
Mitochondrial Dysfunction: A Key Player in Brain Aging and Diseases


Abstract
:1. Introduction
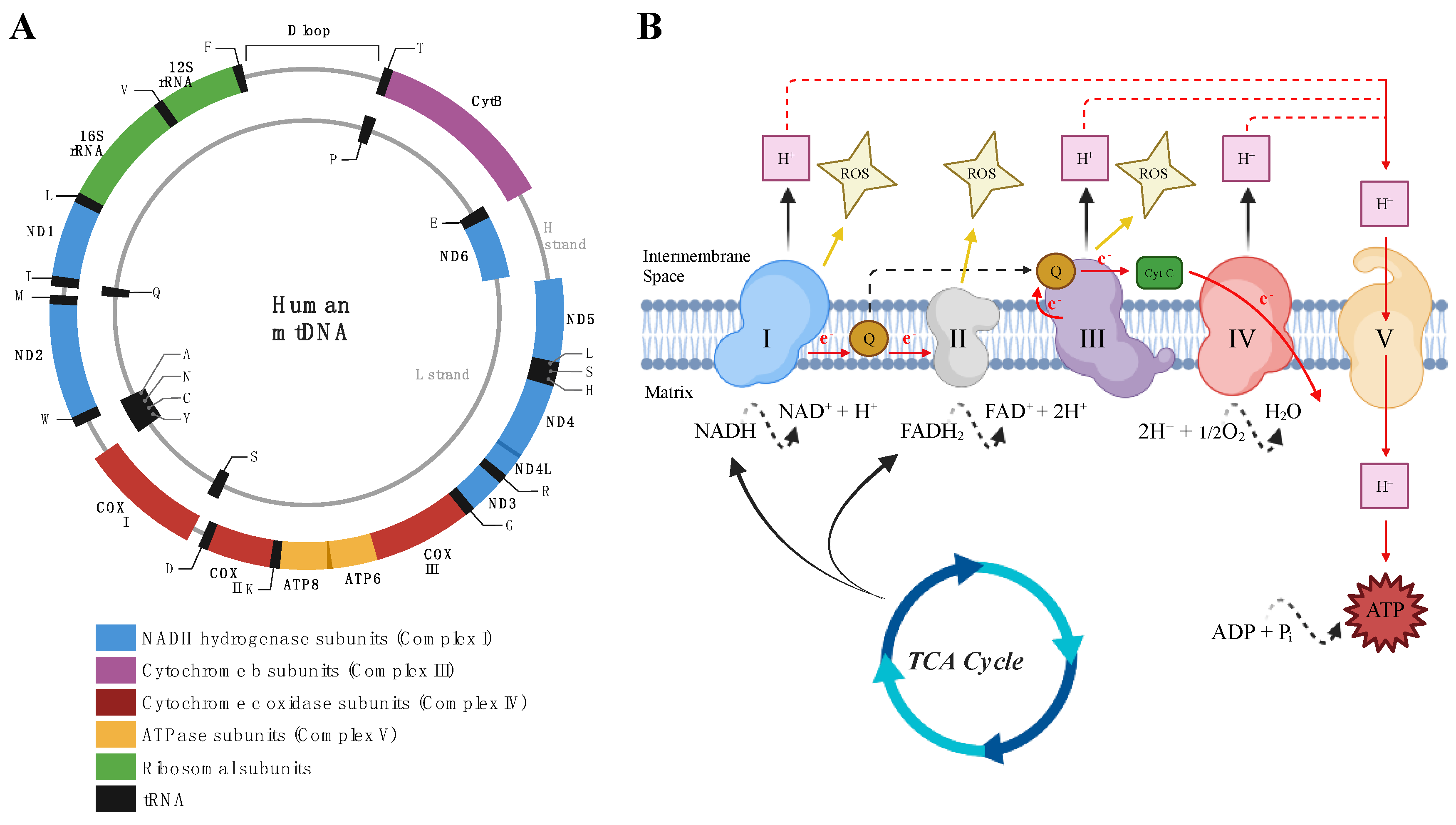
2. Mitochondria
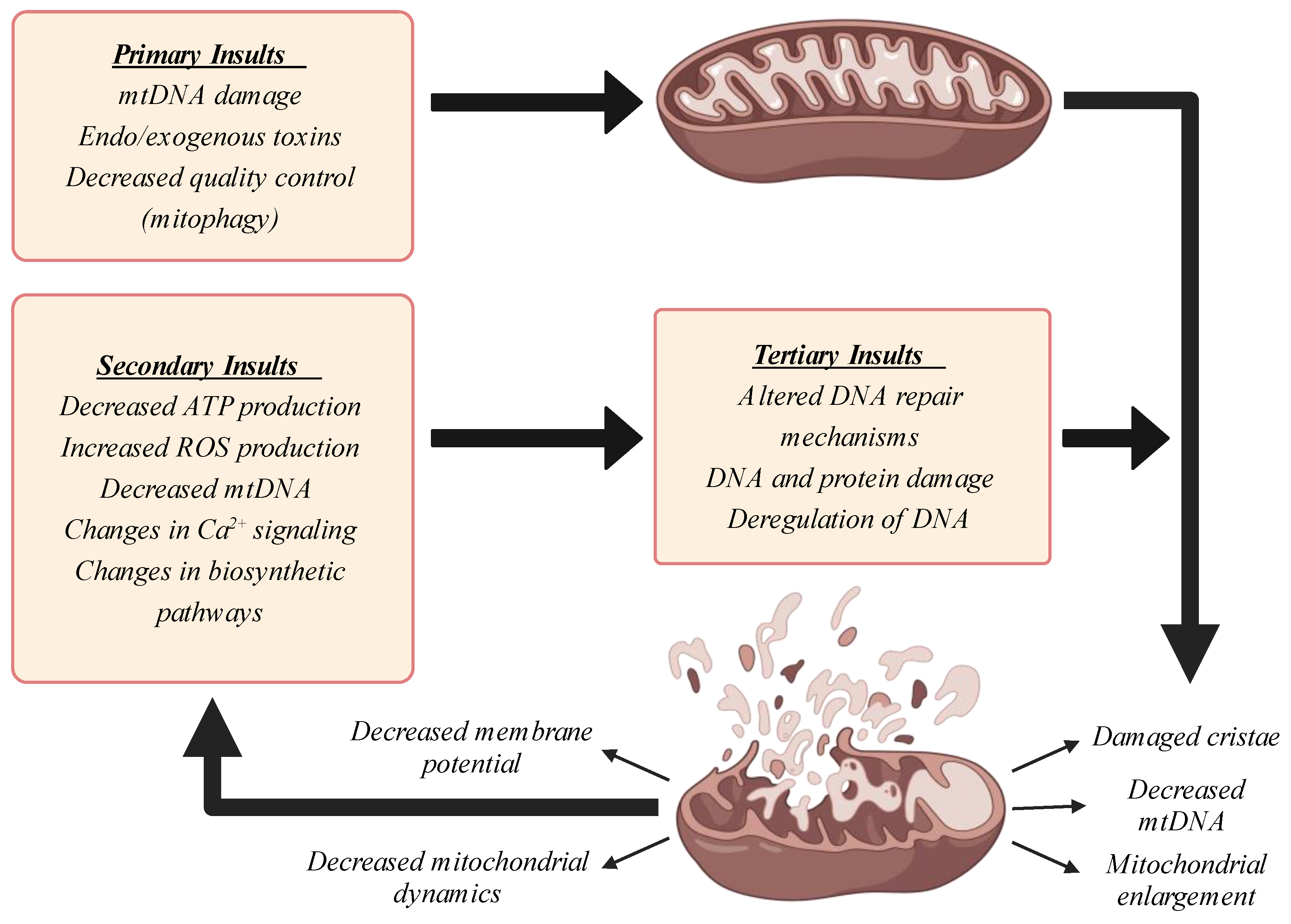
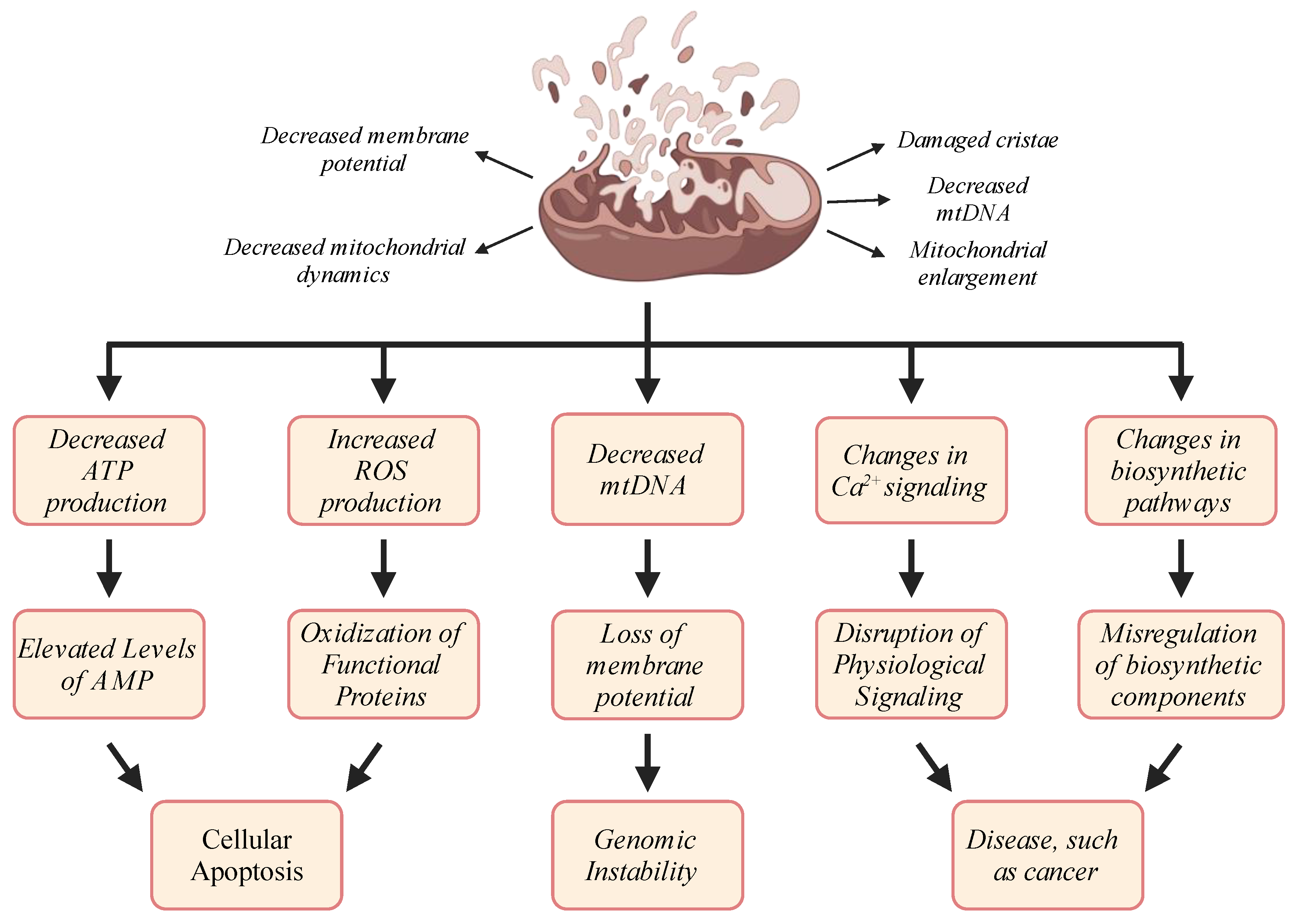
3. Mitochondrial Dysfunction in Aging: Causes
4. Mitochondrial Dysfunction and Inflammation
5. Mitochondrial Dysfunction and Brain Aging
6. Role of Mitochondrial Dysfunction in Brain Aging Disorders
7. Huntington’s Disease
8. Mitochondrial Dysfunction in Models of Huntington’s Disease
9. Parkinson’s Disease
10. Mitochondrial Dysfunction in Models of Parkinson’s Disease
11. Alzheimer’s Disease
12. Mitochondrial Dysfunction in Models of Alzheimer’s Disease
13. Current Treatments of Mitochondrial Dysfunction
14. PGC-1α Expression
15. Caloric Restriction and Fasting Diets
16. Physical Exercise
17. Prevention of Drug-Induced Mitochondrial Toxicity
18. Dietary Supplements
18.1. Antioxidants
18.2. Vitamin D
18.3. Coenzyme Q
18.4. Melatonin
18.5. Herbal Medicine
19. Pharmacologics
20. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| ADAF-1 | apoptotic protease activating factor-1 |
| AET | aerobic exercise training |
| ALP | autophagy-lysosomal pathway |
| ApoE | apolipoprotein E |
| APP | amyloid precursor protein |
| ATP | adenosine triphosphate (energy) |
| Aβ | amyloid-beta |
| AβPP | amyloid beta protein precursor |
| BBB | blood brain barrier |
| Ca2+ | calcium |
| CAT | catalase |
| cGAMP | cyclic dinucleotide 2′3′-Cyclic GMP-AMP |
| cGAS | cyclic GMP-AMP synthase |
| CLPP | Clp protease proteolytic subunit |
| CoQ | coenzyme Q/ubiquinone |
| CoQ10 | coenzyme Q10 |
| CR | caloric restriction |
| DAMP | damage-associated molecular pattern |
| DOPAC | 3,4-Dihydroxyphenylacetic acid |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| FPR | formyl-peptide receptor |
| GHS | glutathione |
| GPx | glutathione peroxidase |
| GSC | germline stem cell |
| H+ | protons |
| H2O | water |
| H2O2 | hydrogen peroxide |
| HD | Huntington’s Disease |
| HO• | hydroxyl radical |
| HTT | huntingtin |
| HVA | homovanillic acid |
| IFN-I | type I interferon |
| IMM | inner mitochondrial membrane |
| IRF-3 | interferon regulatory factor 3 |
| KDC | ketoglutarate dehydrogenase complex |
| KI | knock-in |
| KO | knock-out |
| KRS | Kufor–Rakeb syndrome |
| LONP | lon protease homologue |
| MAO | monoamine oxidase |
| MCU | mitochondrial calcium uniporter |
| mDAMP | mitochondrial damage-associated molecular pattern |
| MFRTA | mitochondrial free-radical theory of aging |
| MI | myocardial infarction |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| mtDNA | mitochondrial DNA |
| MTFMT | methionyl-tRNA formyltransferase |
| mtROS | mitochondrial reactive oxygen species |
| nDNA | nuclear DNA |
| NFT | intracellular neurofibrillary tangle |
| NLR | NOD-like receptor |
| O2•− | superoxide radical |
| OMM | outer mitochondrial membrane |
| oxCL | oxidized cardiolipin |
| OXPHOS | oxidative phosphorylation |
| P2 | purinergic |
| PAMP | pathogen-associated molecular pattern |
| PD | Parkinson’s Disease |
| PDC | pyruvate dehydrogenase complex |
| PGC-1α | Peroxisome proliferator-activated receptor-γ coactivator |
| PINK-1 | PTEN-induced kinase-1 |
| PolgA | mtDNA polymerase-γ |
| PRR | pattern recognition receptor |
| PS1/2 | presenilin 1 or 2 |
| pTAU | hyperphosphorylated tau |
| RA | rheumatoid arthritis |
| RET | resistance exercise training |
| ROS | reactive oxygen species |
| rRNA | ribosomal RNA |
| SNpc | substantia nigra pars compacta |
| SOD | superoxide dismutase |
| TCA | tricarboxylic acid cycle |
| TFAM | Transcription factor A mitochondrial |
| TLR | toll-like receptor |
| tRNA | transfer RNA |
| UPS | Ubiquitin Proteasome System |
| VDAC | voltage-dependent anion channel |
| VDR | vitamin D receptor |
| YAC | yeast artificial chromosome |
| α-Syn | alpha-synuclein |
| 4-HNE | 4-hydroxynonenal |
| 5-Lox | 5-lypoxygenase |
| 6-OHDA | 6-hydroxydopamine |
| 8-OHdG | 8-oxo-2′-deoxyguanosine |
References
- Brown, G.C. Living Too Long. EMBO Rep. 2015, 16, 137–141. [Google Scholar] [CrossRef]
- Holbrook, J.A.; Jarosz-Griffiths, H.H.; Caseley, E.; Lara-Reyna, S.; Poulter, J.A.; Williams-Gray, C.H.; Peckham, D.; McDermott, M.F. Neurodegenerative Disease and the NLRP3 Inflammasome. Front. Pharmacol. 2021, 12, 643254. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.-G. The Role of Mitochondria in Aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.L. Understanding the Odd Science of Aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, P.P. Human Ageing as a Dynamic, Emergent and Malleable Process: From Disease-Oriented to Health-Oriented Approaches. Biogerontology 2020, 21, 125–130. [Google Scholar] [CrossRef] [PubMed]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The Effect of Retarded Growth Upon the Length of Life Span and Upon the Ultimate Body Size: One Figure. J. Nutr. 1935, 10, 63–79. [Google Scholar] [CrossRef]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. Elegans Mutant That Lives Twice as Long as Wild Type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L. The New Biology of Ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Melzer, D.; Pilling, L.C.; Ferrucci, L. The Genetics of Human Ageing. Nat. Rev. Genet. 2020, 21, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.; Kirkwood, T.B.L. Chance, Development, and Aging; Oxford University Press: New York, NY, USA, 2000; ISBN 978-0-19-513361-5. [Google Scholar]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a “Good” Look at Free Radicals in the Aging Process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef]
- Jang, Y.C.; Remmen, H.V. The Mitochondrial Theory of Aging: Insight from Transgenic and Knockout Mouse Models. Exp. Gerontol. 2009, 44, 256–260. [Google Scholar] [CrossRef]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Harman, D. The Biologic Clock: The Mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Stuart, J.A.; Maddalena, L.A.; Merilovich, M.; Robb, E.L. A Midlife Crisis for the Mitochondrial Free Radical Theory of Aging. Longev. Heal. 2014, 3, 4. [Google Scholar] [CrossRef]
- Embley, T.M.; Martin, W. Eukaryotic Evolution, Changes and Challenges. Nature 2006, 440, 623–630. [Google Scholar] [CrossRef]
- Kurland, C.G.; Andersson, S.G.E. Origin and Evolution of the Mitochondrial Proteome. Microbiol. Mol. Biol. Rev. 2000, 64, 786–820. [Google Scholar] [CrossRef]
- Weinrich, T.W.; Kam, J.H.; Ferrara, B.T.; Thompson, E.P.; Mitrofanis, J.; Jeffery, G. A Day in the Life of Mitochondria Reveals Shifting Workloads. Sci. Rep. 2019, 9, 13898. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Allen, J.A.; Shankara, T.; Janus, P.; Buck, S.; Diemer, T.; Held Hales, K.; Hales, D.B. Energized, Polarized, and Actively Respiring Mitochondria Are Required for Acute Leydig Cell Steroidogenesis. Endocrinology 2006, 147, 3924–3935. [Google Scholar] [CrossRef]
- Antico Arciuch, V.G.; Elguero, M.E.; Poderoso, J.J.; Carreras, M.C. Mitochondrial Regulation of Cell Cycle and Proliferation. Antioxid. Redox Signal. 2012, 16, 1150–1180. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Chandel, N.S.; Williamson, E.K.; Schumacker, P.T.; Thompson, C.B. Bcl-xL Regulates the Membrane Potential and Volume Homeostasis of Mitochondria. Cell 1997, 91, 627–637. [Google Scholar] [CrossRef]
- Coppotelli, G.; Ross, J.M. Mitochondria in Ageing and Diseases: The Super Trouper of the Cell. Int. J. Mol. Sci. 2016, 17, 711. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial Dysfunction in Neurodegenerative Diseases and Drug Targets via Apoptotic Signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Ding, L.; Liu, Y. Borrowing Nuclear DNA Helicases to Protect Mitochondrial DNA. Int. J. Mol. Sci. 2015, 16, 10870–10887. [Google Scholar] [CrossRef]
- Gray, M.W. Mitochondrial Evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef]
