatic Protein Acylation as a Carbon Stress Regulated by Sirtuin Deacylases. Mol. Cell 2014, 54, 5–16. [Google Scholar] [CrossRef]
- Wagner, G.R.; Payne, R.M. Widespread and Enzyme-independent Nϵ-Acetylation and Nϵ-Succinylation of Proteins in the Chemical Conditions of the Mitochondrial Matrix. J. Biol. Chem. 2013, 288, 29036–29045. [Google Scholar] [CrossRef]
- Hausinger, R.P. Fe (ii)/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 21–68. [Google Scholar] [CrossRef]
- Alarcon, C.; Wicksteed, B.; Prentki, M.; Corkey, B.E.; Rhodes, C.J. Succinate Is a Preferential Metabolic Stimulus-Coupling Signal for Glucose-Induced Proinsulin Biosynthesis Translation. Diabetes 2002, 51, 2496–2504. [Google Scholar] [CrossRef] [PubMed]
- Smestad, J.; Erber, L.; Chen, Y.; Maher, L.J. Chromatin Succinylation Correlates with Active Gene Expression and Is Perturbed by Defective TCA Cycle Metabolism. iScience 2018, 2, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Xu, H.; Chen, H.; Chen, W.; Denton, T.T.; Zhang, S. Alpha-ketoglutarate dehydrogenase complex-dependent succinylation of proteins in neurons and neuronal cell lines. J. Neurochem. 2015, 134, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, Y.R.; Liu, K.; Yin, Z.; Liu, R.; Xia, Y.; Tan, L.; Yang, P.; Lee, J.-H.; Li, X.-J.; et al. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 2017, 552, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Guo, D.; Yan, D.; Ma, C.; Shao, F.; Wang, Y.; Luo, S.; Lin, L.; Tao, J.; Jiang, Y.; et al. KAT2A succinyltransferase activity-mediated 14-3-3ζ upregulation promotes β-catenin stabilization-dependent glycolysis and proliferation of pancreatic carcinoma cells. Cancer Lett. 2020, 469, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, J.; Chung, M.W.H.; Feng, L.; Sun, H.; Hao, Q. Identification of the yeats domain of gas41 as a ph-dependent reader of histone succinylation. Proc. Natl. Acad. Sci. USA 2018, 115, 2365–2370. [Google Scholar] [CrossRef] [PubMed]
- Kurmi, K.; Hitosugi, S.; Wiese, E.K.; Boakye-Agyeman, F.; Gonsalves, W.I.; Lou, Z.; Karnitz, L.M.; Goetz, M.P.; Hitosugi, T. Carnitine Palmitoyltransferase 1A Has a Lysine Succinyltransferase Activity. Cell Rep. 2018, 22, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, C.; Li, X.; Shen, J.; Xu, Y.; Shi, H.; Mu, X.; Pan, J.; Zhao, T.; Li, M.; et al. CPT1A-mediated succinylation of S100A10 increases human gastric cancer invasion. J. Cell. Mol. Med. 2019, 23, 293–305. [Google Scholar] [CrossRef]
- Chanda, A.; Roze, L.V.; Kang, S.; Artymovich, K.A.; Hicks, G.R.; Raikhel, N.V.; Calvo, A.M.; Linz, J.E. A key role for vesicles in fungal secondary metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 19533–19538. [Google Scholar] [CrossRef]
- Tong, L. Acetyl-coenzyme A carboxylase: Crucial metabolic enzyme and attractive target for drug discovery. Cell. Mol. Life Sci. 2005, 62, 1784–1803. [Google Scholar] [CrossRef]
- Zhang, H.; Li, P.; Ren, S.; Cheng, Z.; Zhao, G.; Zhao, W. ScCobB2-mediated Lysine Desuccinylation Regulates Protein Biosynthesis and Carbon Metabolism in Streptomyces coelicolor. Mol. Cell. Proteom. 2019, 18, 2003–2017. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shi, L.; Yang, S.; Yan, R.; Zhang, D.; Yang, J.; He, L.; Li, W.; Yi, X.; Sun, L.; et al. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 2016, 7, 12235. [Google Scholar] [CrossRef] [PubMed]
