
Revisiting Neuroblastoma: Nrf2, NF-κB and Phox2B as a Promising Network in Neuroblastoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
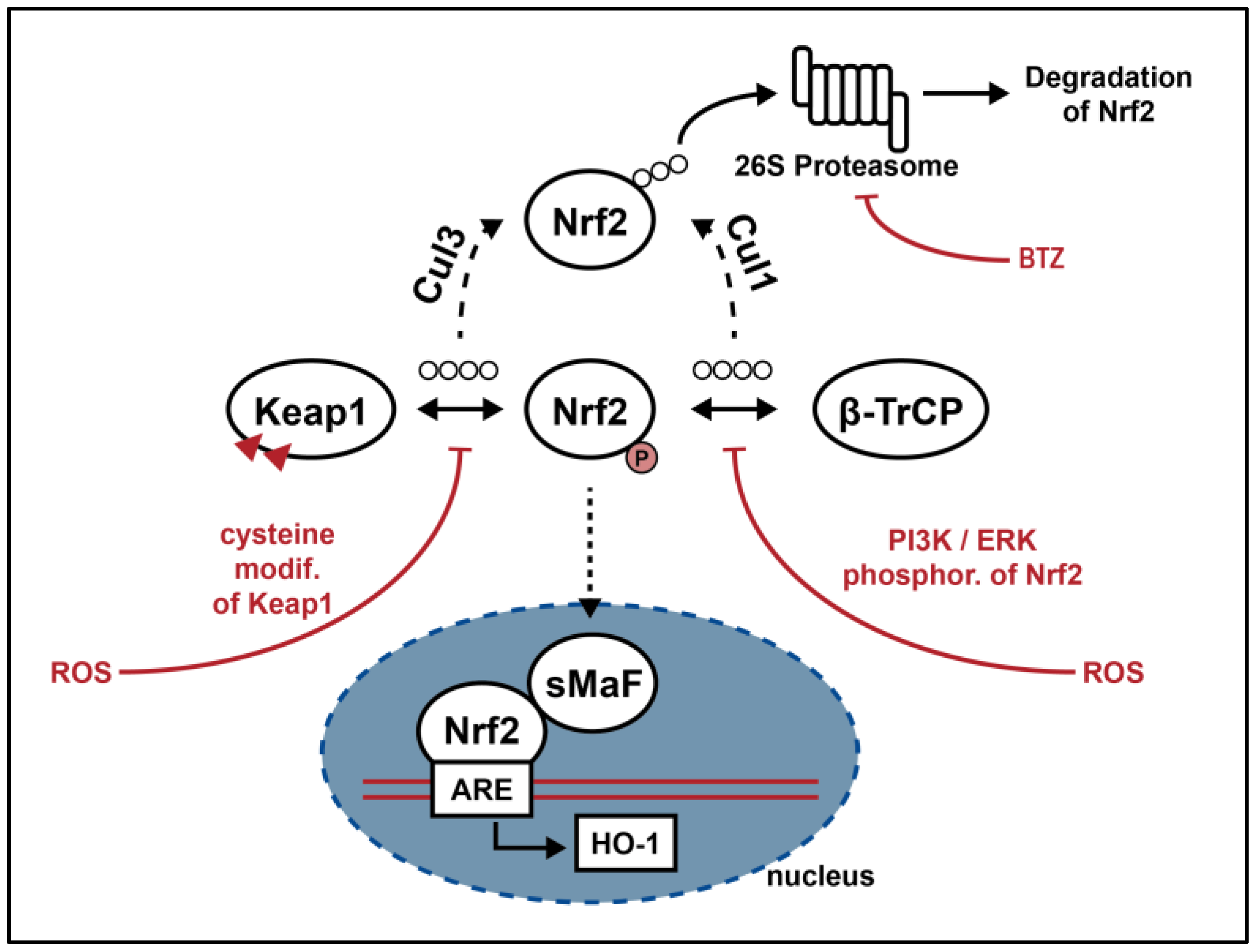
2. Nrf2 Signaling Pathway
2.1. Nrf2 Molecular and Functional Biology
2.2. Nrf2 in Neuroblastoma
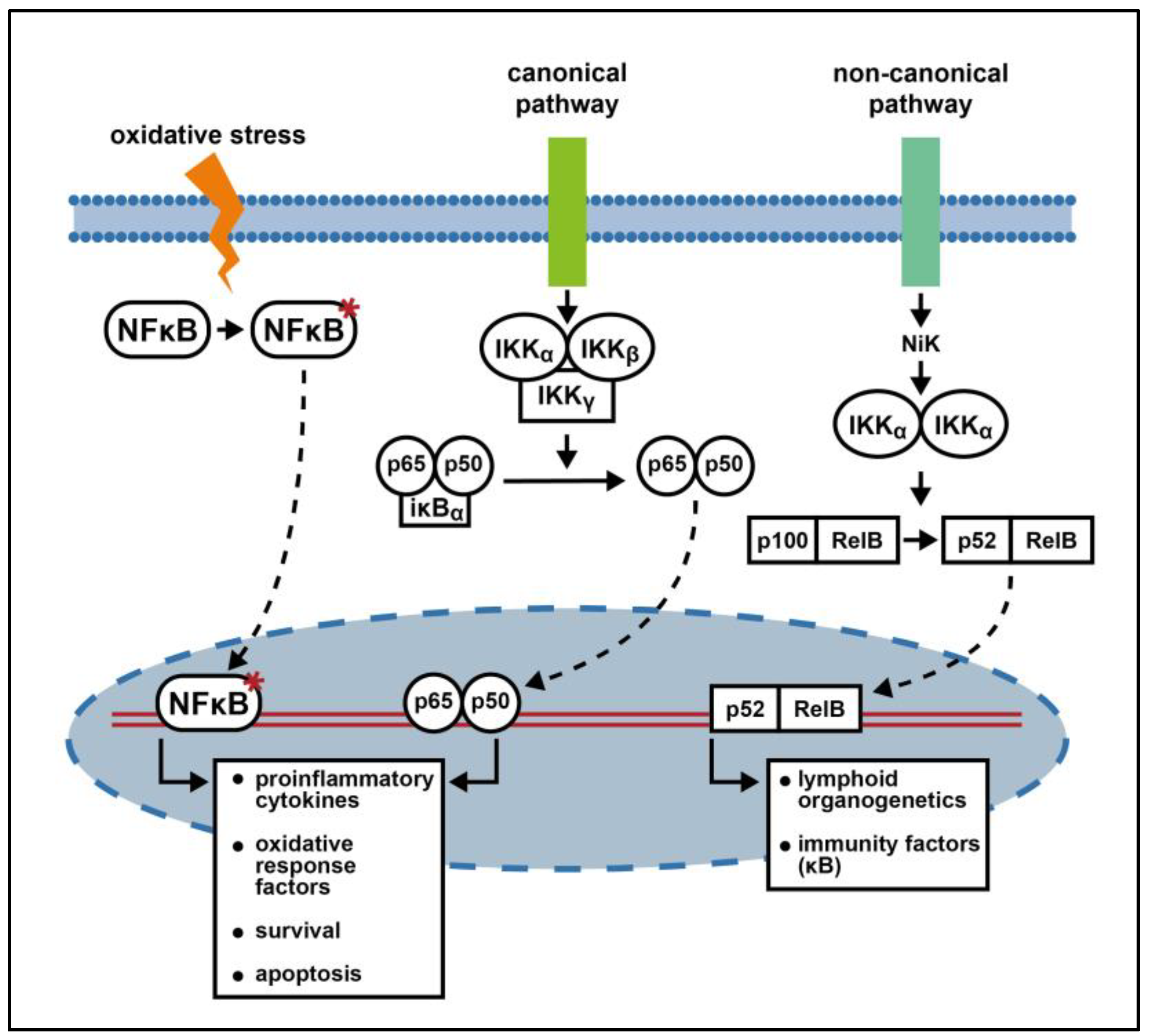
3. NF-kB Signaling Pathway
3.1. NF-kB Molecular and Functional Biology
3.2. NF-κB in Neuroblastoma
4. Phox2B Signaling Pathway
4.1. Phox2B Molecular and Functional Biology
4.2. Phox2B in Neuroblastoma
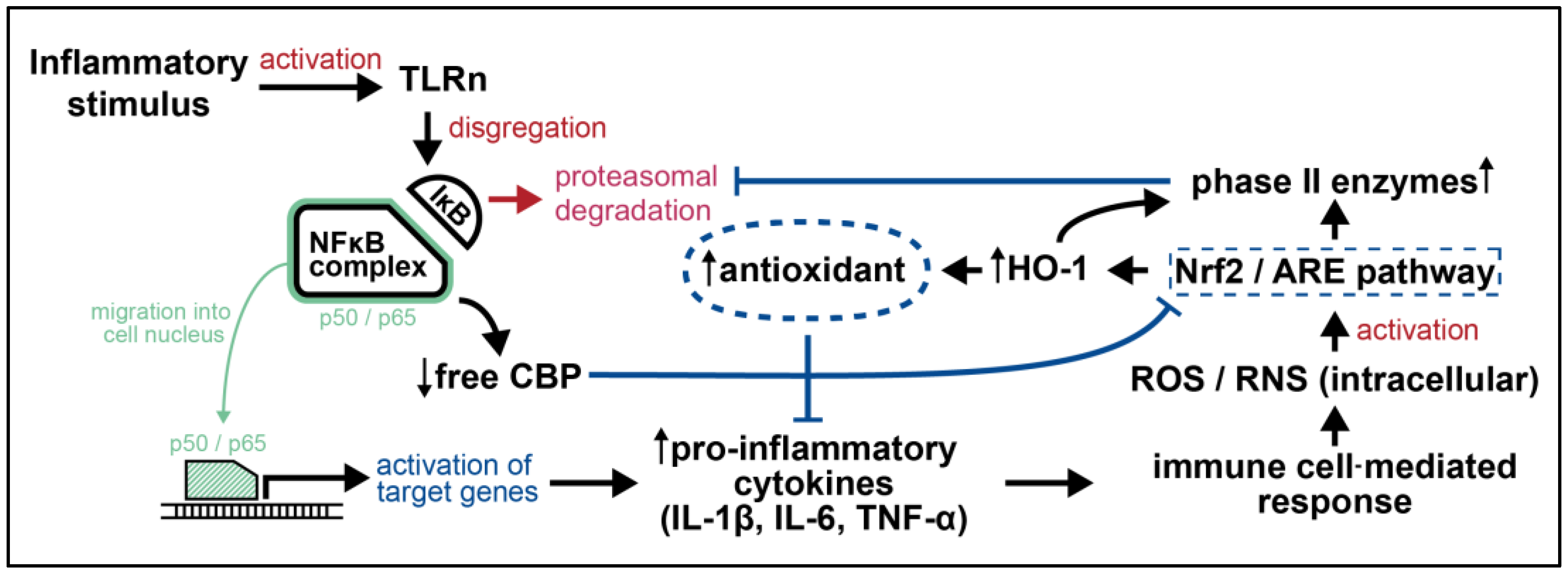
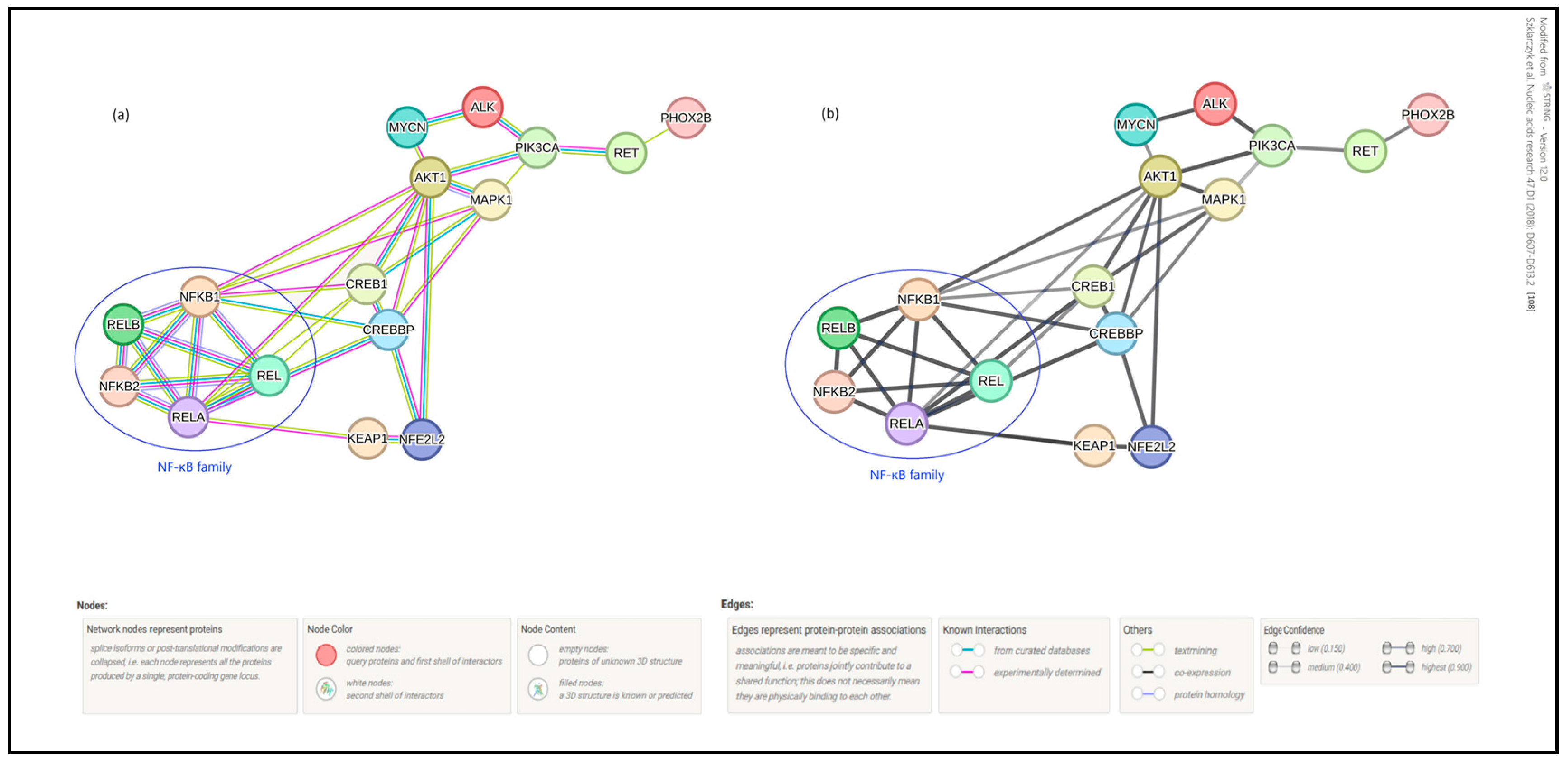
5. Potential Nrf2—NF-κB—Phox2B Crosstalk in Neuroblastoma
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, F.; Kaatsch, P.; Grabow, D.; Spix, C. ‘German Childhood Cancer Registry—Annual Report 2019 (1980–2018); Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz: Mainz, Germany, 2020. [Google Scholar]
- Shimada, H.; Chatten, J.; Newton, W.A.; Sachs, N.; Hamoudi, A.B.; Chiba, T.; Marsden, H.B.; Misugi, K. Histopathologic Prognostic Factors in Neuroblastic Tumors: Definition of Subtypes of Ganglioneuroblastoma and an Age-Linked Classification of Neuroblastomas. JNCI J. Natl. Cancer Inst. 1984, 73, 405–416. [Google Scholar] [CrossRef]
- Zafar, A.; Wang, W.; Liu, G.; Wang, X.; Xian, W.; McKeon, F.; Foster, J.; Zhou, J.; Zhang, R. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med. Res. Rev. 2020, 41, 961–1021. [Google Scholar] [CrossRef]