- Bar-Yaacov, D.; Blumberg, A.; Mishmar, D. Mitochondrial-Nuclear Co-Evolution and Its Effects on OXPHOS Activity and Regulation. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2012, 1819, 1107–1111. [Google Scholar] [CrossRef]
- Lasserre, J.-P.; Dautant, A.; Aiyar, R.S.; Kucharczyk, R.; Glatigny, A.; Tribouillard-Tanvier, D.; Rytka, J.; Blondel, M.; Skoczen, N.; Reynier, P.; et al. Yeast as a System for Modeling Mitochondrial Disease Mechanisms and Discovering Therapies. Dis. Model. Mech. 2015, 8, 509–526. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Haass, C. Mitochondrial Dysfunction in Parkinson’s Disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 29–44. [Google Scholar] [CrossRef]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The Oxidative Phosphorylation System in Mammalian Mitochondria. Adv. Exp. Med. Biol. 2012, 942, 3–37. [Google Scholar] [CrossRef]
- Diaz, F. Cytochrome c Oxidase Deficiency: Patients and Animal Models. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial Calcium: Transport and Modulation of Cellular Processes in Homeostasis and Cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic Reticulum Ca2+ Handling in Excitable Cells in Health and Disease. Pharmacol. Rev. 2011, 63, 700–727. [Google Scholar] [CrossRef] [PubMed]
- Wescott, A.P.; Kao, J.P.Y.; Lederer, W.J.; Boyman, L. Voltage-Energized Calcium-Sensitive ATP Production by Mitochondria. Nat. Metab. 2019, 1, 975–984. [Google Scholar] [CrossRef]
- Rostovtseva, T.; Colombini, M. VDAC Channels Mediate and Gate the Flow of ATP: Implications for the Regulation of Mitochondrial Function. Biophys. J. 1997, 72, 1954–1962. [Google Scholar] [CrossRef]
- Duchen, M.R. Mitochondria and Calcium: From Cell Signalling to Cell Death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef]
- Barry, M.; Bleackley, R.C. Cytotoxic T Lymphocytes: All Roads Lead to Death. Nat. Rev. Immunol. 2002, 2, 401–409. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X. Cytochrome c Promotes Caspase-9 Activation by Inducing Nucleotide Binding to Apaf-1. J. Biol. Chem. 2000, 275, 31199–31203. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as Biosynthetic Factories for Cancer Proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef]
- Son, H.G.; Altintas, O.; Kim, E.J.E.; Kwon, S.; Lee, S.-J.V. Age-Dependent Changes and Biomarkers of Aging in Caenorhabditis Elegans. Aging Cell 2019, 18, e12853. [Google Scholar] [CrossRef]
- Cho, J.; Hur, J.H.; Walker, D.W. The Role of Mitochondria in Drosophila Aging. Exp. Gerontol. 2011, 46, 331–334. [Google Scholar] [CrossRef]
- García, M.L.; Fernández, A.; Solas, M.T. Mitochondria, Motor Neurons and Aging. J. Neurol. Sci. 2013, 330, 18–26. [Google Scholar] [CrossRef]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.-G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Fakouri, N.B.; Hansen, T.L.; Desler, C.; Anugula, S.; Rasmussen, L.J. From Powerhouse to Perpetrator—Mitochondria in Health and Disease. Biology 2019, 8, 35. [Google Scholar] [CrossRef]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxidative Med. Cell. Longev. 2019, 2019, e6175804. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, e360438. [Google Scholar] [CrossRef]
- Esterbauer, H.; Eckl, P.; Ortner, A. Possible Mutagens Derived from Lipids and Lipid Precursors. Mutat. Res./Rev. Genet. Toxicol. 1990, 238, 223–233. [Google Scholar] [CrossRef]
- Zhang, H.; Forman, H.J. 4-Hydroxynonenal-Mediated Signaling and Aging. Free Radic. Biol. Med. 2017, 111, 219–225. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Edgar, D.; Trifunovic, A. The mtDNA Mutator Mouse: Dissecting Mitochondrial Involvement in Aging. Aging 2009, 1, 1028–1032. [Google Scholar] [CrossRef]
- Lawless, C.; Greaves, L.; Reeve, A.K.; Turnbull, D.M.; Vincent, A.E. The Rise and Rise of Mitochondrial DNA Mutations. Open Biol. 2020, 10, 200061. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature Ageing in Mice Expressing Defective Mitochondrial DNA Polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Niu, X.; Trifunovic, A.; Larsson, N.-G.; Canlon, B. Somatic mtDNA Mutations Cause Progressive Hearing Loss in the Mouse. Exp. Cell Res. 2007, 313, 3924–3934. [Google Scholar] [CrossRef]
- Ross, J.M.; Stewart, J.B.; Hagström, E.; Brené, S.; Mourier, A.; Coppotelli, G.; Freyer, C.; Lagouge, M.; Hoffer, B.J.; Olson, L.; et al. Germline Mitochondrial DNA Mutations Aggravate Ageing and Can Impair Brain Development. Nature 2013, 501, 412–415. [Google Scholar] [CrossRef]
- Michikawa, Y.; Mazzucchelli, F.; Bresolin, N.; Scarlato, G.; Attardi, G. Aging-Dependent Large Accumulation of Point Mutations in the Human mtDNA Control Region for Replication. Science 1999, 286, 774–779. [Google Scholar] [CrossRef]
- Münscher, C.; Rieger, T.; Müller-Höcker, J.; Kadenbach, B. The Point Mutation of Mitochondrial DNA Characteristic for MERRF Disease Is Found Also in Healthy People of Different Ages. FEBS Lett. 1993, 317, 27–30. [Google Scholar] [CrossRef]
- Khaidakov, M.; Heflich, R.H.; Manjanatha, M.G.; Myers, M.B.; Aidoo, A. Accumulation of Point Mutations in Mitochondrial DNA of Aging Mice. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2003, 526, 1–7. [Google Scholar] [CrossRef]
- Stefano, G.B.; Bjenning, C.; Wang, F.; Wang, N.; Kream, R.M. Mitochondrial Heteroplasmy. In Mitochondrial Dynamics in Cardiovascular Medicine; Santulli, G., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; pp. 577–594. ISBN 978-3-319-55330-6. [Google Scholar]
- Wei Soong, N.; Hinton, D.R.; Cortopassi, G.; Arnheim, N. Mosaicism for a Specific Somatic Mitochondrial DNA Mutation in Adult Human Brain. Nat. Genet. 1992, 2, 318–323. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Arnheim, N. Detection of a Specific Mitochondrial DNA Deletion in Tissues of Older Humans. Nucleic Acids Res. 1990, 18, 6927–6933. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Flint Beal, M.; Wallace, D.C. Mitochondrial DNA Deletions in Human Brain: Regional Variability and Increase with Advanced Age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef]
- Ross, J.M.; Coppotelli, G.; Hoffer, B.J.; Olson, L. Maternally Transmitted Mitochondrial DNA Mutations Can Reduce Lifespan. Sci. Rep. 2014, 4, 6569. [Google Scholar] [CrossRef]
- López-Lluch, G.; Irusta, P.M.; Navas, P.; de Cabo, R. Mitochondrial Biogenesis and Healthy Aging. Exp. Gerontol. 2008, 43, 813–819. [Google Scholar] [CrossRef]
- Ghosh, S.; Lertwattanarak, R.; Lefort, N.; Molina-Carrion, M.; Joya-Galeana, J.; Bowen, B.P.; de Jesus Garduno-Garcia, J.; Abdul-Ghani, M.; Richardson, A.; DeFronzo, R.A.; et al. Reduction in Reactive Oxygen Species Production by Mitochondria from Elderly Subjects with Normal and Impaired Glucose Tolerance. Diabetes 2011, 60, 2051–2060. [Google Scholar] [CrossRef]
- Garcia, S.; Nissanka, N.; Mareco, E.A.; Rossi, S.; Peralta, S.; Diaz, F.; Rotundo, R.L.; Carvalho, R.F.; Moraes, C.T. Overexpression of PGC-1α in Aging Muscle Enhances a Subset of Young-like Molecular Patterns. Aging Cell 2018, 17, e12707. [Google Scholar] [CrossRef]
- Dillon, L.M.; Williams, S.L.; Hida, A.; Peacock, J.D.; Prolla, T.A.; Lincoln, J.; Moraes, C.T. Increased Mitochondrial Biogenesis in Muscle Improves Aging Phenotypes in the mtDNA Mutator Mouse. Hum. Mol. Genet. 2012, 21, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of Mitochondrial Fission and Fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, Pathophysiological Roles, and Analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D.C. Functions and Dysfunctions of Mitochondrial Dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Amartuvshin, O.; Lin, C.-H.; Hsu, S.-C.; Kao, S.-H.; Chen, A.; Tang, W.-C.; Chou, H.-L.; Chang, D.-L.; Hsu, Y.-Y.; Hsiao, B.-S.; et al. Aging Shifts Mitochondrial Dynamics toward Fission to Promote Germline Stem Cell Loss. Aging Cell 2020, 19, e13191. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389.e6. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 Deficiency Links Age-Related Sarcopenia and Impaired Autophagy to Activation of an Adaptive Mitophagy Pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Iqbal, S.; Ostojic, O.; Singh, K.; Joseph, A.-M.; Hood, D.A. Expression of Mitochondrial Fission and Fusion Regulatory Proteins in Skeletal Muscle during Chronic Use and Disuse. Muscle Nerve 2013, 48, 963–970. [Google Scholar] [CrossRef]