- Hang, T.; Chen, W.; Wu, M.; Zhan, L.; Wang, C.; Jia, N.; Zhang, X.; Zang, J. Structural insights into the molecular mechanism underlying Sirt5-catalyzed desuccinylation of histone peptides. Biochem. J. 2019, 476, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Hu, H.; Qu, S.; Wang, J.; Hua, C.; Zhang, J.; Wei, P.; He, X.; Hao, J.; Liu, P.; et al. SIRT5 deacylates metabolism-related proteins and attenuates hepatic steatosis in ob/ob mice. EBioMedicine 2018, 36, 347–357. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.; et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. USA 2016, 113, 4320–4325. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tian, M.; Sun, R.; Zhang, M.; Zhou, L.; Jin, L.; Chen, L.; Zhou, W.; Duan, K.; Chen, Y.; et al. SIRT 5 inhibits peroxisomal ACOX 1 to prevent oxidative damage and is downregulated in liver cancer. Embo Rep. 2018, 19, e45124. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Meyer, J.G.; Cai, W.; Softic, S.; Li, M.E.; Verdin, E.; Newgard, C.; Schilling, B.; Kahn, C.R. Regulation of UCP1 and Mitochondrial Metabolism in Brown Adipose Tissue by Reversible Succinylation. Mol. Cell 2019, 74, 844–857.e7. [Google Scholar] [CrossRef] [PubMed]
- Xiangyun, Y.; Xiaomin, N.; Linping, G.; Yunhua, X.; Ziming, L.; Yongfeng, Y.; Zhiwei, C.; Shun, L. Desuccinylation of pyruvate kinase M2 by SIRT5 contributes to antioxidant response and tumor growth. Oncotarget 2017, 8, 6984–6993. [Google Scholar] [CrossRef]
- Kumar, S.; Lombard, D.B. Generation and purification of catalytically active recombinant sirtuin5 (sirt5) protein. In Histone Deacetylases Methods and Protocols; Humana: New York, NY, USA, 2016; pp. 241–257. [Google Scholar]
- Yang, X.; Wang, Z.; Li, X.; Liu, B.; Liu, M.; Liu, L.; Chen, S.; Ren, M.; Wang, Y.; Yu, M.; et al. SHMT2 Desuccinylation by SIRT5 Drives Cancer Cell Proliferation. Cancer Res 2018, 78, 372–386. [Google Scholar] [CrossRef]
- Polletta, L.; Vernucci, E.; Carnevale, I.; Arcangeli, T.; Rotili, D.; Palmerio, S.; Steegborn, C.; Nowak, T.; Schutkowski, M.; Pellegrini, L.; et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 2015, 11, 253–270. [Google Scholar] [CrossRef]
- Zhang, Y.; Bharathi, S.S.; Rardin, M.J.; Lu, J.; Maringer, K.V.; Sims-Lucas, S.; Prochownik, E.V.; Gibson, B.W.; Goetzman, E.S. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J. Biol. Chem. 2017, 292, 10239–10249. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, J.; Sun, R.; Tao, X.; Wang, X.; Kang, Q.; Wang, H.; Zhang, L.; Liu, P.; Zhang, J.; et al. SIRT5 deficiency suppresses mitochondrial ATP production and promotes AMPK activation in response to energy stress. PLoS ONE 2019, 14, e0211796. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.-F.; Xu, H.-B.; Wang, J.-Y.; Lin, Q.; Ruan, Z.; Liu, F.-B.; Jin, W.; Huang, H.-H.; Chen, X. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem. Biophys. Res. Commun. 2013, 441, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, F.; Sun, R.; Chen, X.; Zhang, M.; Xu, Q.; Wang, Y.; Wang, S.; Xiong, Y.; Guan, K.; et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. Embo Rep. 2016, 17, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Marmorstein, R. Structural Basis for Sirtuin Activity and Inhibition. J. Biol. Chem. 2012, 287, 42428–42435. [Google Scholar] [CrossRef] [PubMed]