- Aygun, N. Biological and Genetic Features of Neuroblastoma and Their Clinical Importance. Curr. Pediatr. Rev. 2018, 14, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Berthold, F.; Spix, C.; Kaatsch, P.; Lampert, F. Incidence, Survival, and Treatment of Localized and Metastatic Neuroblastoma in Germany 1979–2015. Pediatr. Drugs 2017, 19, 577–593. [Google Scholar] [CrossRef]
- Park, J.R.; Bagatell, R.; London, W.B.; Maris, J.M.; Cohn, S.L.; Mattay, K.M.; Hogarty, M.; on behalf of the COG Neuroblastoma Committee. Children’s Oncology Group’s 2013 blueprint for research: Neuroblastoma. Pediatr. Blood Cancer 2013, 60, 985–993. [Google Scholar] [CrossRef]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef]
- Berlanga, P.; Cañete, A.; Castel, V. Advances in emerging drugs for the treatment of neuroblastoma. Expert Opin. Emerg. Drugs 2016, 22, 63–75. [Google Scholar] [CrossRef]
- Megison, M.L.; Gillory, L.A.; Beierle, E.A. Cell Survival Signaling in Neuroblastoma. Anti-Cancer Agents Med. Chem. 2013, 13, 563–575. [Google Scholar] [CrossRef]
- Louis, C.U.; Shohet, J.M. Neuroblastoma: Molecular pathogenesis and therapy. Annu. Rev. Med. 2015, 66, 49–63. [Google Scholar] [CrossRef]
- Ma, Y.; Feng, J.; Zhao, J.; Ding, D.; Tian, F.; Chen, L.; Zheng, J.; Xiao, X. PHOX2B as a Reliable Marker for Neuroblastoma in Tissue and Cytology Specimens. J. Neuropathol. Exp. Neurol. 2021, 80, 1108–1116. [Google Scholar] [CrossRef]