- Konopka, A.R.; Suer, M.K.; Wolff, C.A.; Harber, M.P. Markers of Human Skeletal Muscle Mitochondrial Biogenesis and Quality Control: Effects of Age and Aerobic Exercise Training. J. Gerontol. Ser. A 2014, 69, 371–378. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.-P.; Picard, M.; Pelletier, F.S.-J.; Sgarioto, N.; Auger, M.-J.; Vallée, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial Morphology Is Altered in Atrophied Skeletal Muscle of Aged Mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef]
- Faitg, J.; Leduc-Gaudet, J.-P.; Reynaud, O.; Ferland, G.; Gaudreau, P.; Gouspillou, G. Effects of Aging and Caloric Restriction on Fiber Type Composition, Mitochondrial Morphology and Dynamics in Rat Oxidative and Glycolytic Muscles. Front. Physiol. 2019, 10, 420. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The Pathways of Mitophagy for Quality Control and Clearance of Mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin Is Recruited Selectively to Impaired Mitochondria and Promotes Their Autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.-M.; et al. Mitochondrial Dysfunction in Drosophila PINK1 Mutants Is Complemented by Parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila Pink1 Is Required for Mitochondrial Function and Interacts Genetically with Parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Innokentev, A.; Kanki, T. Regulatory Mechanisms of Mitochondrial Autophagy: Lessons from Yeast. Front. Plant Sci. 2019, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.M.; Olson, L.; Coppotelli, G. Mitochondrial Dysfunction and Protein Homeostasis in Aging: Insights from a Premature-Aging Mouse Model. Biomolecules 2024, 14, 162. [Google Scholar] [CrossRef] [PubMed]
- Markaki, M.; Metaxakis, A.; Tavernarakis, N. Chapter 2—The Role of Autophagy in Aging: Molecular Mechanisms. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: New York, NY, USA, 2017; pp. 123–138. ISBN 978-0-12-812146-7. [Google Scholar]
- Pyo, J.-O.; Yoo, S.-M.; Ahn, H.-H.; Nah, J.; Hong, S.-H.; Kam, T.-I.; Jung, S.; Jung, Y.-K. Overexpression of Atg5 in Mice Activates Autophagy and Extends Lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [PubMed]
- Deshwal, S.; Fiedler, K.U.; Langer, T. Mitochondrial Proteases: Multifaceted Regulators of Mitochondrial Plasticity. Annu. Rev. Biochem. 2020, 89, 501–528. [Google Scholar] [CrossRef]
- Quirós, P.M.; Langer, T.; López-Otín, C. New Roles for Mitochondrial Proteases in Health, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef]
- Ross, J.M.; Olson, L.; Coppotelli, G. Mitochondrial and Ubiquitin Proteasome System Dysfunction in Ageing and Disease: Two Sides of the Same Coin? Int. J. Mol. Sci. 2015, 16, 19458–19476. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.W.; et al. Mitochondrial Dynamics Controls T Cell Fate Through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial Control of Inflammation. Nat. Rev. Immunol. 2023, 23, 159–173. [Google Scholar] [CrossRef]
- de Oliveira, L.G.; Angelo, Y.d.S.; Iglesias, A.H.; Peron, J.P.S. Unraveling the Link Between Mitochondrial Dynamics and Neuroinflammation. Front. Immunol. 2021, 12, 624919. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile Inflammation: Sensing and Reacting to Damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging. An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Matzinger, P. The Danger Model: A Renewed Sense of Self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Quintana, F.J.; Cohen, I.R. Heat Shock Proteins as Endogenous Adjuvants in Sterile and Septic Inflammation. J. Immunol. 2005, 175, 2777–2782. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of Chromatin Protein HMGB1 by Necrotic Cells Triggers Inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Banoth, B.; Cassel, S.L. Mitochondria in Innate Immune Signaling. Transl. Res. 2018, 202, 52–68. [Google Scholar] [CrossRef]
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.-M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 2019, 29, 1261–1273.e6. [Google Scholar] [CrossRef]
- Madsen, P.M.; Pinto, M.; Patel, S.; McCarthy, S.; Gao, H.; Taherian, M.; Karmally, S.; Pereira, C.V.; Dvoriantchikova, G.; Ivanov, D.; et al. Mitochondrial DNA Double-Strand Breaks in Oligodendrocytes Cause Demyelination, Axonal Injury, and CNS Inflammation. J. Neurosci. 2017, 37, 10185–10199. [Google Scholar] [CrossRef]
- Sasai, M.; Linehan, M.M.; Iwasaki, A. Bifurcation of Toll-Like Receptor 9 Signaling by Adaptor Protein 3. Science 2010, 329, 1530–1534. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome During Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Cai, X.; Chiu, Y.-H.; Chen, Z.J. The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Lei, Y.; Guerra Martinez, C.; Torres-Odio, S.; Bell, S.L.; Birdwell, C.E.; Bryant, J.D.; Tong, C.W.; Watson, R.O.; West, L.C.; West, A.P. Elevated Type I Interferon Responses Potentiate Metabolic Dysfunction, Inflammation, and Accelerated Aging in mtDNA Mutator Mice. Sci. Adv. 2021, 7, eabe7548. [Google Scholar] [CrossRef]
- Crouser, E.D.; Shao, G.; Julian, M.W.; Macre, J.E.; Shadel, G.S.; Tridandapani, S.; Huang, Q.; Wewers, M.D. Monocyte Activation by Necrotic Cells Is Promoted by Mitochondrial Proteins and Formyl Peptide Receptors. Crit. Care Med. 2009, 37, 2000–2009. [Google Scholar] [CrossRef]
- Chaung, W.W.; Wu, R.; Ji, Y.; Dong, W.; Wang, P. Mitochondrial Transcription Factor A Is a Proinflammatory Mediator in Hemorrhagic Shock. Int. J. Mol. Med. 2012, 30, 199–203. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial Metabolism of Reactive Oxygen Species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Wu, J.; Yan, Z.; Schwartz, D.E.; Yu, J.; Malik, A.B.; Hu, G. Activation of NLRP3 Inflammasome in Alveolar Macrophages Contributes to Mechanical Stretch-Induced Lung Inflammationand Injury. J. Immunol. 2013, 190, 3590–3599. [Google Scholar] [CrossRef]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides Released by Apoptotic Cells Act as a Find-Me Signal for Phagocytic Clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef]
- Gombault, A.; Baron, L.; Couillin, I. ATP Release and Purinergic Signaling in NLRP3 Inflammasome Activation. Front. Immunol. 2013, 3, 414. [Google Scholar] [CrossRef]
- Grimolizzi, F.; Arranz, L. Multiple Faces of Succinate beyond Metabolism in Blood. Haematologica 2018, 103, 1586–1592. [Google Scholar] [CrossRef]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwärzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the Succinate Receptor GPR91 on Dendritic Cells Enhances Immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef]
- Tannahill, G.; Curtis, A.; Adamik, J.; Palsson-McDermott, E.; McGettrick, A.; Goel, G.; Frezza, C.; Bernard, N.; Kelly, B.; Foley, N.; et al. Succinate Is a Danger Signal That Induces IL-1β via HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Pizzuto, M.; Pelegrin, P. Cardiolipin in Immune Signaling and Cell Death. Trends Cell Biol. 2020, 30, 892–903. [Google Scholar] [CrossRef]
- Claypool, S.M.; Koehler, C.M. The Complexity of Cardiolipin in Health and Disease. Trends Biochem. Sci. 2012, 37, 32–41. [Google Scholar] [CrossRef]
- Wan, M.; Hua, X.; Su, J.; Thiagarajan, D.; Frostegård, A.G.; Haeggström, J.Z.; Frostegård, J. Oxidized but Not Native Cardiolipin Has Pro-Inflammatory Effects, Which Are Inhibited by Annexin A5. Atherosclerosis 2014, 235, 592–598. [Google Scholar] [CrossRef]
- Ayyub, S.A.; Varshney, U. Translation Initiation in Mammalian Mitochondria- a Prokaryotic Perspective. RNA Biol. 2020, 17, 165–175. [Google Scholar] [CrossRef]