- Albaugh, B.N.; Arnold, K.M.; Denu, J.M. KAT(ching) Metabolism by the Tail: Insight into the Links between Lysine Acetyltransferases and Metabolism. ChemBioChem 2011, 12, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Horie, S.; Isobe, M.; Suga, T. Changes in CoA Pools in Hepatic Peroxisomes of the Rat, under Various Conditions. J. Biochem. 1986, 99, 1345–1352. [Google Scholar] [CrossRef]
- Alexson, S.E.; Nedergaard, J.; Cannon, B. Inhibition of acetyl-carnitine oxidation in rat brown-adipose-tissue mitochondria by erucoyl-carnitine is due to sequestration of CoA. Biochim. Biophys. Acta BBA—Lipids Lipid Metab. 1985, 834, 149–158. [Google Scholar] [CrossRef]
- Alexson, S.; Nedergaard, J.; Cannon, B. Partial protection erucoyl-carnitine inhibition in hamster brown-adipose-tissue mitochondria is due to high CoA levels: A comparison with rat brown-adipose-tissue mitochondria. Comp. Biochem. Physiol. B 1986, 83, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Burch, J.S.; Marcero, J.R.; Maschek, J.A.; Cox, J.E.; Jackson, L.K.; Medlock, A.E.; Phillips, J.D.; Dailey, H.A., Jr. Glutamine via α-ketoglutarate dehydrogenase provides succinyl-coa for heme synthesis during erythropoiesis. Blood J. Am. Soc. Hematol. 2018, 132, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Seyfried, T.N. Mitochondrial Substrate-Level Phosphorylation as Energy Source for Glioblastoma: Review and Hypothesis. ASN Neuro 2018, 10, 1759091418818261. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C. Which way does the citric acid cycle turn during hypoxia? The critical role of α-ketoglutarate dehydrogenase complex. J. Neurosci. Res. 2013, 91, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Gerencser, A.A.; Mandi, M.; Mathe, K.; Töröcsik, B.; Doczi, J.; Turiak, L.; Kiss, G.; Konràd, C.; Vajda, S. Forward operation of adenine nucleotide translocase during f0f1-atpase reversal: Critical role of matrix substrate-level phosphorylation. FASEB J. 2010, 24, 2405. [Google Scholar] [CrossRef] [PubMed]
- Gut, P.; Matilainen, S.; Meyer, J.G.; Pällijeff, P.; Richard, J.; Carroll, C.J.; Euro, L.; Jackson, C.B.; Isohanni, P.; Minassian, B.A.; et al. SUCLA2 mutations cause global protein succinylation contributing to the pathomechanism of a hereditary mitochondrial disease. Nat. Commun. 2020, 11, 5927. [Google Scholar] [CrossRef] [PubMed]
- Schölz, C.; Lyon, D.; Refsgaard, J.C.; Jensen, L.J.; Choudhary, C.; Weinert, B.T. Avoiding abundance bias in the functional annotation of posttranslationally modified proteins. Nat. Methods 2015, 12, 1003–1004. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.J.; Smith, A.D.; Hasan, N.M.; Sabat, G.; Fahien, L.A. Feasibility of pathways for transfer of acyl groups from mitochondria to the cytosol to form short chain acyl-coas in the pancreatic beta cell. J. Biol. Chem. 2007, 282, 30596–30606. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; Mooijer, P.A.; Zhang, Z.; Reddy, J.K.; Spector, A.A.; Wanders, R.J. Identification of the peroxisomal β-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J. Lipid Res. 2001, 42, 1987–1995. [Google Scholar] [CrossRef]