- Kobayashi, M.; Yamamoto, M. Molecular Mechanisms Activating the Nrf2-Keap1 Pathway of Antioxidant Gene Regulation. Antioxidants Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2017, 69, 393–402. [Google Scholar] [CrossRef]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Furfaro, A.L.; Piras, S.; Domenicotti, C.; Fenoglio, D.; De Luigi, A.; Salmona, M.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; et al. Role of Nrf2, HO-1 and GSH in Neuroblastoma Cell Resistance to Bortezomib. PLoS ONE 2016, 11, e0152465. [Google Scholar] [CrossRef]
- Sekhar, K.R.; Rachakonda, G.; Freeman, M.L. Cysteine-based regulation of the CUL3 adaptor protein Keap1. Toxicol. Appl. Pharmacol. 2010, 244, 21–26. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Su, Z.-Y.; Kong, A.-N.T. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP Promotes Glycogen Synthase Kinase 3-Dependent Degradation of the Nrf2 Transcription Factor in a Keap1-Independent Manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.; Sternberg, P.; Cai, J. Essential Roles of the PI3 Kinase/Akt Pathway in Regulating Nrf2-Dependent Anti-oxidant Functions in the RPE. Invest. Ophthalmol. Vis. Sci. 2008, 49, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Kwak, M.-K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.M.; Kleeberger, S.R.; Bream, J.H.; Fallon, P.G.; Kensler, T.W.; Yamamoto, M.; Reddy, S.P. Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling. Oncogene 2008, 27, 5821–5832. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Impacts Cellular Bioenergetics by Controlling Substrate Availability for Mitochondrial Respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, A.; Onda, K.; Kawahara, H.; Uchiyama, Y.; Nakayama, H.; Omi, T.; Nagaoka, M.; Matsui, H.; Hirano, T. Sofalcone, a gastric mucosa protective agent, increases vascular endothelial growth factor via the Nrf2-heme-oxygenase-1 dependent pathway in gastric epithelial cells. Biochem. Biophys. Res. Commun. 2010, 398, 581–584. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 Protein Up-regulates Antiapoptotic Protein Bcl-2 and Prevents Cellular Apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A. TRPM2 in Cancer. Cell Calcium 2019, 80, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, Z.; Salvador, G.; Liu, X.; Oteiza, P. Zinc and the modulation of Nrf2 in human neuroblastoma cells. Free. Radic. Biol. Med. 2020, 155, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Surh, Y.-J. The Role of Nrf2 in Cellular Innate Immune Response to Inflammatory Injury. Toxicol. Res. 2009, 25, 159–173. [Google Scholar] [CrossRef]
- High-Risk Neuroblastoma Standard Clinical Practice Recommendations. Available online: https://siope.eu/media/documents/escp-high-risk-neuroblastoma-standard-clinical-practice-recommendations.pdf (accessed on 1 January 2024).