- Chen, K.; Bao, Z.; Gong, W.; Tang, P.; Yoshimura, T.; Wang, J.M. Regulation of Inflammation by Members of the Formyl-Peptide Receptor Family. J. Autoimmun. 2017, 85, 64–77. [Google Scholar] [CrossRef]
- Raoof, M.; Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial Peptides Are Potent Immune Activators That Activate Human Neutrophils via FPR-1. J. Trauma. 2010, 68, 1328–1332; discussion 1332–1334. [Google Scholar] [CrossRef]
- Wenceslau, C.F.; McCarthy, C.G.; Szasz, T.; Goulopoulou, S.; Webb, R.C. Mitochondrial N-Formyl Peptides Induce Cardiovascular Collapse and Sepsis-like Syndrome. Am. J. Physiol.-Heart Circ. Physiol. 2015, 308, H768–H777. [Google Scholar] [CrossRef]
- Wenceslau, C.F.; Szasz, T.; McCarthy, C.G.; Baban, B.; NeSmith, E.; Webb, R.C. Mitochondrial N-Formyl Peptides Cause Airway Contraction and Lung Neutrophil Infiltration via Formyl Peptide Receptor Activation. Pulm. Pharmacol. Ther. 2016, 37, 49–56. [Google Scholar] [CrossRef]
- Pullerits, R.; Bokarewa, M.; Jonsson, I.-M.; Verdrengh, M.; Tarkowski, A. Extracellular Cytochrome c, a Mitochondrial Apoptosis-Related Protein, Induces Arthritis. Rheumatology 2005, 44, 32–39. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, J.; Ran, Z. Emerging Views of Mitophagy in Immunity and Autoimmune Diseases. Autophagy 2019, 16, 3–17. [Google Scholar] [CrossRef]
- Montava-Garriga, L.; Ganley, I.G. Outstanding Questions in Mitophagy: What We Do and Do Not Know. J. Mol. Biol. 2020, 432, 206–230. [Google Scholar] [CrossRef]
- Peters, R. Ageing and the Brain. Postgrad. Med. J. 2006, 82, 84–88. [Google Scholar] [CrossRef]
- Sams, E.C. Oligodendrocytes in the Aging Brain. Neuronal Signal. 2021, 5, NS20210008. [Google Scholar] [CrossRef]
- Chiareli, R.A.; Carvalho, G.A.; Marques, B.L.; Mota, L.S.; Oliveira-Lima, O.C.; Gomes, R.M.; Birbrair, A.; Gomez, R.S.; Simão, F.; Klempin, F.; et al. The Role of Astrocytes in the Neurorepair Process. Front. Cell Dev. Biol. 2021, 9, 665795. [Google Scholar] [CrossRef]
- Dheen, S.T.; Kaur, C.; Ling, E.-A. Microglial Activation and Its Implications in the Brain Diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak-Zmijewska, A.; Dudkowska, M.; Krzystyniak, A.; Mosieniak, G.; Wesierska, M.; Wlodarczyk, J. Cellular Senescence in Brain Aging. Front. Aging Neurosci. 2021, 13, 646924. [Google Scholar] [CrossRef]
- Stan, A.D.; Ghose, S.; Gao, X.-M.; Roberts, R.C.; Lewis-Amezcua, K.; Hatanpaa, K.J.; Tamminga, C.A. Human Postmortem Tissue: What Quality Markers Matter? Brain Res. 2006, 1123, 1–11. [Google Scholar] [CrossRef]
- Holtze, S.; Gorshkova, E.; Braude, S.; Cellerino, A.; Dammann, P.; Hildebrandt, T.B.; Hoeflich, A.; Hoffmann, S.; Koch, P.; Terzibasi Tozzini, E.; et al. Alternative Animal Models of Aging Research. Front. Mol. Biosci. 2021, 8, 660959. [Google Scholar] [CrossRef]
- Azevedo, F.A.C.; Carvalho, L.R.B.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.L.; Leite, R.E.P.; Filho, W.J.; Lent, R.; Herculano-Houzel, S. Equal Numbers of Neuronal and Nonneuronal Cells Make the Human Brain an Isometrically Scaled-up Primate Brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic Reprogramming during Neuronal Differentiation from Aerobic Glycolysis to Neuronal Oxidative Phosphorylation. eLife 2016, 5, e13374. [Google Scholar] [CrossRef]
- Yuan, Y.; Huo, H.; Fang, T. Effects of Metabolic Energy on Synaptic Transmission and Dendritic Integration in Pyramidal Neurons. Front. Comput. Neurosci. 2018, 12, 79. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial Bioenergetic Deficit Precedes Alzheimer’s Pathology in Female Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Weber, D.; Stuetz, W.; Toussaint, O.; Debacq-Chainiaux, F.; Dollé, M.E.T.; Jansen, E.; Gonos, E.S.; Franceschi, C.; Sikora, E.; Hervonen, A.; et al. Associations between Specific Redox Biomarkers and Age in a Large European Cohort: The MARK-AGE Project. Oxidative Med. Cell. Longev. 2017, 2017, e1401452. [Google Scholar] [CrossRef]
- Guo, C.; Li, X.; Wang, R.; Yu, J.; Ye, M.; Mao, L.; Zhang, S.; Zheng, S. Association between Oxidative DNA Damage and Risk of Colorectal Cancer: Sensitive Determination of Urinary 8-Hydroxy-2′-Deoxyguanosine by UPLC-MS/MS Analysis. Sci. Rep. 2016, 6, 32581. [Google Scholar] [CrossRef]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA Damage by Lipid Peroxidation Products: Implications in Cancer, Inflammation and Autoimmunity. AIMS Genet. 2017, 4, 103–137. [Google Scholar] [CrossRef]
- Dubey, A.; Forster, M.J.; Lal, H.; Sohal, R.S. Effect of Age and Caloric Intake on Protein Oxidation in Different Brain Regions and on Behavioral Functions of the Mouse. Arch. Biochem. Biophys. 1996, 333, 189–197. [Google Scholar] [CrossRef]
- Ross, J.M.; Coppotelli, G.; Branca, R.M.; Kim, K.M.; Lehtiö, J.; Sinclair, D.A.; Olson, L. Voluntary Exercise Normalizes the Proteomic Landscape in Muscle and Brain and Improves the Phenotype of Progeroid Mice. Aging Cell 2019, 18, e13029. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Florenzano, F.; Atlante, A.; Bobba, A.; Nicolin, V.; Nori, S.L.; Calissano, P. Morphological and Bioenergetic Demands Underlying the Mitophagy in Post-Mitotic Neurons: The Pink–Parkin Pathway. Front. Aging Neurosci. 2014, 6, 18. [Google Scholar] [CrossRef]
- Perluigi, M.; Domenico, F.D.; Giorgi, A.; Schininà, M.E.; Coccia, R.; Cini, C.; Bellia, F.; Cambria, M.T.; Cornelius, C.; Butterfield, D.A.; et al. Redox Proteomics in Aging Rat Brain: Involvement of Mitochondrial Reduced Glutathione Status and Mitochondrial Protein Oxidation in the Aging Process. J. Neurosci. Res. 2010, 88, 3498–3507. [Google Scholar] [CrossRef]
- Navarro, A.; Boveris, A. Rat Brain and Liver Mitochondria Develop Oxidative Stress and Lose Enzymatic Activities on Aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1244–R1249. [Google Scholar] [CrossRef]
- Kriebel, M.; Ebel, J.; Battke, F.; Griesbach, S.; Volkmer, H. Interference with Complex IV as a Model of Age-Related Decline in Synaptic Connectivity. Front. Mol. Neurosci. 2020, 13, 43. [Google Scholar] [CrossRef]
- Manczak, M.; Jung, Y.; Park, B.S.; Partovi, D.; Reddy, P.H. Time-Course of Mitochondrial Gene Expressions in Mice Brains: Implications for Mitochondrial Dysfunction, Oxidative Damage, and Cytochrome c in Aging. J. Neurochem. 2005, 92, 494–504. [Google Scholar] [CrossRef]
- Sterky, F.H.; Hoffman, A.F.; Milenkovic, D.; Bao, B.; Paganelli, A.; Edgar, D.; Wibom, R.; Lupica, C.R.; Olson, L.; Larsson, N.-G. Altered Dopamine Metabolism and Increased Vulnerability to MPTP in Mice with Partial Deficiency of Mitochondrial Complex I in Dopamine Neurons. Hum. Mol. Genet. 2012, 21, 1078–1089. [Google Scholar] [CrossRef]
- Andriollo-Sanchez, M.; Hininger-Favier, I.; Meunier, N.; Venneria, E.; O’Connor, J.M.; Maiani, G.; Coudray, C.; Roussel, A.M. Age-Related Oxidative Stress and Antioxidant Parameters in Middle-Aged and Older European Subjects: The ZENITH Study. Eur. J. Clin. Nutr. 2005, 59, S58–S62. [Google Scholar] [CrossRef]
- Rebrin, I.; Forster, M.J.; Sohal, R.S. Effects of Age and Caloric Intake on Glutathione Redox State in Different Brain Regions of C57BL/6 and DBA/2 Mice. Brain Res. 2007, 1127, 10–18. [Google Scholar] [CrossRef]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative Stress Induces Degradation of Mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef]
- Wei, Y.-H. Mitochondrial DNA Alterations as Ageing-Associated Molecular Events. Mutat. Res./DNAging 1992, 275, 145–155. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Adler, D.; DeMaria, M.; Mechtouf, N.; Teachenor, R.; Cardin, G.B.; Desprez, P.-Y.; Campisi, J.; Rodier, F. Mitochondrial DNA Damage Induces Apoptosis in Senescent Cells. Cell Death Dis. 2013, 4, e727. [Google Scholar] [CrossRef]
- Barja, G.; Herrero, A. Oxidative Damage to Mitochondrial DNA Is Inversely Related to Maximum Life Span in the Heart and Brain of Mammals. FASEB J. 2000, 14, 312–318. [Google Scholar] [CrossRef]