- Prip-Buus, C.; Thuillier, L.; Abadi, N.; Prasad, C.; Dilling, L.; Klasing, J.; Demaugre, F.; Greenberg, C.R.; Haworth, J.C.; Droin, V.; et al. Molecular and Enzymatic Characterization of a Unique Carnitine Palmitoyltransferase 1A Mutation in the Hutterite Community. Mol. Genet. Metab. 2001, 73, 46–54. [Google Scholar] [CrossRef]
- Ijlst, L.; Mandel, H.; Oostheim, W.; Ruiter, J.P.; Gutman, A.; Wanders, R.J. Molecular basis of hepatic carnitine palmitoyltransferase I deficiency. J. Clin. Investig. 1998, 102, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Schmidt, T.; Situ, A.J.; Johnson, J.O.; Lee, P.R.; Chen, K.-L.; Bott, L.C.; Fadó, R.; Harmison, G.H.; Parodi, S. Mutation in cpt1c associated with pure autosomal dominant spastic paraplegia. JAMA Neurol. 2015, 72, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Lamhonwah, A.-M.; Skaug, J.; Scherer, S.W.; Tein, I. A third human carnitine/organic cation transporter (OCTN3) as a candidate for the 5q31 Crohn’s disease locus (IBD5). Biochem. Biophys. Res. Commun. 2003, 301, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Januszewicz, E.; Pająk, B.; Gajkowska, B.; Samluk, Ł.; Djavadian, R.L.; Hinton, B.T.; Nałęcz, K.A. Organic cation/carnitine transporter OCTN3 is present in astrocytes and is up-regulated by peroxisome proliferators-activator receptor agonist. Int. J. Biochem. Cell Biol. 2009, 41, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P.; de Schryver, E.; Young, S.G.; Zwijsen, A.; Fransen, M.; Espeel, M.; Baes, M.; Van Ael, E. Slc25a17 Gene Trapped Mice: PMP34 Plays a Role in the Peroxisomal Degradation of Phytanic and Pristanic Acid. Front. Cell Dev. Biol. 2020, 8, 144. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef]
- Vamecq, J.; de Hoffmann, E.; Van Hoof, F. The microsomal dicarboxylyl-coA synthetase. Biochem. J. 1985, 230, 683–693. [Google Scholar] [CrossRef]
- Vamecq, J.; Draye, J.-P. Peroxisomal and mitochondrial βoxidation of monocarboxylyl-coA, ω-hydroxymonocarboxylyl-coa and dicarboxylyl-coa esters in tissues from untreated and clofibrate-treated rats. J. Biochem. 1989, 106, 216–222. [Google Scholar] [CrossRef]
- Chinopoulos, C. The Mystery of Extramitochondrial Proteins Lysine Succinylation. Int. J. Mol. Sci. 2021, 22, 6085. [Google Scholar] [CrossRef]
- Wang, G.; Xu, L.; Yu, H.; Gao, J.; Guo, L. Systematic analysis of the lysine succinylome in the model medicinal mushroom Ganoderma lucidum. BMC Genom. 2019, 20, 585. [Google Scholar] [CrossRef]
- Wang, J.; Li, L.; Chai, R.; Zhang, Z.; Qiu, H.; Mao, X.; Hao, Z.; Wang, Y.; Sun, G. Succinyl-proteome profiling of Pyricularia oryzae, a devastating phytopathogenic fungus that causes rice blast disease. Sci. Rep. 2019, 9, 3490. [Google Scholar] [CrossRef]
- Zheng, H.; He, Y.; Zhou, X.; Qian, G.; Lv, G.; Shen, Y.; Liu, J.; Li, D.; Li, X.; Liu, W. Systematic Analysis of the Lysine Succinylome in Candida albicans. J. Proteome Res. 2016, 15, 3793–3801. [Google Scholar] [CrossRef] [PubMed]
- Frankovsky, J.; Keresztesová, B.; Bellová, J.; Kunová, N.; Čanigová, N.; Hanakova, K.; Bauer, J.A.; Ondrovičová, G.; Lukáčová, V.; Siváková, B. The yeast mitochondrial succinylome: Implications for regulation of mitochondrial nucleoids. J. Biol. Chem. 2021, 297, 101155. [Google Scholar] [CrossRef]