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.J.; Liu, Y.; Han, S.; Yang, C. Brusatol, an NRF2 inhibitor for future cancer therapeutic. Cell Biosci. 2019, 9, 1–3. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Ramos, V.d.M.; Gasparotto, J.; Figueiró, F.; Dias, A.d.F.; Rostirolla, D.C.; Somensi, N.; da Rosa, H.T.; Grun, L.K.; Barbé-Tuana, F.M.; Gelain, D.P.; et al. Retinoic acid downregulates thiol antioxidant defences and homologous recombination while promotes A549 cells sensitization to cisplatin. Cell. Signal. 2019, 62, 109356. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Fu, S.; Zhou, A.; Su, Y.; Gao, X.; Zhang, Y.; Huang, B.; Du, J.; Liu, D. Camptothecin Regulates Microglia Polarization and Exerts Neuroprotective Effects via Activating AKT/Nrf2/HO-1 and Inhibiting NF-κB Pathways In Vivo and In Vitro. Front. Immunol. 2021, 12, 619761. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Ma, S.; Zhuo, R.; Xu, L.; Jia, S.; Yang, P.; Yao, Y.; Cao, H.; Ma, L.; Pan, J.; et al. Suppression of endoplasmic reticulum stress-dependent autophagy enhances cynaropicrin-induced apoptosis via attenuation of the P62/Keap1/Nrf2 pathways in neuroblastoma. Front. Pharmacol. 2022, 13, 977622. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.; Ravanan, P. Bardoxolone methyl induces neuritogenesis in Neuro2a cells. Pharmacol. Rep. 2018, 70, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.; Talwar, P.; D’Hellencourt, C.L.; Ravanan, P. CDDO and ATRA Instigate Differentiation of IMR32 Human Neuroblastoma Cells. Front. Mol. Neurosci. 2017, 10, 310. [Google Scholar] [CrossRef]
- Odarenko, K.V.; Salomatina, O.V.; Chernikov, I.V.; Salakhutdinov, N.F.; Zenkova, M.A.; Markov, A.V. Soloxolone Methyl Reduces the Stimulatory Effect of Leptin on the Aggressive Phenotype of Murine Neuro2a Neuroblastoma Cells via the MAPK/ERK1/2 Pathway. Pharmaceuticals 2023, 16, 1369. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zhang, J.; Chang, N. Epigenetic modification of Nrf2 by sulforaphane increases the antioxidative and anti-inflammatory capacity in a cellular model of Alzheimer’s disease. Eur. J. Pharmacol. 2018, 824, 1–10. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Hyun, Y.J.; Zhen, A.X.; Cho, S.J.; Ahn, M.J.; Yi, J.M.; Hyun, J.W. Luteolin promotes apoptotic cell death via upregulation of Nrf2 expression by DNA demethylase and the interaction of Nrf2 with p53 in human colon cancer cells. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shu, L.; Zhang, C.; Li, W.; Wu, R.; Guo, Y.; Yang, Y.; Kong, A. Histone Methyltransferase Setd7 Regulates Nrf2 Signaling Pathway by Phenethyl Isothiocyanate and Ursolic Acid in Human Prostate Cancer Cells. Mol. Nutr. Food Res. 2018, 62, e1700840. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ghazi, T.; Chuturgoon, A.A. Fumonisin B1 alters global m6A RNA methylation and epigenetically regulates Keap1-Nrf2 signaling in human hepatoma (HepG2) cells. Arch. Toxicol. 2021, 95, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Kudo, N.; Yoshida, M.; Miyamoto, S. A nuclear export signal in the N-terminal regulatory domain of IκBα controls cytoplasmic localization of inactive NF-κB/IκBα complexes. Proc. Natl. Acad. Sci. USA 2000, 97, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Solan, N.J.; Miyoshi, H.; Carmona, E.M.; Bren, G.D.; Paya, C.V. RelB Cellular Regulation and Transcriptional Activity Are Regulated by p100. J. Biol. Chem. 2002, 277, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Jardin, F. NFkB Pathway and Hodgkin Lymphoma. Biomedicines 2022, 10, 2153. [Google Scholar] [CrossRef] [PubMed]
- KEGG PATHWAY: NF-Kappa B Signaling Pathway—Reference Pathway’. Available online: https://www.kegg.jp/pathway/map=map04064&keyword=nfkb (accessed on 8 January 2024).
- Savinova, O.V.; Hoffmann, A.; Ghosh, G. The Nfkb1 and Nfkb2 Proteins p105 and p100 Function as the Core of High-Molecular-Weight Heterogeneous Complexes. Mol. Cell 2009, 34, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Kayagaki, N.; Yan, M.; Seshasayee, D.; Wang, H.; Lee, W.; French, D.M.; Grewal, I.S.; Cochran, A.G.; Gordon, N.C.; Yin, J.; et al. BAFF/BLyS Receptor 3 Binds the B Cell Survival Factor BAFF Ligand through a Discrete Surface Loop and Promotes Processing of NF-κB2. Immunity 2002, 17, 515–524. [Google Scholar] [CrossRef]
- Fagiani, F.; Catanzaro, M.; Buoso, E.; Basagni, F.; Di Marino, D.; Raniolo, S.; Amadio, M.; Frost, E.H.; Corsini, E.; Racchi, M.; et al. Targeting Cytokine Release Through the Differential Modulation of Nrf2 and NF-κB Pathways by Electrophilic/Non-Electrophilic Compounds. Front. Pharmacol. 2020, 11, 1256. [Google Scholar] [CrossRef]
