
Unveiling the Ovarian Cell Characteristics and Molecular Mechanism of Prolificacy in Goats via Single-Nucleus Transcriptomics Data Analysis

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Preparation and Animal Tissue Collection
2.2. Single-Cell RNA-Seq Library Construction and Sequencing
2.3. Data Processing and Downstream Analysis
2.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.5. Identification of TFs by SCENIC
2.6. Pesudotemperal Trajectory Analysis
2.7. Analysis of Cell–Cell Communication
2.8. Identification DEGs
2.9. Gene Enrichment Analysis
3. Results
3.1. Single-Nucleus Transcriptome Profiling of Goat Ovaries Elucidated Ovarian Cell Types and Gene Expression Signatures
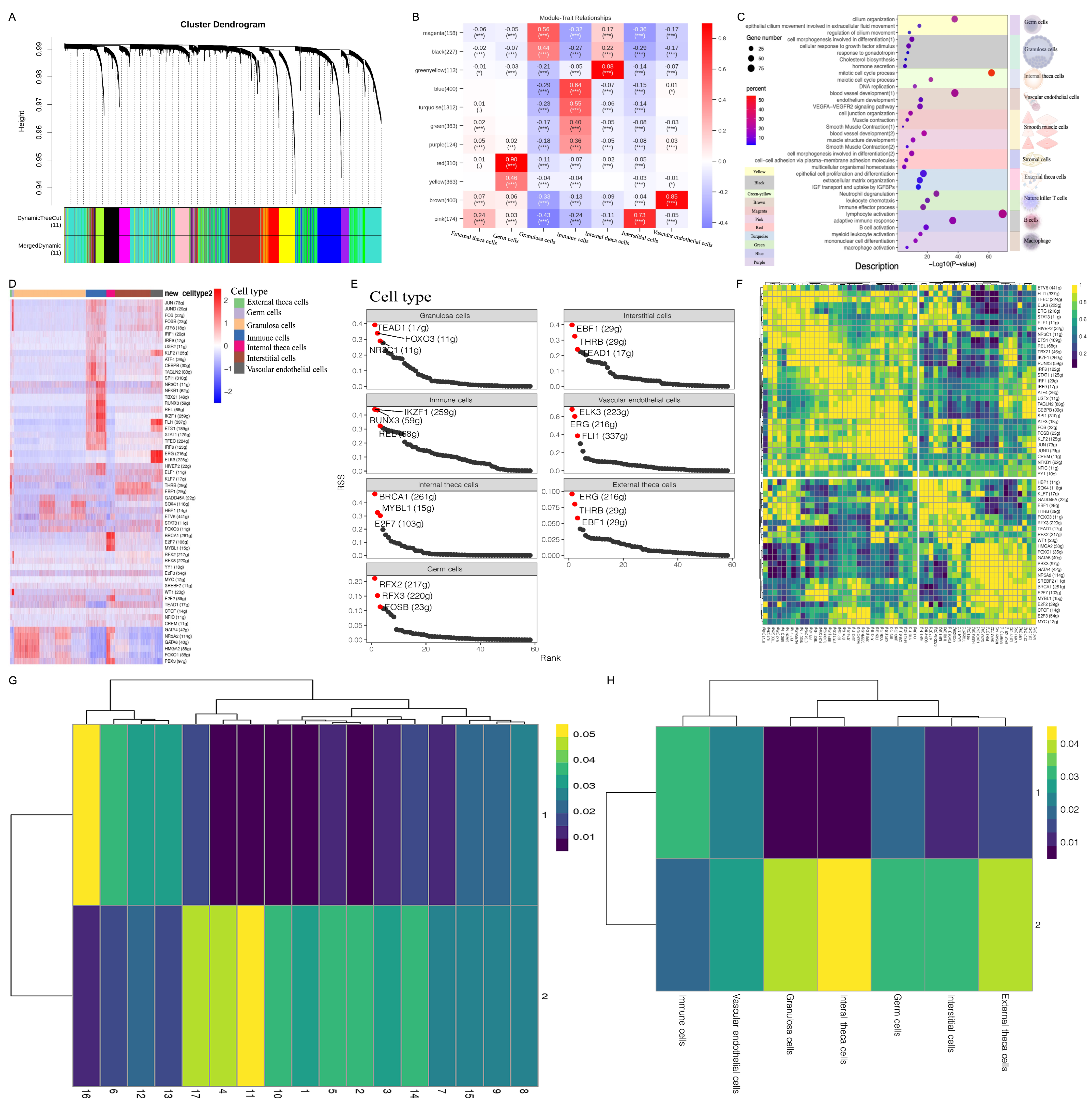
3.2. WGCNA Revealed the Biological Functions and Hub Regulatory Networks of Ovarian Cell Types
3.3. SCENIC Analysis Revealed Key Transcription Factors (TFs) Regulating Ovarian-Specific Cell Types
3.4. Niche Regulation of Ovary Cell Type Revealed by Cell–Cell Communication
3.5. GC Subtype Identification and Transcriptional Signature Analysis of Goat Ovaries
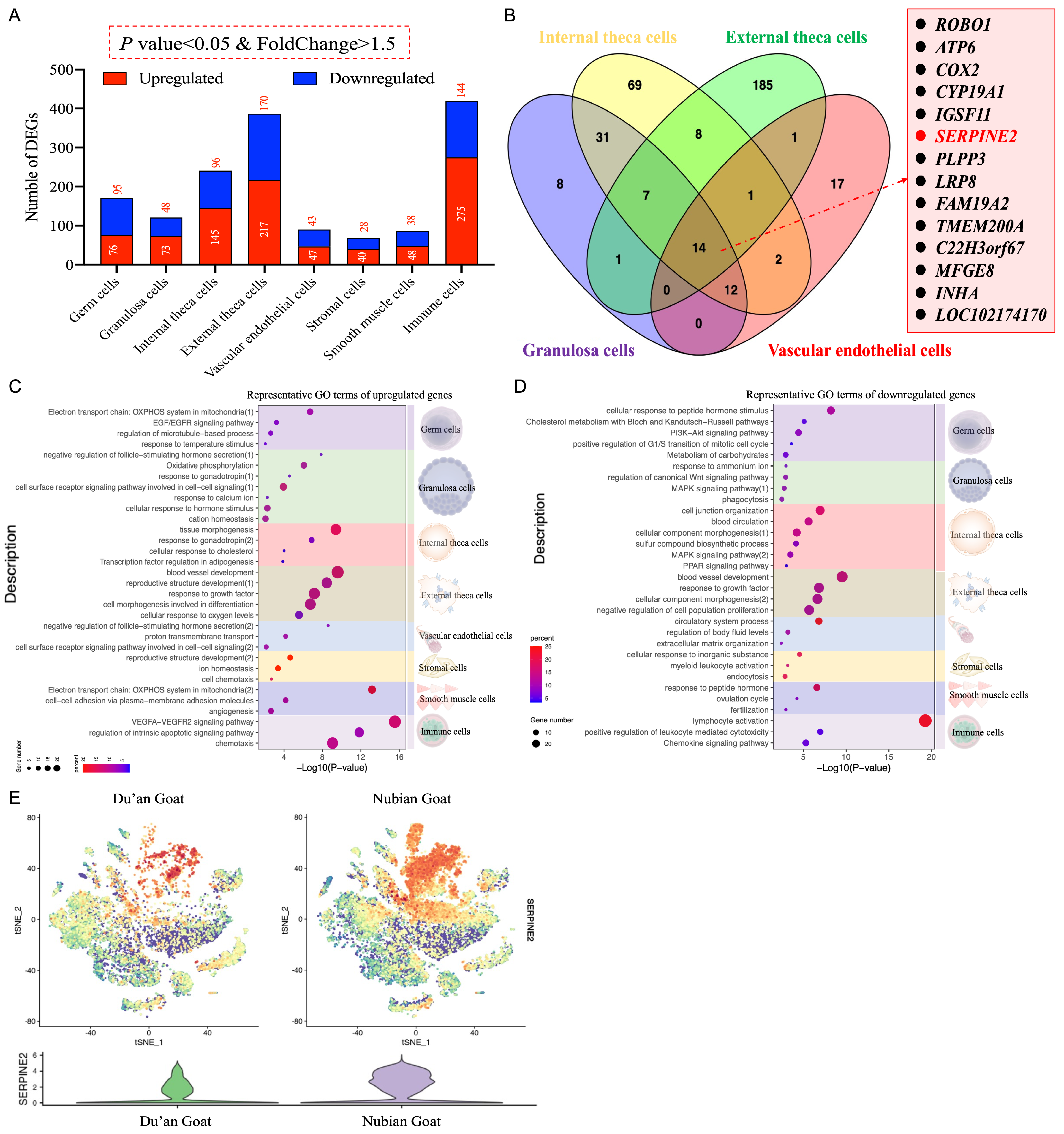
3.6. Differences in Ovarian Cell Expression Profiles between the Polytocous and Monotocous Goats
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, J.; Li, X.; Cao, Y.H.; Pokharel, K.; Hu, X.J.; Chen, Z.H.; Xu, S.S.; Peippo, J.; Honkatukia, M.; Kantanen, J.; et al. Comparative mRNA and miRNA expression in European mouflon (Ovis musimon) and sheep (Ovis aries) provides novel insights into the genetic mechanisms for female reproductive success. Heredity 2019, 122, 172–186. [Google Scholar] [CrossRef]
- Warriach, H.M.; McGill, D.M.; Bush, R.D.; Wynn, P.C.; Chohan, K.R. A Review of Recent Developments in Buffalo Reproduction—A Review. Asian Australas. J. Anim. 2015, 28, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, X.; Di, R.; Liu, Q.; Hu, W.; He, X.; Yu, J.; Zhang, X.; Zhang, J.; Broniowska, K.; et al. Metabolic Effects of FecB Gene on Follicular Fluid and Ovarian Vein Serum in Sheep (Ovis aries). Int. J. Mol. Sci. 2018, 19, 539. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Zhou, J.; Kang, N.; Liu, W.; Yu, L.; Zhang, Z.; Zhang, Y.; Yue, Q.; Yang, Q.; Zhang, X.; et al. Temporal and spatial dynamics mapping reveals follicle development regulated by different stromal cell populations. bioRxiv 2022. [Google Scholar] [CrossRef]
- Gougeon, A. Human ovarian follicular development: From activation of resting follicles to preovulatory maturation. Ann. Endocrinol. 2010, 71, 132–143. [Google Scholar] [CrossRef]