- Liu, J.; Shangguan, Y.; Tang, D.; Dai, Y. Histone succinylation and its function on the nucleosome. J. Cell. Mol. Med. 2021, 25, 7101–7109. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Ding, D.; Tian, G.; Kwan, K.C.J.; Liu, Z.; Ishibashi, T.; Li, X.D. Semisynthesis of site-specifically succinylated histone reveals that succinylation regulates nucleosome unwrapping rate and DNA accessibility. Nucleic Acids Res. 2020, 48, 9538–9549. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, Z.; Wang, Y.; Li, Y.; Zhu, J.; Hu, R.; Yang, Y.; Liu, M. Quantitative Proteomic Analysis for High- and Low-Aflatoxin-Yield Aspergillus flavus Strains Isolated from Natural Environments. Front. Microbiol. 2021, 12, 741875. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Chen, J.; Yang, Y.; Shen, C.; Xu, D.; Wang, J.; Yan, D.; He, Y.; Zheng, B. Quantitative succinyl-proteome profiling of Chinese hickory (Carya cathayensis) during the grafting process. BMC Plant Biol. 2019, 19, 467. [Google Scholar] [CrossRef]
- Meng, X.; Mujahid, H.; Zhang, Y.; Peng, X.; Redoña, E.D.; Wang, C.; Peng, Z. Comprehensive Analysis of the Lysine Succinylome and Protein Co-modifications in Developing Rice Seeds. Mol. Cell. Proteom. 2019, 18, 2359–2372. [Google Scholar] [CrossRef]
- Zhou, H.; Finkemeier, I.; Guan, W.; Tossounian, M.A.; Wei, B.; Young, D.; Huang, J.; Messens, J.; Yang, X.; Zhu, J. Oxidative stress-triggered interactions between the succinyl-and acetyl-proteomes of rice leaves. Plant Cell Environ. 2018, 41, 1139–1153. [Google Scholar] [CrossRef]
- Xie, L.; Liu, W.; Li, Q.; Chen, S.; Xu, M.; Huang, Q.; Zeng, J.; Zhou, M.; Xie, J. First Succinyl-Proteome Profiling of Extensively Drug-Resistant Mycobacterium tuberculosis Revealed Involvement of Succinylation in Cellular Physiology. J. Proteome Res. 2015, 14, 107–119. [Google Scholar] [CrossRef]
- Yang, K.; Tian, J.; Keller, N.P. Post-translational modifications drive secondary metabolite biosynthesis in aspergillus: A review. Environ. Microbiol. 2022, 24, 2857–2881. [Google Scholar] [CrossRef] [PubMed]
- Qoronfleh, M.W.; Benton, B.; Ignacio, R.; Kaboord, B. Selective Enrichment of Membrane Proteins by Partition Phase Separation for Proteomic Studies. J. Biomed. Biotechnol. 2003, 2003, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ning, Q.; Chai, H.; Ma, Z. Accurate in silico identification of protein succinylation sites using an iterative semi-supervised learning technique. J. Theor. Biol. 2015, 374, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ding, Y.-X.; Ding, J.; Lei, Y.-H.; Wu, L.-Y.; Deng, N.-Y. Isuc-pseaac: Predicting lysine succinylation in proteins by incorporating peptide position-specific propensity. Sci. Rep. 2015, 5, 10184. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-D.; Shi, S.-P.; Wen, P.-P.; Qiu, J.-D. SuccFind: A novel succinylation sites online prediction tool via enhanced characteristic strategy. Bioinformatics 2015, 31, 3748–3750. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.-C. Isuc-pseopt: Identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal. Biochem. 2016, 497, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.-C. Psuc-lys: Predict lysine succinylation sites in proteins with pseaac and ensemble random forest approach. J. Theor. Biol. 2016, 394, 223–230. [Google Scholar] [CrossRef] [PubMed]
- López, Y.; Dehzangi, A.; Lal, S.P.; Taherzadeh, G.; Michaelson, J.; Sattar, A.; Tsunoda, T.; Sharma, A. SucStruct: Prediction of succinylated lysine residues by using structural properties of amino acids. Anal. Biochem. 2017, 527, 24–32. [Google Scholar] [CrossRef]