- Zhang, T.; Ma, C.; Zhang, Z.; Zhang, H.; Hu, H. NF-κB signaling in inflammation and cancer. Medcomm 2021, 2, 618–653. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, M.; Candido, M.F.; Valera, E.T.; Brassesco, M.S. The multifaceted NF-kB: Are there still prospects of its inhibition for clinical intervention in pediatric central nervous system tumors? Cell. Mol. Life Sci. 2021, 78, 6161–6200. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; McAllister-Lucas, L.M.; Shao, F.; Schumacher, K.R.; Feng, Z.; Porter, A.G.; Castle, V.P.; Opipari, A.W. NF-κB Activation Mediates Doxorubicin-induced Cell Death in N-type Neuroblastoma Cells. J. Biol. Chem. 2001, 276, 48921–48929. [Google Scholar] [CrossRef] [PubMed]
- Brandetti, E.; Focaccetti, C.; Pezzolo, A.; Ognibene, M.; Folgiero, V.; Cotugno, N.; Benvenuto, M.; Palma, P.; Manzari, V.; Rossi, P.; et al. Enhancement of Neuroblastoma NK-Cell-Mediated Lysis through NF-kB p65 Subunit-Induced Expression of FAS and PVR, the Loss of Which Is Associated with Poor Patient Outcome. Cancers 2021, 13, 4368. [Google Scholar] [CrossRef] [PubMed]
- Posadas, I.; Santos, P.; Ceña, V. Acetaminophen Induces Human Neuroblastoma Cell Death through NFKB Activation. PLoS ONE 2012, 7, e50160. [Google Scholar] [CrossRef] [PubMed]
- Karacay, B.; Sanlioglu, S.; Griffith, T.S.; Sandler, A.; Bonthius, D.J. Inhibition of the NF-κB pathway enhances TRAIL-mediated apoptosis in neuroblastoma cells. Cancer Gene Ther. 2004, 11, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, S.; Forloni, M.; Cifaldi, L.; Antonucci, C.; Citti, A.; Boldrini, R.; Pezzullo, M.; Castellano, A.; Russo, V.; van der Bruggen, P.; et al. IRF1 and NF-kB Restore MHC Class I-Restricted Tumor Antigen Processing and Presentation to Cytotoxic T Cells in Aggressive Neuroblastoma. PLoS ONE 2012, 7, e46928. [Google Scholar] [CrossRef] [PubMed]
- Forloni, M.; Albini, S.; Limongi, M.Z.; Cifaldi, L.; Boldrini, R.; Nicotra, M.R.; Giannini, G.; Natali, P.G.; Giacomini, P.; Fruci, D. NF-κB, and not MYCN, Regulates MHC Class I and Endoplasmic Reticulum Aminopeptidases in Human Neuroblastoma Cells. Cancer Res. 2010, 70, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Appaiah, H.N.; Anjanappa, M.; Gilley, D.; Tanaka, H.; Badve, S.; Crooks, P.A.; Mathews, W.; Sweeney, C.; Bhat-Nakshatri, P. NF-κB-dependent and -independent epigenetic modulation using the novel anti-cancer agent DMAPT. Cell Death Dis. 2015, 6, e1608. [Google Scholar] [CrossRef]
- Pattyn, A.; Morin, X.; Cremer, H.; Goridis, C.; Brunet, J.-F. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature 1999, 399, 366–370. [Google Scholar] [CrossRef]
- Ooi, C.Y.; Carter, D.R.; Liu, B.; Mayoh, C.; Beckers, A.; Lalwani, A.; Nagy, Z.; De Brouwer, S.; Decaesteker, B.; Hung, T.-T.; et al. Network Modeling of microRNA–mRNA Interactions in Neuroblastoma Tumorigenesis Identifies miR-204 as a Direct Inhibitor of MYCN. Cancer Res. 2018, 78, 3122–3134. [Google Scholar] [CrossRef] [PubMed]
- Perri, P.; Ponzoni, M.; Corrias, M.V.; Ceccherini, I.; Candiani, S.; Bachetti, T. A Focus on Regulatory Networks Linking MicroRNAs, Transcription Factors and Target Genes in Neuroblastoma. Cancers 2021, 13, 5528. [Google Scholar] [CrossRef] [PubMed]
- Flora, A.; Lucchetti, H.; Benfante, R.; Goridis, C.; Clementi, F.; Fornasari, D. SP Proteins and PHOX2B Regulate the Expression of the HumanPHOX2aGene. J. Neurosci. 2001, 21, 7037–7045. [Google Scholar] [CrossRef] [PubMed]
- Di Zanni, E.; Bianchi, G.; Ravazzolo, R.; Raffaghello, L.; Ceccherini, I.; Bachetti, T. Targeting ofPHOX2Bexpression allows the identification of drugs effective in counteracting neuroblastoma cell growth. Oncotarget 2017, 8, 72133–72146. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Amiel, J.; Laudier, B.; Attié-Bitach, T.; Trang, H.; de Pontual, L.; Gener, B.; Trochet, D.; Etchevers, H.; Ray, P.; Simonneau, M.; et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat. Genet. 2003, 33, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Khazi, D.; Carlisle, A.J.; Winter, C.L.; Rappaport, E.; Maris, J.M. Germline PHOX2B Mutation in Hereditary Neuroblastoma. Am. J. Hum. Genet. 2004, 75, 727–730. [Google Scholar] [CrossRef]
- Zhao, J.; Zhu, Y.; Xie, X.; Yao, Y.; Zhang, J.; Zhang, R.; Huang, L.; Cheng, J.; Xia, H.; He, J.; et al. Pleiotropic effect of common PHOX2B variants in Hirschsprung disease and neuroblastoma. Aging 2019, 11, 1252–1261. [Google Scholar] [CrossRef]