- Ge, T.; Wen, Y.F.; Li, B.; Huang, X.Y.; Jiang, S.H.; Zhang, E.P. Single-cell sequencing reveals the reproductive variations between primiparous and multiparous Hu ewes. J. Anim. Sci. Biotechnol. 2023, 14, 144. [Google Scholar] [CrossRef]
- Mcgee, E.A.; Hsueh, A. Initial and cyclic recruitment of ovarian follicles. Endocr. Rev. 2000, 21, 200–214. [Google Scholar]
- Li, J.; Ye, Y.; Zhang, R.; Zhang, L.; Hu, X.; Han, D.; Chen, J.; He, X.; Wang, G.; Yang, X. Robo1/2 regulate follicle atresia through manipulating granulosa cell apoptosis in mice. Sci. Rep. 2015, 5, 9720. [Google Scholar] [CrossRef]
- de Mello Bianchi, P.H.; Serafini, P.; Monteiro da Rocha, A.; Assad Hassun, P.; Alves da Motta, E.L.; Sampaio Baruselli, P.; Chada Baracat, E. Review: Follicular waves in the human ovary: A new physiological paradigm for novel ovarian stimulation protocols. Reprod. Sci. 2010, 17, 1067–1076. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, Z.; Qin, Q.; Nisenblat, V.; Yan, L. Transcriptome Landscape of Human Folliculogenesis Reveals Oocyte and Granulosa Cell Interactions. Mol. Cell 2018, 72, 1021–1034.e4. [Google Scholar] [CrossRef]
- Spencer, T.E.; Wells, K.D.; Lee, K.; Telugu, B.P.; Hansen, P.J.; Bartol, F.F.; Blomberg, L.; Schook, L.B.; Dawson, H.; Lunney, J.K.; et al. Future of biomedical, agricultural, and biological systems research using domesticated animals. Biol. Reprod. 2022, 106, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, R.; Yin, C.H.; Kee, K. Studying human reproductive biology through single-cell analysis and in vitro differentiation of stem cells into germ cell-like cells. Hum. Reprod. Update 2020, 26, 670–688. [Google Scholar] [CrossRef] [PubMed]
- La, H.; Yoo, H.; Lee, E.J.; Thang, N.X.; Choi, H.J.; Oh, J.; Park, J.H.; Hong, K. Insights from the Applications of Single-Cell Transcriptomic Analysis in Germ Cell Development and Reproductive Medicine. Int. J. Mol. Sci. 2021, 22, 823. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.C.; You, X.; Meng, K.; Sha, R.L.; Wang, Z.Z.; Yuan, N.Y.; Peng, Q.; Li, Z.G.; Xie, Z.Q.; Chen, R.J.; et al. Single-Cell RNA Sequencing Reveals Heterogeneity of Myf5-Derived Cells and Altered Myogenic Fate in the Absence of SRSF2. Adv. Sci. 2022, 9, e2105775. [Google Scholar] [CrossRef] [PubMed]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019, 8, 281–291. [Google Scholar] [CrossRef]