- López, Y.; Sharma, A.; Dehzangi, A.; Lal, S.P.; Taherzadeh, G.; Sattar, A.; Tsunoda, T. Success: Evolutionary and structural properties of amino acids prove effective for succinylation site prediction. BMC Genom. 2018, 19, 105–114. [Google Scholar] [CrossRef]
- Dehzangi, A.; López, Y.; Lal, S.P.; Taherzadeh, G.; Michaelson, J.; Sattar, A.; Tsunoda, T.; Sharma, A. PSSM-Suc: Accurately predicting succinylation using position specific scoring matrix into bigram for feature extraction. J. Theor. Biol. 2017, 425, 97–102. [Google Scholar] [CrossRef]
- Dehzangi, A.; López, Y.; Lal, S.P.; Taherzadeh, G.; Sattar, A.; Tsunoda, T.; Sharma, A. Improving succinylation prediction accuracy by incorporating the secondary structure via helix, strand and coil, and evolutionary information from profile bigrams. PLoS ONE 2018, 13, e0191900. [Google Scholar] [CrossRef]
- Ning, Q.; Zhao, X.; Bao, L.; Ma, Z.; Zhao, X. Detecting Succinylation sites from protein sequences using ensemble support vector machine. BMC Bioinform. 2018, 19, 237. [Google Scholar] [CrossRef]
- Ai, H.; Wu, R.; Zhang, L.; Wu, X.; Ma, J.; Hu, H.; Huang, L.; Chen, W.; Zhao, J.; Liu, H. Psuc-pserat: Predicting lysine succinylation in proteins by exploiting the ratios of sequence coupling and properties. J. Comput. Biol. 2017, 24, 1050–1059. [Google Scholar] [CrossRef]
- Qiu, W.-R.; Sun, B.-Q.; Xiao, X.; Xu, Z.-C.; Chou, K.-C. Iptm-mlys: Identifying multiple lysine ptm sites and their different types. Bioinformatics 2016, 32, 3116–3123. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Khatun, M.S.; Kurata, H. Large-Scale Assessment of Bioinformatics Tools for Lysine Succinylation Sites. Cells 2019, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Kurata, H. GPSuc: Global Prediction of Generic and Species-specific Succinylation Sites by aggregating multiple sequence features. PLoS ONE 2018, 13, e0200283. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Yang, S.; Zhou, Y.; Mollah, M.N.H. SuccinSite: A computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties. Mol. BioSyst. 2016, 12, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Khatun, M.S.; Mollah, N.H.; Yong, C.; Guo, D. A systematic identification of species-specific protein succinylation sites using joint element features information. Int. J. Nanomed. 2017, 12, 6303–6315. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-Y.; Hsu, J.B.-K.; Lee, T.-Y. Characterization and Identification of Lysine Succinylation Sites based on Deep Learning Method. Sci. Rep. 2019, 9, 16175. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Jia, C.; Li, F.; Song, J. Inspector: A lysine succinylation predictor based on edited nearest-neighbor undersampling and adaptive synthetic oversampling. Anal. Biochem. 2020, 593, 113592. [Google Scholar] [CrossRef]
- Thapa, N.; Chaudhari, M.; McManus, S.; Roy, K.; Newman, R.H.; Saigo, H.; Kc, D.B. DeepSuccinylSite: A deep learning based approach for protein succinylation site prediction. BMC Bioinform. 2020, 21, 63. [Google Scholar] [CrossRef] [PubMed]