- Di Lascio, S.; Benfante, R.; Cardani, S.; Fornasari, D. Research Advances on Therapeutic Approaches to Congenital Central Hypoventilation Syndrome (CCHS). Front. Neurosci. 2021, 14, 615666. [Google Scholar] [CrossRef] [PubMed]
- van Limpt, V.; Chan, A.; Schramm, A.; Eggert, A.; Versteeg, R. Phox2B mutations and the Delta–Notch pathway in neuroblastoma. Cancer Lett. 2005, 228, 59–63. [Google Scholar] [CrossRef]
- Hung, Y.P.; Lee, J.P.; Bellizzi, A.M.; Hornick, J.L. PHOX2B reliably distinguishes neuroblastoma among small round blue cell tumours. Histopathology 2017, 71, 786–794. [Google Scholar] [CrossRef]
- Stutterheim, J.; Gerritsen, A.; Zappeij-Kannegieter, L.; Kleijn, I.; Dee, R.; Hooft, L.; van Noesel, M.M.; Bierings, M.; Berthold, F.; Versteeg, R.; et al. PHOX2B Is a Novel and Specific Marker for Minimal Residual Disease Testing in Neuroblastoma. J. Clin. Oncol. 2008, 26, 5443–5449. [Google Scholar] [CrossRef] [PubMed]
- Longo, L.; Borghini, S.; Schena, F.; Parodi, S.; Albino, D.; Bachetti, T.; Da Prato, L.; Truini, M.; Gambini, C.; Tonini, G.P.; et al. PHOX2A and PHOX2B genes are highly co-expressed in human neuroblastoma. Int. J. Oncol. 1992, 33, 985–991. [Google Scholar] [CrossRef]
- Trochet, D.; Bourdeaut, F.; Janoueix-Lerosey, I.; Deville, A.; de Pontual, L.; Schleiermacher, G.; Coze, C.; Philip, N.; Frébourg, T.; Munnich, A.; et al. Germline Mutations of the Paired–Like Homeobox 2B (PHOX2B) Gene in Neuroblastoma. Am. J. Hum. Genet. 2004, 74, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Di Lascio, S.; Bachetti, T.; Saba, E.; Ceccherini, I.; Benfante, R.; Fornasari, D. Transcriptional dysregulation and impairment of PHOX2B auto-regulatory mechanism induced by polyalanine expansion mutations associated with congenital central hypoventilation syndrome. Neurobiol. Dis. 2012, 50, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Di Lascio, S.; Benfante, R.; Di Zanni, E.; Cardani, S.; Adamo, A.; Fornasari, D.; Ceccherini, I.; Bachetti, T. Structural and functional differences inPHOX2Bframeshift mutations underlie isolated or syndromic congenital central hypoventilation syndrome. Hum. Mutat. 2017, 39, 219–236. [Google Scholar] [CrossRef] [PubMed]
- Revet, I.; Huizenga, G.; Chan, A.; Koster, J.; Volckmann, R.; van Sluis, P.; Øra, I.; Versteeg, R.; Geerts, D. The MSX1 homeobox transcription factor is a downstream target of PHOX2B and activates the Delta–Notch pathway in neuroblastoma. Exp. Cell Res. 2008, 314, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.-X.; Zhang, D.; Zhao, H.; Hu, R.; Dong, Z.; Yang, R.; Zhu, S.; Xia, Q.; Ding, H.-F.; Cui, H. Phox2B correlates with MYCN and is a prognostic marker for neuroblastoma development. Oncol. Lett. 2015, 9, 2507–2514. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reiff, T.; Tsarovina, K.; Majdazari, A.; Schmidt, M.; del Pino, I.; Rohrer, H. Neuroblastoma Phox2b Variants Stimulate Proliferation and Dedifferentiation of Immature Sympathetic Neurons. J. Neurosci. 2010, 30, 905–915. [Google Scholar] [CrossRef]
- Naftali, O.; Maman, S.; Meshel, T.; Sagi-Assif, O.; Ginat, R.; Witz, I.P. PHOX2B is a suppressor of neuroblastoma metastasis. Oncotarget 2016, 7, 10627–10637. [Google Scholar] [CrossRef] [PubMed]
- Maman, S.; Edry-Botzer, L.; Sagi-Assif, O.; Meshel, T.; Yuan, W.; Lu, W.; Witz, I.P. The metastatic microenvironment: Lung-derived factors control the viability of neuroblastoma lung metastasis. Int. J. Cancer 2013, 133, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- de Pontual, L.; Trochet, D.; Bourdeaut, F.; Thomas, S.; Etchevers, H.; Chompret, A.; Minard, V.; Valteau, D.; Brugieres, L.; Munnich, A.; et al. Methylation-associated PHOX2B gene silencing is a rare event in human neuroblastoma. Eur. J. Cancer 2007, 43, 2366–2372. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iyer, N.G.; Özdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [PubMed]
- Yerra, V.G.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Sedighi, M.; Baluchnejadmojarad, T.; Afshin-Majd, S.; Amiri, M.; Aminzade, M.; Roghani, M. Anti-aging Klotho Protects SH-SY5Y Cells Against Amyloid β1–42 Neurotoxicity: Involvement of Wnt1/pCREB/Nrf2/HO-1 Signaling. J. Mol. Neurosci. 2020, 71, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Alvariño, R.; Alonso, E.; Tabudravu, J.N.; Pérez-Fuentes, N.; Alfonso, A.; Botana, L.M. Tavarua Deoxyriboside A and Jasplakinolide as Potential Neuroprotective Agents: Effects on Cellular Models of Oxidative Stress and Neuroinflammation. ACS Chem. Neurosci. 2020, 12, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Jaafaru, M.S.; Nordin, N.; Rosli, R.; Shaari, K.; Bako, H.Y.; Saad, N.; Noor, N.M.; Razis, A.F.A. Neuroprotective effects of glucomoringin-isothiocyanate against H2O2-Induced cytotoxicity in neuroblastoma (SH-SY5Y) cells. NeuroToxicology 2019, 75, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Holla, V.R.; Elamin, Y.Y.; Bailey, A.M.; Johnson, A.M.; Litzenburger, B.C.; Khotskaya, Y.B.; Sanchez, N.S.; Zeng, J.; Shufean, A.; Shaw, K.R.; et al. ALK: A tyrosine kinase target for cancer therapy. Mol. Case Stud. 2017, 3, a001115. [Google Scholar] [CrossRef] [PubMed]