- Tosches, M.A.; Yamawaki, T.M.; Naumann, R.K.; Jacobi, A.A.; Tushev, G.; Laurent, G. Evolution of pallium, hippocampus, and cortical cell types revealed by single-cell transcriptomics in reptiles. Science 2018, 360, 881–888. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.; Moerman, T.; Wouters, J.; Aerts, S. SCENIC: Single-Cell Regulatory Network Inference and Clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polanski, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Guo, L.; Li, H.; Li, J.; Dong, F.; Yi, Z.Y.; Ouyang, Y.C.; Hou, Y.; Wang, Z.B.; Sun, Q.Y.; et al. NEK5 regulates cell cycle progression during mouse oocyte maturation and preimplantation embryonic development. Mol. Reprod. Dev. 2019, 86, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Davie, K.; Janssens, J.; Koldere, D.; Waegeneer, M.; Pech, U.; Kreft, U.; Aibar, S.; Makhzami, S.; Christiaens, V.; González-Blas, C. A Single-Cell Transcriptome Atlas of the Aging Drosophila Brain. Cell 2018, 174, 1–17. [Google Scholar] [CrossRef]
- Ojima, F.; Saito, Y.; Tsuchiya, Y.; Ogoshi, M.; Fukamachi, H.; Inagaki, K.; Otsuka, F.; Takeuchi, S.; Takahashi, S. Runx3 regulates folliculogenesis and steroidogenesis in granulosa cells of immature mice. Cell Tissue Res. 2019, 375, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Liu, Z.; Li, B.; Teng, Y.; Zhang, J.; Tang, Y.; Sun, S.C.; Liu, H. Involvement of FoxO1 in the effects of follicle-stimulating hormone on inhibition of apoptosis in mouse granulosa cells. Cell Death Dis. 2014, 5, e1475. [Google Scholar] [CrossRef] [PubMed]
- Herndon, M.K.; Law, N.C.; Donaubauer, E.M.; Kyriss, B.; Hunzicker-Dunn, M. Forkhead box O member FOXO1 regulates the majority of follicle-stimulating hormone responsive genes in ovarian granulosa cells. Mol. Cell. Endocrinol. 2016, 434, 116–126. [Google Scholar] [CrossRef]
- Anttonen, M.; Parviainen, H.; Kyronlahti, A.; Bielinska, M.; Wilson, D.B.; Ritvos, O.; Heikinheimo, M. GATA-4 is a granulosa cell factor employed in inhibin-alpha activation by the TGF-beta pathway. J. Mol. Endocrinol. 2006, 36, 557–568. [Google Scholar] [CrossRef]
- Zhao, Z.H.; Li, C.Y.; Meng, T.G.; Wang, Y.; Liu, W.B.; Li, A.; Cai, Y.J.; Hou, Y.; Schatten, H.; Wang, Z.B.; et al. Single-cell RNA sequencing reveals regulation of fetal ovary development in the monkey (Macaca fascicularis). Cell Discov. 2020, 6, 97. [Google Scholar] [CrossRef]
- Ghanim, A.; Chantelle, R.; Amanda, T.; Tim, O.; Jim, M.F. The Role of BMP Signalling Pathway in the Regulation of Ovarian Follicle Development. Biol. Reprod. 2008, 78 (Suppl. S1), 289–290. [Google Scholar]