- Simon, D.K.; Lin, M.T.; Zheng, L.; Liu, G.-J.; Ahn, C.H.; Kim, L.M.; Mauck, W.M.; Twu, F.; Beal, M.F.; Johns, D.R. Somatic Mitochondrial DNA Mutations in Cortex and Substantia Nigra in Aging and Parkinson’s Disease. Neurobiol. Aging 2004, 25, 71–81. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef]
- Stauch, K.L.; Purnell, P.R.; Fox, H.S. Aging Synaptic Mitochondria Exhibit Dynamic Proteomic Changes While Maintaining Bioenergetic Function. Aging 2014, 6, 320–334. [Google Scholar] [CrossRef]
- Oettinghaus, B.; Schulz, J.M.; Restelli, L.M.; Licci, M.; Savoia, C.; Schmidt, A.; Schmitt, K.; Grimm, A.; Morè, L.; Hench, J.; et al. Synaptic Dysfunction, Memory Deficits and Hippocampal Atrophy Due to Ablation of Mitochondrial Fission in Adult Forebrain Neurons. Cell Death Differ. 2016, 23, 18–28. [Google Scholar] [CrossRef]
- Brown, M.R.; Geddes, J.W.; Sullivan, P.G. Brain Region-Specific, Age-Related, Alterations in Mitochondrial Responses to Elevated Calcium. J. Bioenerg. Biomembr. 2004, 36, 401–406. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative Diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a007476. [Google Scholar] [CrossRef] [PubMed]
- Vonsattel, J.P.; DiFiglia, M. Huntington Disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef]
- Reddy, P.H.; Mao, P.; Manczak, M. Mitochondrial Structural and Functional Dynamics in Huntington’s Disease. Brain Res. Rev. 2009, 61, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Beal, M.F.; Ferrante, R.J. Mitochondrial Loss, Dysfunction and Altered Dynamics in Huntington’s Disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [PubMed]
- Flønes, I.H.; Fernandez-Vizarra, E.; Lykouri, M.; Brakedal, B.; Skeie, G.O.; Miletic, H.; Lilleng, P.K.; Alves, G.; Tysnes, O.-B.; Haugarvoll, K.; et al. Neuronal Complex I Deficiency Occurs throughout the Parkinson’s Disease Brain, but Is Not Associated with Neurodegeneration or Mitochondrial DNA Damage. Acta Neuropathol. 2018, 135, 409–425. [Google Scholar] [CrossRef]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s Disease Mutations in PINK1 Result in Decreased Complex I Activity and Deficient Synaptic Function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Strobbe, D.; Robinson, A.A.; Harvey, K.; Rossi, L.; Ferraina, C.; de Biase, V.; Rodolfo, C.; Harvey, R.J.; Campanella, M. Distinct Mechanisms of Pathogenic DJ-1 Mutations in Mitochondrial Quality Control. Front. Mol. Neurosci. 2018, 11, 68. [Google Scholar] [CrossRef]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s Disease: Occurrence, Determinants, and Strategies toward Intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, K.E.; Duchen, M. Modelling Mitochondrial Dysfunction in Alzheimer’s Disease Using Human Induced Pluripotent Stem Cells. World J. Stem Cells 2019, 11, 236–253. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid Beta, Mitochondrial Dysfunction and Synaptic Damage: Implications for Cognitive Decline in Aging and Alzheimer’s Disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: Progress and Perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Dennis, J.; Khan, S.M. Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis. 2016, 7, 201–214. [Google Scholar] [CrossRef]
- Warby, S.C.; Montpetit, A.; Hayden, A.R.; Carroll, J.B.; Butland, S.L.; Visscher, H.; Collins, J.A.; Semaka, A.; Hudson, T.J.; Hayden, M.R. CAG Expansion in the Huntington Disease Gene Is Associated with a Specific and Targetable Predisposing Haplogroup. Am. J. Hum. Genet. 2009, 84, 351–366. [Google Scholar] [CrossRef]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 52–61. [Google Scholar] [CrossRef]
- Nopoulos, P.C. Huntington Disease: A Single-Gene Degenerative Disorder of the Striatum. Dialogues Clin. Neurosci. 2016, 18, 91–98. [Google Scholar] [CrossRef]
- Bossy-Wetzel, E.; Petrilli, A.; Knott, A.B. Mutant Huntingtin and Mitochondrial Dysfunction. Trends Neurosci. 2008, 31, 609–616. [Google Scholar] [CrossRef]
- Tellez-Nagel, I.; Johnson, A.B.; Terry, R.D. Studies on Brain Biopsies of Patients with Huntington’s Chorea. J. Neuropathol. Exp. Neurol. 1974, 33, 308–332. [Google Scholar] [CrossRef]
- Oliveira, J.M.A.; Jekabsons, M.B.; Chen, S.; Lin, A.; Rego, A.C.; Gonçalves, J.; Ellerby, L.M.; Nicholls, D.G. Mitochondrial Dysfunction in Huntington’s Disease: The Bioenergetics of Isolated and in Situ Mitochondria from Transgenic Mice. J. Neurochem. 2007, 101, 241–249. [Google Scholar] [CrossRef]
- Li, J.Y.; Popovic, N.; Brundin, P. The Use of the R6 Transgenic Mouse Models of Huntington’s Disease in Attempts to Develop Novel Therapeutic Strategies. NeuroRx 2005, 2, 447–464. [Google Scholar] [CrossRef]
- Levine, M.S.; Cepeda, C.; Hickey, M.A.; Fleming, S.M.; Chesselet, M.-F. Genetic Mouse Models of Huntington’s and Parkinson’s Diseases: Illuminating but Imperfect. Trends Neurosci. 2004, 27, 691–697. [Google Scholar] [CrossRef]
- Rattray, I.; Smith, E.J.; Crum, W.R.; Walker, T.A.; Gale, R.; Bates, G.P.; Modo, M. Correlations of Behavioral Deficits with Brain Pathology Assessed through Longitudinal MRI and Histopathology in the R6/1 Mouse Model of Huntington’s Disease. PLoS ONE 2013, 8, e84726. [Google Scholar] [CrossRef]
- Breuer, M.E.; Willems, P.H.G.M.; Russel, F.G.M.; Koopman, W.J.H.; Smeitink, J.A.M. Modeling Mitochondrial Dysfunctions in the Brain: From Mice to Men. J. Inherit. Metab. Dis. 2012, 35, 193–210. [Google Scholar] [CrossRef]
- Burtscher, J.; Di Pardo, A.; Maglione, V.; Schwarzer, C.; Squitieri, F. Mitochondrial Respiration Changes in R6/2 Huntington’s Disease Model Mice during Aging in a Brain Region Specific Manner. Int. J. Mol. Sci. 2020, 21, 5412. [Google Scholar] [CrossRef]
- Aidt, F.H.; Nielsen, S.M.B.; Kanters, J.; Pesta, D.; Nielsen, T.T.; Nørremølle, A.; Hasholt, L.; Christiansen, M.; Hagen, C.M. Dysfunctional Mitochondrial Respiration in the Striatum of the Huntington’s Disease Transgenic R6/2 Mouse Model. PLoS Curr. 2013, 5, ecurrents.hd.d8917b4862929772c5a2f2a34ef1c201. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.G.; Agopyan, N.; Gutekunst, C.-A.; Leavitt, B.R.; LePiane, F.; Singaraja, R.; Smith, D.J.; Bissada, N.; McCutcheon, K.; Nasir, J.; et al. A YAC Mouse Model for Huntington’s Disease with Full-Length Mutant Huntingtin, Cytoplasmic Toxicity, and Selective Striatal Neurodegeneration. Neuron 1999, 23, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Slow, E.J.; van Raamsdonk, J.; Rogers, D.; Coleman, S.H.; Graham, R.K.; Deng, Y.; Oh, R.; Bissada, N.; Hossain, S.M.; Yang, Y.-Z.; et al. Selective Striatal Neuronal Loss in a YAC128 Mouse Model of Huntington Disease. Hum. Mol. Genet. 2003, 12, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Warby, S.C.; Hayden, M.R. Selective Degeneration in YAC Mouse Models of Huntington Disease. Brain Res. Bull. 2007, 72, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Bečanović, K.; Asghar, M.; Gadawska, I.; Sachdeva, S.; Walker, D.; Lazarowski, E.R.; Franciosi, S.; Park, K.H.J.; Côté, H.C.F.; Leavitt, B.R. Age-Related Mitochondrial Alterations in Brain and Skeletal Muscle of the YAC128 Model of Huntington Disease. NPJ Aging Mech. Dis. 2021, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.M.A.; Chen, S.; Almeida, S.; Riley, R.; Gonçalves, J.; Oliveira, C.R.; Hayden, M.R.; Nicholls, D.G.; Ellerby, L.M.; Rego, A.C. Mitochondrial-Dependent Ca2+ Handling in Huntington’s Disease Striatal Cells: Effect of Histone Deacetylase Inhibitors. J. Neurosci. 2006, 26, 11174–11186. [Google Scholar] [CrossRef]
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s Disease. Subcell. Biochem. 2012, 65, 389–455. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Starkov, A.A.; Beal, M.F.; Thomas, B. Mitochondrial Dysfunction in the Limelight of Parkinson’s Disease Pathogenesis. Biochim. Biophys. Acta 2009, 1792, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in Humans Due to a Product of Meperidine-Analog Synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease—Cause or Consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef]
- Grünewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain–Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much More than Mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef]
- Darios, F.; Corti, O.; Lücking, C.B.; Hampe, C.; Muriel, M.-P.; Abbas, N.; Gu, W.-J.; Hirsch, E.C.; Rooney, T.; Ruberg, M.; et al. Parkin Prevents Mitochondrial Swelling and Cytochrome c Release in Mitochondria-Dependent Cell Death. Hum. Mol. Genet. 2003, 12, 517–526. [Google Scholar] [CrossRef]
- Narendra, D.P.; Youle, R.J. Targeting Mitochondrial Dysfunction: Role for PINK1 and Parkin in Mitochondrial Quality Control. Antioxid. Redox Signal. 2011, 14, 1929–1938. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 Stabilized by Mitochondrial Depolarization Recruits Parkin to Damaged Mitochondria and Activates Latent Parkin for Mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Aleyasin, H.; Rousseaux, M.W.C.; Marcogliese, P.C.; Hewitt, S.J.; Irrcher, I.; Joselin, A.P.; Parsanejad, M.; Kim, R.H.; Rizzu, P.; Callaghan, S.M.; et al. DJ-1 Protects the Nigrostriatal Axis from the Neurotoxin MPTP by Modulation of the AKT Pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 3186–3191. [Google Scholar] [CrossRef]
- Park, J.-S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s Disease-Associated Human ATP13A2 (PARK9) Deficiency Causes Zinc Dyshomeostasis and Mitochondrial Dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815. [Google Scholar] [CrossRef]
- Mullin, S.; Schapira, A. α-Synuclein and Mitochondrial Dysfunction in Parkinson’s Disease. Mol. Neurobiol. 2013, 47, 587–597. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 Regulates Mitochondrial Dynamics and Function through Direct Interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [PubMed]
- Davidzon, G.; Greene, P.; Mancuso, M.; Klos, K.J.; Ahlskog, J.E.; Hirano, M.; DiMauro, S. Early-Onset Familial Parkinsonism Due to POLG Mutations. Ann. Neurol. 2006, 59, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.; Wang, C.; Tsai, C.; Yeh, Y.; Lee, Y.S.; Wu, Y. POLG R964C and GBA L444P Mutations in Familial Parkinson’s Disease: Case Report and Literature Review. Brain Behav. 2019, 9, e01281. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.; Karuppagounder, S.S.; Neifert, S.; Kang, B.G.; Wang, H.; Dawson, V.L.; Dawson, T.M. The Absence of Parkin Does Not Promote Dopamine or Mitochondrial Dysfunction in PolgAD257A/D257A Mitochondrial Mutator Mice. J. Neurosci. 2022, 42, 9263–9277. [Google Scholar] [CrossRef] [PubMed]
- Hauser, D.N.; Primiani, C.T.; Langston, R.G.; Kumaran, R.; Cookson, M.R. The Polg Mutator Phenotype Does Not Cause Dopaminergic Neurodegeneration in DJ-1-Deficient Mice. eNeuro 2015, 2, ENEURO.0075-14.2015. [Google Scholar] [CrossRef]
- Zeng, X.-S.; Geng, W.-S.; Jia, J.-J. Neurotoxin-Induced Animal Models of Parkinson Disease: Pathogenic Mechanism and Assessment. ASN Neuro 2018, 10, 1759091418777438. [Google Scholar] [CrossRef]
- Macaya, A.; Munell, F.; Gubits, R.M.; Burke, R.E. Apoptosis in Substantia Nigra Following Developmental Striatal Excitotoxic Injury. Proc. Natl. Acad. Sci. USA 1994, 91, 8117–8121. [Google Scholar] [CrossRef]
- Wang, Z.; Chan, S.-W.; Zhao, H.; Miu, K.-K.; Chan, W.-Y. Outlook of PINK1/Parkin Signaling in Molecular Etiology of Parkinson’s Disease, with Insights into Pink1 Knockout Models. Zool. Res. 2023, 44, 559–576. [Google Scholar] [CrossRef]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic Animal Models of Parkinson’s Disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef]
- Andres-Mateos, E.; Perier, C.; Zhang, L.; Blanchard-Fillion, B.; Greco, T.M.; Thomas, B.; Ko, H.S.; Sasaki, M.; Ischiropoulos, H.; Przedborski, S.; et al. DJ-1 Gene Deletion Reveals That DJ-1 Is an Atypical Peroxiredoxin-like Peroxidase. Proc. Natl. Acad. Sci. USA 2007, 104, 14807–14812. [Google Scholar] [CrossRef]
- Beal, M.F. Parkinson’s Disease: A Model Dilemma. Nature 2010, 466, S8–S10. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive Parkinsonism in Mice with Respiratory-Chain-Deficient Dopamine Neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef]
- Branch, S.Y.; Chen, C.; Sharma, R.; Lechleiter, J.D.; Li, S.; Beckstead, M.J. Dopaminergic Neurons Exhibit an Age-Dependent Decline in Electrophysiological Parameters in the MitoPark Mouse Model of Parkinson’s Disease. J. Neurosci. 2016, 36, 4026–4037. [Google Scholar] [CrossRef]
- Galter, D.; Pernold, K.; Yoshitake, T.; Lindqvist, E.; Hoffer, B.; Kehr, J.; Larsson, N.-G.; Olson, L. MitoPark Mice Mirror the Slow Progression of Key Symptoms and L-DOPA Response in Parkinson’s Disease. Genes Brain Behav. 2010, 9, 173–181. [Google Scholar] [CrossRef]
- Castellani, R.; Hirai, K.; Aliev, G.; Drew, K.L.; Nunomura, A.; Takeda, A.; Cash, A.D.; Obrenovich, M.E.; Perry, G.; Smith, M.A. Role of Mitochondrial Dysfunction in Alzheimer’s Disease. J. Neurosci. Res. 2002, 70, 357–360. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.; Casadesus, G.; Perry, G.; Zhu, X. Impaired Mitochondrial Biogenesis Contributes to Mitochondrial Dysfunction in Alzheimer’s Disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.; Zhu, X. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Weidling, I.; Swerdlow, R.H. Mitochondrial Dysfunction and Stress Responses in Alzheimer’s Disease. Biology 2019, 8, 39. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Kipiani, K.; Yu, F.; Wille, E.; Katz, M.; Calingasan, N.Y.; Gouras, G.K.; Lin, M.T.; Beal, M.F. Coenzyme Q10 Decreases Amyloid Pathology and Improves Behavior in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-β Overproduction Causes Abnormal Mitochondrial Dynamics via Differential Modulation of Mitochondrial Fission/Fusion Proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-Amyloid Peptides Induce Mitochondrial Dysfunction and Oxidative Stress in Astrocytes and Death of Neurons through Activation of NADPH Oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial Medicine for Aging and Neurodegenerative Diseases. Neuromol. Med. 2008, 10, 291–315. [Google Scholar] [CrossRef]
- Duncan, T.; Valenzuela, M. Alzheimer’s Disease, Dementia, and Stem Cell Therapy. Stem Cell Res. Ther. 2017, 8, 111. [Google Scholar] [CrossRef]
- Hall, A.M.; Roberson, E.D. Mouse Models of Alzheimer’s Disease. Brain Res. Bull. 2012, 88, 3–12. [Google Scholar] [CrossRef]
- Businaro, R.; Ippoliti, F.; Ricci, S.; Canitano, N.; Fuso, A. Alzheimer’s Disease Promotion by Obesity: Induced Mechanisms—Molecular Links and Perspectives. Curr. Gerontol. Geriatr. Res. 2012, 2012, 986823. [Google Scholar] [CrossRef] [PubMed]
- Mattar, J.M.; Majchrzak, M.; Iannucci, J.; Bartman, S.; Robinson, J.K.; Grammas, P. Sex Differences in Metabolic Indices and Chronic Neuroinflammation in Response to Prolonged High-Fat Diet in ApoE4 Knock-In Mice. Int. J. Mol. Sci. 2022, 23, 3921. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Jiang, P.; Guan, X.-M.; Singh, G.; Trumbauer, M.E.; Yu, H.; Chen, H.Y.; Van der Ploeg, L.H.T.; Zheng, H. Mutant Human Presenilin 1 Protects Presenilin 1 Null Mouse against Embryonic Lethality and Elevates Aβ1–42/43 Expression. Neuron 1998, 20, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Bachstetter, A.D.; Van Eldik, L.J. Comprehensive Behavioral Characterization of an APP/PS-1 Double Knock-in Mouse Model of Alzheimer’s Disease. Alzheimer’s Res. Ther. 2013, 5, 28. [Google Scholar] [CrossRef]
- Chen, L.; Xu, S.; Wu, T.; Shao, Y.; Luo, L.; Zhou, L.; Ou, S.; Tang, H.; Huang, W.; Guo, K.; et al. Studies on APP Metabolism Related to Age-Associated Mitochondrial Dysfunction in APP/PS1 Transgenic Mice. Aging 2019, 11, 10242–10251. [Google Scholar] [CrossRef]
- Dixit, S.; Fessel, J.P.; Harrison, F.E. Mitochondrial Dysfunction in the APP/PSEN1 Mouse Model of Alzheimer’s Disease and a Novel Protective Role for Ascorbate. Free. Radic. Biol. Med. 2017, 112, 515–523. [Google Scholar] [CrossRef]