- Ning, Q.; Ma, Z.; Zhao, X.; Yin, M. SSKM_Succ: A Novel Succinylation Sites Prediction Method Incorporating K-Means Clustering with a New Semi-Supervised Learning Algorithm. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 19, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Ning, W.; Xu, H.; Jiang, P.; Cheng, H.; Deng, W.; Guo, Y.; Xue, Y. HybridSucc: A Hybrid-learning Architecture for General and Species-specific Succinylation Site Prediction. Genom. Proteom. Bioinform. 2020, 18, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, M.; Qin, X.; Liu, G. Succinylation site prediction based on protein sequences using the ifs-lightgbm (bo) model. Comput. Math. Methods Med. 2020, 2020, 8858489. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, H.; Yan, Z.; Zhao, J.; Han, J. MDCAN-Lys: A Model for Predicting Succinylation Sites Based on Multilane Dense Convolutional Attention Network. Biomolecules 2021, 11, 872. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Shen, Q.; Zhang, G.; Wang, P.; Yu, Z.-G. LSTMCNNsucc: A Bidirectional LSTM and CNN-Based Deep Learning Method for Predicting Lysine Succinylation Sites. BioMed Res. Int. 2021, 2021, 9923112. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Rahman, A.; Hasan, A.M.; Ahmad, S.; Shovan, S.M. Computational identification of multiple lysine PTM sites by analyzing the instance hardness and feature importance. Sci. Rep. 2021, 11, 18882. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fungus | Enzymes | Function | References |
|---|---|---|---|
| Trichophyton rubrum | There are 18 proteins annotated as acetyltransferases (TERG_00136T0, TERG_00160T0, TERG_00920T0, TERG_00960T0, TERG_02546T0, TERG_03442T0, TERG_03711T0, TERG_04055T0, TERG_04687T0, TERG_04983T0, TERG_05174T0, TERG_05450T0, TERG_05561T0, TERG_06411T0, TERG_06572T0, TERG_07217T0, TERG_07375T0, and TERG_07548T0 | Additional experiments are required to investigate whether one or several of these acetyltransferases possess succinyltransferase activities, or if there is another succinyltransferase in T. rubrum. | [48] |
| Yeast | In yeast, five Sir2 proteins have been acknowledged (Sir2p and Hst1–4) | Can catalyze other forms of lysine acylation. In yeast, Hst2 exhibits a higher affinity for binding propionyl-lysine and butyryl-lysine compared to acetyl-lysine | [117,118,119] |
| Trichophyton rubrum | T. rubrum, five potential deacylases have been designated as Sir2 family histone deacetylases (TERG_03010T0, TERG_03268T0, TERG_05234T0, TERG_06970T0, and TERG_07330T0). | Further investigations are required to explore the activities of these Sir2 proteins. They are curious about whether one of these deacetylases might be involved in desuccinylase activities or if there is another desuccinylase present in these dermatophytes | [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adejor, J.; Tumukunde, E.; Li, G.; Lin, H.; Xie, R.; Wang, S. Impact of Lysine Succinylation on the Biology of Fungi. Curr. Issues Mol. Biol. 2024, 46, 1020-1046. https://doi.org/10.3390/cimb46020065
Adejor J, Tumukunde E, Li G, Lin H, Xie R, Wang S. Impact of Lysine Succinylation on the Biology of Fungi. Current Issues in Molecular Biology. 2024; 46(2):1020-1046. https://doi.org/10.3390/cimb46020065
Chicago/Turabian StyleAdejor, John, Elisabeth Tumukunde, Guoqi Li, Hong Lin, Rui Xie, and Shihua Wang. 2024. "Impact of Lysine Succinylation on the Biology of Fungi" Current Issues in Molecular Biology 46, no. 2: 1020-1046. https://doi.org/10.3390/cimb46020065