- Kissel, H.; Timokhina, I.; Hardy, M.P.; Rothschild, G.; Tajima, Y.; Soares, V.; Angeles, M.; Whitlow, S.R.; Manova, K.; Besmer, P. Point mutation in Kit receptor tyrosine kinase reveals essential roles for Kit signaling in spermatogenesis and oogenesis without affecting other Kit responses. EMBO J. 2014, 19, 1312–1326. [Google Scholar] [CrossRef]
- Wang, L.Q.; Liu, J.C.; Chen, C.L.; Cheng, S.F.; Sun, X.F.; Zhao, Y.; Yin, S.; Hou, Z.M.; Pan, B.; Ding, C. Regulation of primordial follicle recruitment by cross-talk between the Notch and phosphatase and tensin homologue (PTEN)/AKT pathways. Reprod. Fertil. Dev. 2014, 28, 700–712. [Google Scholar] [CrossRef]
- Makker, A.; Goel, M.M.; Mahdi, A.A. PI3K/PTEN/Akt and TSC/mTOR signaling pathways, ovarian dysfunction, and infertility: An update. J. Mol. Endocrinol. 2014, 53, R103–R118. [Google Scholar] [CrossRef]
- Yu, C.; Zhou, J.J.; Fan, H.Y. Studying the Functions of TGF-β Signaling in the Ovary. In TGF-β Signaling; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016. [Google Scholar]
- Gu, C.; Liu, S.; Wu, Q.; Zhang, L.; Guo, F. Integrative single-cell analysis of transcriptome, DNA methylome and chromatin accessibility in mouse oocytes. Cell Res. 2019, 29, 110–123. [Google Scholar] [CrossRef]
- Mcfee, R.M.; Rozell, T.G.; Cupp, A.S. The balance of proangiogenic and antiangiogenic VEGFA isoforms regulate follicle development. Cell Tissue Res. 2012, 349, 635–647. [Google Scholar] [CrossRef]
- Wagner, M.; Yoshihara, M.; Douagi, I.; Damdimopoulos, A.; Damdimopoulou, P. Single-cell analysis of human ovarian cortex identifies distinct cell populations but no oogonial stem cells. Nat. Commun. 2020, 11, 1147. [Google Scholar] [CrossRef]
- Zhao, Z.H.; Wang, X.Y.; Schatten, H.; Sun, Q.Y. Single cell RNA sequencing techniques and applications in research of ovary development and related diseases. Reprod. Toxicol. 2022, 107, 97–103. [Google Scholar] [CrossRef]
- Wang, S.; Zheng, Y.; Li, J.; Yu, Y.; Zhang, W.; Song, M.; Liu, Z.; Min, Z.; Hu, H.; Jing, Y.; et al. Single-Cell Transcriptomic Atlas of Primate Ovarian Aging. Cell 2020, 180, 585–600.e19. [Google Scholar] [CrossRef]
- Meinsohn, M.C.; Saatcioglu, H.D.; Wei, L.; Li, Y.; Horn, H.; Chauvin, M.; Kano, M.; Nguyen, N.M.P.; Nagykery, N.; Kashiwagi, A.; et al. Single-cell sequencing reveals suppressive transcriptional programs regulated by MIS/AMH in neonatal ovaries. Proc. Natl. Acad. Sci. USA 2021, 118, e2100920118. [Google Scholar] [CrossRef]