- Kulic, L.; Wollmer, M.A.; Rhein, V.; Pagani, L.; Kuehnle, K.; Cattepoel, S.; Tracy, J.; Eckert, A.; Nitsch, R.M. Combined Expression of Tau and the Harlequin Mouse Mutation Leads to Increased Mitochondrial Dysfunction, Tau Pathology and Neurodegeneration. Neurobiol. Aging 2011, 32, 1827–1838. [Google Scholar] [CrossRef]
- Götz, J.; Chen, F.; Barmettler, R.; Nitsch, R.M. Tau Filament Formation in Transgenic Mice Expressing P301L Tau*. J. Biol. Chem. 2001, 276, 529–534. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Aβ and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Sun, J.; Hon, S.; Nylander, A.N.; Xia, W.; Feng, Y.; Wang, X.; Lemere, C.A. L-3-n-Butylphthalide Improves Cognitive Impairment and Reduces Amyloid-β in a Transgenic Model of Alzheimer’s Disease. J. Neurosci. 2010, 30, 8180–8189. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, J.; Roy Chowdhury, S.; Snow, W.M.; Perez, C.; Cadonic, C.; Fernyhough, P.; Albensi, B.C. Early Onset of Sex-Dependent Mitochondrial Deficits in the Cortex of 3xTg Alzheimer’s Mice. Cells 2020, 9, 1541. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and Function in Lipid Metabolism, Neurobiology, and Alzheimer’s Diseases. Neurobiol. Dis. 2014, 72PA, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Liraz, O.; Boehm-Cagan, A.; Michaelson, D.M. ApoE4 Induces Aβ42, Tau, and Neuronal Pathology in the Hippocampus of Young Targeted Replacement apoE4 Mice. Mol. Neurodegener. 2013, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Schmukler, E.; Solomon, S.; Simonovitch, S.; Goldshmit, Y.; Wolfson, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Altered Mitochondrial Dynamics and Function in APOE4-Expressing Astrocytes. Cell Death Dis. 2020, 11, 578. [Google Scholar] [CrossRef] [PubMed]
- Wenz, T. Mitochondria and PGC-1α in Aging and Age-Associated Diseases. J. Aging Res. 2011, 2011, 810619. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1α Expression Decreases in the Alzheimer Disease Brain as a Function of Dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef]
- Omodei, D.; Fontana, L. Calorie Restriction and Prevention of Age-Associated Chronic Disease. FEBS Lett. 2011, 585, 1537–1542. [Google Scholar] [CrossRef]
- Lass, A.; Sohal, B.H.; Weindruch, R.; Forster, M.J.; Sohal, R.S. Caloric Restriction Prevents Age-Associated Accrual of Oxidative Damage to Mouse Skeletal Muscle Mitochondria. Free Radic. Biol. Med. 1998, 25, 1089–1097. [Google Scholar] [CrossRef]
- Sohal, R.S.; Weindruch, R. Oxidative Stress, Caloric Restriction, and Aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef]
- Guarente, L. Mitochondria—A Nexus for Aging, Calorie Restriction, and Sirtuins? Cell 2008, 132, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; de Cabo, R.; Anderson, R.M. Caloric Restriction Improves Health and Survival of Rhesus Monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric Restriction Delays Disease Onset and Mortality in Rhesus Monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.M.; Herbert, R.L.; Longo, D.L.; Allison, D.B.; Young, J.E.; Bryant, M.; et al. Impact of Caloric Restriction on Health and Survival in Rhesus Monkeys from the NIA Study. Nature 2012, 489, 318–321. [Google Scholar] [CrossRef]
- Waziry, R.; Ryan, C.P.; Corcoran, D.L.; Huffman, K.M.; Kobor, M.S.; Kothari, M.; Graf, G.H.; Kraus, V.B.; Kraus, W.E.; Lin, D.T.S.; et al. Effect of Long-Term Caloric Restriction on DNA Methylation Measures of Biological Aging in Healthy Adults from the CALERIE Trial. Nat. Aging 2023, 3, 248–257. [Google Scholar] [CrossRef]
- Ruetenik, A.; Barrientos, A. Dietary Restriction, Mitochondrial Function and Aging: From Yeast to Humans. Biochim. Biophys. Acta 2015, 1847, 1434–1447. [Google Scholar] [CrossRef]
- Goodrick, C.L.; Ingram, D.K.; Reynolds, M.A.; Freeman, J.R.; Cider, N. Effects of Intermittent Feeding upon Body Weight and Lifespan in Inbred Mice: Interaction of Genotype and Age. Mech. Ageing Dev. 1990, 55, 69–87. [Google Scholar] [CrossRef]
- Gudden, J.; Arias Vasquez, A.; Bloemendaal, M. The Effects of Intermittent Fasting on Brain and Cognitive Function. Nutrients 2021, 13, 3166. [Google Scholar] [CrossRef]
- Dias, G.P.; Murphy, T.; Stangl, D.; Ahmet, S.; Morisse, B.; Nix, A.; Aimone, L.J.; Aimone, J.B.; Kuro-O, M.; Gage, F.H.; et al. Intermittent Fasting Enhances Long-Term Memory Consolidation, Adult Hippocampal Neurogenesis, and Expression of Longevity Gene Klotho. Mol. Psychiatry 2021, 26, 6365–6379. [Google Scholar] [CrossRef]
- Bang, E.; Fincher, A.S.; Nader, S.; Murchison, D.A.; Griffith, W.H. Late-Onset, Short-Term Intermittent Fasting Reverses Age-Related Changes in Calcium Buffering and Inhibitory Synaptic Transmission in Mouse Basal Forebrain Neurons. J. Neurosci. 2022, 42, 1020–1034. [Google Scholar] [CrossRef]
- de Cabo, R.; Mattson, M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Deota, S.; Lin, T.; Chaix, A.; Williams, A.; Le, H.; Calligaro, H.; Ramasamy, R.; Huang, L.; Panda, S. Diurnal Transcriptome Landscape of a Multi-Tissue Response to Time-Restricted Feeding in Mammals. Cell Metab. 2023, 35, 150–165.e4. [Google Scholar] [CrossRef]
- Reimers, C.D.; Knapp, G.; Reimers, A.K. Does Physical Activity Increase Life Expectancy? A Review of the Literature. J. Aging Res. 2012, 2012, 243958. [Google Scholar] [CrossRef]
- Nilsson, M.I.; Bourgeois, J.M.; Nederveen, J.P.; Leite, M.R.; Hettinga, B.P.; Bujak, A.L.; May, L.; Lin, E.; Crozier, M.; Rusiecki, D.R.; et al. Lifelong Aerobic Exercise Protects against Inflammaging and Cancer. PLoS ONE 2019, 14, e0210863. [Google Scholar] [CrossRef]
- Jeppesen, T.D. Aerobic Exercise Training in Patients With mtDNA-Related Mitochondrial Myopathy. Front. Physiol. 2020, 11, 349. [Google Scholar] [CrossRef]
- Nilsson, M.I.; Tarnopolsky, M.A. Mitochondria and Aging—The Role of Exercise as a Countermeasure. Biology 2019, 8, 40. [Google Scholar] [CrossRef]
- Rabin, J.S.; Klein, H.; Kirn, D.R.; Schultz, A.P.; Yang, H.-S.; Hampton, O.; Jiang, S.; Buckley, R.F.; Viswanathan, A.; Hedden, T.; et al. Associations of Physical Activity and β-Amyloid with Longitudinal Cognition and Neurodegeneration in Clinically Normal Older Adults. JAMA Neurol. 2019, 76, 1203–1210. [Google Scholar] [CrossRef]
- Choe, Y.M.; Suh, G.-H.; Lee, B.C.; Choi, I.-G.; Kim, H.S.; Kim, J.W.; Hwang, J.; Yi, D.; Kim, J.W. High-Intensity Walking in Midlife Is Associated with Improved Memory in Physically Capable Older Adults. Alzheimers Res. Ther. 2023, 15, 143. [Google Scholar] [CrossRef]
- Adlard, P.A.; Perreau, V.M.; Pop, V.; Cotman, C.W. Voluntary Exercise Decreases Amyloid Load in a Transgenic Model of Alzheimer’s Disease. J. Neurosci. 2005, 25, 4217–4221. [Google Scholar] [CrossRef]
- Will, Y.; Shields, J.E.; Wallace, K.B. Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions. Biology 2019, 8, 32. [Google Scholar] [CrossRef]
- Will, Y.; Dykens, J. Mitochondrial Toxicity Assessment in Industry—A Decade of Technology Development and Insight. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1061–1067. [Google Scholar] [CrossRef]
- Čolak, E.; Ignjatović, S.; Radosavljević, A.; Žorić, L. The Association of Enzymatic and Non-Enzymatic Antioxidant Defense Parameters with Inflammatory Markers in Patients with Exudative Form of Age-Related Macular Degeneration. J. Clin. Biochem. Nutr. 2017, 60, 100–107. [Google Scholar] [CrossRef]
- Fusco, D.; Colloca, G.; Monaco, M.R.L.; Cesari, M. Effects of Antioxidant Supplementation on the Aging Process. Clin. Interv. Aging 2007, 2, 377–387. [Google Scholar]
- Colizzi, C. The Protective Effects of Polyphenols on Alzheimer’s Disease: A Systematic Review. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 184–196. [Google Scholar] [CrossRef]
- Polyphenols in Human Health and Disease—1st Edition. Available online: https://shop.elsevier.com/books/polyphenols-in-human-health-and-disease/watson/978-0-12-398456-2 (accessed on 8 February 2024).