- Park, C.J.; Lin, P.C.; Zhou, S.; Barakat, R.; Bashir, S.T.; Choi, J.M.; Cacioppo, J.A.; Oakley, O.R.; Duffy, D.M.; Lydon, J.P.; et al. Progesterone Receptor Serves the Ovary as a Trigger of Ovulation and a Terminator of Inflammation. Cell Rep. 2020, 31, 107496. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Zhao, Y.; Ma, D.; Zhao, M.; Li, N.; Men, Y.; Zhang, Y.; Chu, H.; Lei, C.; et al. scRNA-seq of ovarian follicle granulosa cells from different fertility goats reveals distinct expression patterns. Reprod. Domest. Anim. 2021, 56, 801–811. [Google Scholar] [CrossRef]
- Allen, B.M. The Embryonic development of the Ovary and Testis of the Mammals. Dev. Dyn. 2010, 3, 89–154. [Google Scholar] [CrossRef]
- Contreras-Solis, I.; Catala, M.; Soto-Heras, S.; Roura, M.; Paramio, M.T.; Izquierdo, D. Effect of follicle size on hormonal status of follicular fluid, oocyte ATP content, and in vitro embryo production in prepubertal sheep. Domest. Anim. Endocrinol. 2021, 75, 106582. [Google Scholar] [CrossRef]
- Hoque, S.A.M.; Umehara, T.; Kawai, T.; Shimada, M. Adverse effect of superoxide-induced mitochondrial damage in granulosa cells on follicular development in mouse ovaries. Free Radic. Biol. Med. 2021, 163, 344–355. [Google Scholar] [CrossRef]
- Manabe, N.; Inoue, N.; Miyano, T.; Sakamaki, K.; Sugimoto, M.; Miyamoto, H. Follicle Selection in Mammalian Ovaries: Regulatory Mechanisms of Granulosa Cell Apoptosis during Follicular Atresia. In The Ovary, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 369–385. [Google Scholar]
- Bhardwaj, J.K.; Paliwal, A.; Saraf, P.; Sachdeva, S.N. Role of autophagy in follicular development and maintenance of primordial follicular pool in the ovary. J. Cell Physiol. 2022, 237, 1157–1170. [Google Scholar] [CrossRef]
- Yadav, P.K.; Gupta, A.; Sharma, A.; Yadav, A.K.; Chaube, S. Fate of the germ cells in mammalian ovary: A review. J. Reprod. Health Med. 2020, 3, 1–7. [Google Scholar] [CrossRef]
- Weil, S.; Vendola, K.; Zhou, J.; Bondy, C.A. Androgen and Follicle-Stimulating Hormone Interactions in Primate Ovarian Follicle Development. J. Clin. Endocrinol. Metab. 1999, 84, 2591–2596. [Google Scholar] [CrossRef]
- Heather, J.; Baker, P.J.; Margaret, A.; Charlton, H.M.; Gary, J.; Lynne, F.; Rajendra, K.T.; O’Shaughnessy, P. Regulation of Sertoli cell number and activity by follicle-stimulating hormone and androgen during postnatal development in the mouse. Endocrinology 2004, 145, 318–329. [Google Scholar]
- Stricker, R.; Eberhart, R.; Chevailler, M.C.; Quinn, F.A.; Bischof, P.; Stricker, R. Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT® analyzer. Clin. Chem. Lab. Med. 2006, 44, 883–887. [Google Scholar] [CrossRef]
- Gong, J.G.; Campbell, B.K.; Bramley, T.A.; Gutierrez, C.G.; Peters, A.R.; Webb, R. Suppression in the secretion of follicle-stimulating hormone and luteinizing hormone, and ovarian follicle development in heifers continuously infused with a gonadotropin-releasing hormone agonist. Biol. Reprod. 1996, 55, 68–74. [Google Scholar] [CrossRef]
- Durlinger, A.; Gruijters, M.; Piet, K.; Bas, K.; Rajendra, K.T.; Matzuk, M.M.; Rose, U.M.; De, J.F.H.; Uilenbroek, J.; Anton, G.J. Anti-Müllerian hormone attenuates the effects of FSH on follicle development in the mouse ovary. Endocrinology 2001, 142, 4891–4899. [Google Scholar] [CrossRef]
- Cecconi, S.; Rossi, G.; Coticchio, G.; Macchiarelli, G.; Borini, A.; Canipari, R. Influence of thyroid hormone on mouse preantral follicle development in vitro. Fertil. Steril. 2004, 81 (Suppl. S1), 919–924. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.; Lima, L.F.; Alves, A.; Celestino, J.; Matos, M.; Lima-Verde, I.B.; Bernuci, M.P.; Lopes, C.; Báo, S.; Campello, C. Interaction between melatonin and follicle-stimulating hormone promotes in vitro development of caprine preantral follicles. Domest. Anim. Endocrinol. 2013, 44, 1–9. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Wei, Y.; Gao, X.; Song, Y.; Huang, Y.; Jiang, Q. Unveiling the Ovarian Cell Characteristics and Molecular Mechanism of Prolificacy in Goats via Single-Nucleus Transcriptomics Data Analysis. Curr. Issues Mol. Biol. 2024, 46, 2301-2319. https://doi.org/10.3390/cimb46030147
Zhang S, Wei Y, Gao X, Song Y, Huang Y, Jiang Q. Unveiling the Ovarian Cell Characteristics and Molecular Mechanism of Prolificacy in Goats via Single-Nucleus Transcriptomics Data Analysis. Current Issues in Molecular Biology. 2024; 46(3):2301-2319. https://doi.org/10.3390/cimb46030147
Chicago/Turabian StyleZhang, Sanbao, Yirong Wei, Xiaotong Gao, Ying Song, Yanna Huang, and Qinyang Jiang. 2024. "Unveiling the Ovarian Cell Characteristics and Molecular Mechanism of Prolificacy in Goats via Single-Nucleus Transcriptomics Data Analysis" Current Issues in Molecular Biology 46, no. 3: 2301-2319. https://doi.org/10.3390/cimb46030147
APA StyleZhang, S., Wei, Y., Gao, X., Song, Y., Huang, Y., & Jiang, Q. (2024). Unveiling the Ovarian Cell Characteristics and Molecular Mechanism of Prolificacy in Goats via Single-Nucleus Transcriptomics Data Analysis. Current Issues in Molecular Biology, 46(3), 2301-2319. https://doi.org/10.3390/cimb46030147