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Wimalawansa, S.J. Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging. Biology 2019, 8, 30. [Google Scholar] [CrossRef]
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672. [Google Scholar] [CrossRef]
- Berridge, M.J. Vitamin D Deficiency Accelerates Ageing and Age-related Diseases: A Novel Hypothesis. J. Physiol. 2017, 595, 6825–6836. [Google Scholar] [CrossRef]
- Terock, J.; Bonk, S.; Frenzel, S.; Wittfeld, K.; Garvert, L.; Hosten, N.; Nauck, M.; Völzke, H.; Van der Auwera, S.; Grabe, H.J. Vitamin D Deficit Is Associated with Accelerated Brain Aging in the General Population. Psychiatry Res. Neuroimaging 2022, 327, 111558. [Google Scholar] [CrossRef]
- Wimalawansa, S. Vitamin D: What Clinicians Need to Know. Sri Lanka J. Diabetes Endocrinol. Metab. 2012, 2, 73–88. [Google Scholar] [CrossRef]
- Varela-López, A.; Giampieri, F.; Battino, M.; Quiles, J.L. Coenzyme Q and Its Role in the Dietary Therapy against Aging. Molecules 2016, 21, 373. [Google Scholar] [CrossRef]
- De Barcelos, I.P.; Haas, R.H. CoQ10 and Aging. Biology 2019, 8, 28. [Google Scholar] [CrossRef]
- Chang, P.-S.; Chou, H.-H.; Lai, T.-J.; Yen, C.-H.; Pan, J.-C.; Lin, P.-T. Investigation of Coenzyme Q10 Status, Serum Amyloid-β, and Tau Protein in Patients with Dementia. Front. Aging Neurosci. 2022, 14, 910289. [Google Scholar] [CrossRef]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 Administration Increases Brain Mitochondrial Concentrations and Exerts Neuroprotective Effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef]
- Spindler, M.; Beal, M.F.; Henchcliffe, C. Coenzyme Q10 Effects in Neurodegenerative Disease. Neuropsychiatr. Dis. Treat. 2009, 5, 597–610. [Google Scholar]
- Lerner, A.B.; Case, J.D.; Takahashi, Y.; Lee, T.H.; Mori, W. Isolation of Melatonin, the Pineal Gland Factor that Lightens Melanocytes 1. J. Am. Chem. Soc. 1958, 80, 2587. [Google Scholar] [CrossRef]
- Tordjman, S.; Chokron, S.; Delorme, R.; Charrier, A.; Bellissant, E.; Jaafari, N.; Fougerou, C. Melatonin: Pharmacology, Functions and Therapeutic Benefits. Curr. Neuropharmacol. 2017, 15, 434–443. [Google Scholar] [CrossRef]
- Masters, A.; Pandi-Perumal, S.R.; Seixas, A.; Girardin, J.-L.; McFarlane, S.I. Melatonin, the Hormone of Darkness: From Sleep Promotion to Ebola Treatment. Brain Disord. Ther. 2014, 4, 1000151. [Google Scholar] [CrossRef]
- Bucana, C.D.; Nadakavukaren, M.J.; Frehn, J.L. Novel Features of Hamster Pinealocyte Ultrastructure. Tissue Cell 1974, 6, 85–93. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef]
- Khatoon, R.; Rasheed, M.Z.; Rawat, M.; Alam, M.M.; Tabassum, H.; Parvez, S. Effect of Melatonin on Aβ42 Induced Changes in the Mitochondrial Function Related to Alzheimer’s Disease in Drosophila melanogaster. Neurosci. Lett. 2019, 711, 134376. [Google Scholar] [CrossRef]
- Martín, M.; Macías, M.; Escames, G.; Reiter, R.J.; Agapito, M.T.; Ortiz, G.G.; Acuña-Castroviejo, D. Melatonin-Induced Increased Activity of the Respiratory Chain Complexes I and IV Can Prevent Mitochondrial Damage Induced by Ruthenium Red In Vivo. J. Pineal Res. 2000, 28, 242–248. [Google Scholar] [CrossRef]
- Roy, J.; Wong, K.Y.; Aquili, L.; Uddin, M.S.; Heng, B.C.; Tipoe, G.L.; Wong, K.H.; Fung, M.L.; Lim, L.W. Role of Melatonin in Alzheimer’s Disease: From Preclinical Studies to Novel Melatonin-Based Therapies. Front. Neuroendocrinol. 2022, 65, 100986. [Google Scholar] [CrossRef]
- Firenzuoli, F.; Gori, L. Herbal Medicine Today: Clinical and Research Issues. Evid.-Based Complement. Altern. Med. 2007, 4, 37–40. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, C.; Hou, J.; Su, P.; Yang, Y.; Xia, B.; Zhao, X.; He, R.; Wang, L.; Cao, C.; et al. Red Ginseng Extract Improves Skeletal Muscle Energy Metabolism and Mitochondrial Function in Chronic Fatigue Mice. Front. Pharmacol. 2022, 13, 1077249. [Google Scholar] [CrossRef]
- Shin, S.J.; Jeon, S.G.; Kim, J.; Jeong, Y.; Kim, S.; Park, Y.H.; Lee, S.-K.; Park, H.H.; Hong, S.B.; Oh, S.; et al. Red Ginseng Attenuates Aβ-Induced Mitochondrial Dysfunction and Aβ-Mediated Pathology in an Animal Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3030. [Google Scholar] [CrossRef]
- Mor, D.E.; Sohrabi, S.; Kaletsky, R.; Keyes, W.; Tartici, A.; Kalia, V.; Miller, G.W.; Murphy, C.T. Metformin Rescues Parkinson’s Disease Phenotypes Caused by Hyperactive Mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 26438–26447. [Google Scholar] [CrossRef]
- Kulkarni, A.S.; Brutsaert, E.F.; Anghel, V.; Zhang, K.; Bloomgarden, N.; Pollak, M.; Mar, J.C.; Hawkins, M.; Crandall, J.P.; Barzilai, N. Metformin Regulates Metabolic and Nonmetabolic Pathways in Skeletal Muscle and Subcutaneous Adipose Tissues of Older Adults. Aging Cell 2018, 17, e12723. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef]
- Dharmalingam, M.; Sriram, U.; Baruah, M.P. Liraglutide: A Review of Its Therapeutic Use as a Once Daily GLP-1 Analog for the Management of Type 2 Diabetes Mellitus. Indian. J. Endocrinol. Metab. 2011, 15, 9–17. [Google Scholar] [CrossRef]
- Vaittinen, M.; Ilha, M.; Herbers, E.; Wagner, A.; Virtanen, K.A.; Pietiläinen, K.H.; Pirinen, E.; Pihlajamäki, J. Liraglutide Demonstrates a Therapeutic Effect on Mitochondrial Dysfunction in Human SGBS Adipocytes In Vitro. Diabetes Res. Clin. Pract. 2023, 199, 110635. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Hölscher, C. Liraglutide Can Reverse Memory Impairment, Synaptic Loss and Reduce Plaque Load in Aged APP/PS1 Mice, a Model of Alzheimer’s Disease. Neuropharmacology 2014, 76 Pt A, 57–67. [Google Scholar] [CrossRef]
- Femminella, G.D.; Frangou, E.; Love, S.B.; Busza, G.; Holmes, C.; Ritchie, C.; Lawrence, R.; McFarlane, B.; Tadros, G.; Ridha, B.H.; et al. Evaluating the Effects of the Novel GLP-1 Analogue Liraglutide in Alzheimer’s Disease: Study Protocol for a Randomised Controlled Trial (ELAD Study). Trials 2019, 20, 191. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ramirez, G.; Tang, R.; Paul, C.K.X.; Nair, M.; Henderson, S.; Morimoto, B.; Liu, J.; Kaasgaard, T.; Boyd, B.J.; et al. Modeling Digestion, Absorption, and Ketogenesis after Administration of Tricaprilin Formulations to Humans. Eur. J. Pharm. Biopharm. 2023, 182, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T.; Morimoto, B.H.; Cummings, J.L.; Farlow, M.R.; Walker, J. A Placebo-Controlled, Parallel-Group, Randomized Clinical Trial of AC-1204 in Mild-to-Moderate Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 75, 547–557. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorder | Genes Involved | Affected Mitochondrial Complex(es) | Associated Mitochondrial Dysfunction | Reference(s) |
|---|---|---|---|---|
| Huntington’s Disease | HTT | II, III, IV | Reduced functioning in mitochondrial ETC components; reduced ATP production; decreased Ca2+ loading capacity; altered fission/fusion dynamics; mitochondrial bioenergetic deficits; decreased mtDNA copy number; impaired mitochondrial membrane depolarization | [174,175,176,177] |
| Parkinson’s Disease | PARKIN, PINK1, DJ-1, ATP13A2, SNCA, LRRK2 | I, II, IV | Reduced functioning in mitochondrial ETC components; decrease in mitochondrial membrane potential; decreased ATP production; increased ROS production; abnormal mitochondrial morphology; decreased mitochondrial import; decreased mtDNA copy number; increased mitophagy | [33,178,179,180,181] |
| Alzheimer’s Disease | APP, PS1, PS2, APOE4 | IV | Reduced functioning in mitochondrial ETC components; abnormal mitochondrial morphology; decreased ATP production; increased ROS production; decreased expression and activation of mitochondrial enzymes; altered fission/fusion dynamics; decreased mitochondrial membrane potential and mtDNA copy number; abnormal mitophagy | [172,182,183,184,185,186,187] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartman, S.; Coppotelli, G.; Ross, J.M. Mitochondrial Dysfunction: A Key Player in Brain Aging and Diseases. Curr. Issues Mol. Biol. 2024, 46, 1987-2026. https://doi.org/10.3390/cimb46030130
Bartman S, Coppotelli G, Ross JM. Mitochondrial Dysfunction: A Key Player in Brain Aging and Diseases. Current Issues in Molecular Biology. 2024; 46(3):1987-2026. https://doi.org/10.3390/cimb46030130
Chicago/Turabian StyleBartman, Sydney, Giuseppe Coppotelli, and Jaime M. Ross. 2024. "Mitochondrial Dysfunction: A Key Player in Brain Aging and Diseases" Current Issues in Molecular Biology 46, no. 3: 1987-2026. https://doi.org/10.3390/cimb46030130