- Nakagawara, A.; Li, Y.; Izumi, H.; Muramori, K.; Inada, H.; Nishi, M. Neuroblastoma. Jpn. J. Clin. Oncol. 2018, 48, 214–241. [Google Scholar] [CrossRef]
- Heukamp, L.C.; Thor, T.; Schramm, A.; De Preter, K.; Kumps, C.; De Wilde, B.; Odersky, A.; Peifer, M.; Lindner, S.; Spruessel, A.; et al. Targeted Expression of Mutated ALK Induces Neuroblastoma in Transgenic Mice. Sci. Transl. Med. 2012, 4, 141ra91. [Google Scholar] [CrossRef]
- Lopez-Delisle, L.; Pierre-Eugène, C.; Louis-Brennetot, C.; Surdez, D.; Raynal, V.; Baulande, S.; Boeva, V.; Grossetête-Lalami, S.; Combaret, V.; Peuchmaur, M.; et al. Activated ALK signals through the ERK–ETV5–RET pathway to drive neuroblastoma oncogenesis. Oncogene 2018, 37, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Cage, T.A.; Chanthery, Y.; Chesler, L.; Grimmer, M.; Knight, Z.; Shokat, K.; Weiss, W.A.; Gustafson, W.C. Downregulation of MYCN through PI3K Inhibition in Mouse Models of Pediatric Neural Cancer. Front. Oncol. 2015, 5, 111. [Google Scholar] [CrossRef]
- Guo, Y.; Guo, D.; Zhang, S.; Zhang, Y.; He, X.; Jiang, X.; Chan, A.M.-L.; Zou, L.; Sun, J.; Zhao, H. Inhibition of PI3 kinase isoform p110α suppresses neuroblastoma growth and induces the reduction of Anaplastic Lymphoma Kinase. Cell Biosci. 2022, 12, 1–15. [Google Scholar] [CrossRef]
- Opel, D.; Poremba, C.; Simon, T.; Debatin, K.-M.; Fulda, S. Activation of Akt Predicts Poor Outcome in Neuroblastoma. Cancer Res. 2007, 67, 735–745. [Google Scholar] [CrossRef]
- Heiss, A.; Ammer, H.; Eisinger, D.A. δ-Opioid receptor-stimulated Akt signaling in neuroblastoma×glioma (NG108-15) hybrid cells involves receptor tyrosine kinase-mediated PI3K activation. Exp. Cell Res. 2009, 315, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Mi, T.; Wu, X.; Wang, Z.; Zhang, Z.; Liu, J.; Wang, Z.; Wang, J.; Li, M.; Ren, C.; et al. BI-D1870 Induces Mitotic Dysfunction and Apoptosis in Neuroblastoma by Regulating the PI3K-Akt-mTORC1 Signal Axis. Cancers 2023, 15, 2023. [Google Scholar] [CrossRef]
- Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 2008, 124, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Di Zanni, E.; Fornasari, D.; Ravazzolo, R.; Ceccherini, I.; Bachetti, T. Identification of novel pathways and molecules able to down-regulate PHOX2B gene expression by in vitro drug screening approaches in neuroblastoma cells. Exp. Cell Res. 2015, 336, 43–57. [Google Scholar] [CrossRef]
- Tetri, L.H.; Kolla, V.; Golden, R.L.; Iyer, R.; Croucher, J.L.; Choi, J.; Macfarland, S.P.; Naraparaju, K.; Guan, P.; Nguyen, F.; et al. RET receptor expression and interaction with TRK receptors in neuroblastomas. Oncol. Rep. 2020, 44, 263–272. [Google Scholar] [CrossRef]
- Müller, C.M.; Haase, M.G.; Kemnitz, I.; Fitze, G. Genetic mosaicism of a frameshift mutation in the RET gene in a family with Hirschsprung disease. Gene 2014, 541, 51–54. [Google Scholar] [CrossRef]
- Lambertz, I.; Kumps, C.; Claeys, S.; Lindner, S.; Beckers, A.; Janssens, E.; Carter, D.R.; Cazes, A.; Cheung, B.B.; De Mariano, M.; et al. Upregulation of MAPK Negative Feedback Regulators and RET in Mutant ALK Neuroblastoma: Implications for Targeted Treatment. Clin. Cancer Res. 2015, 21, 3327–3339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Su, Z.-Y.; Khor, T.O.; Shu, L.; Kong, A.-N.T. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem. Pharmacol. 2013, 85, 1398–1404. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [Google Scholar] [CrossRef]
- Lombardo, S.D.; Presti, M.; Mangano, K.; Petralia, M.C.; Basile, M.S.; Libra, M.; Candido, S.; Fagone, P.; Mazzon, E.; Nicoletti, F.; et al. Prediction of PD-L1 Expression in Neuroblastoma via Computational Modeling. Brain Sci. 2019, 9, 221. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peggion, S.; Najem, S.; Kolman, J.P.; Reinshagen, K.; Pagerols Raluy, L. Revisiting Neuroblastoma: Nrf2, NF-κB and Phox2B as a Promising Network in Neuroblastoma. Curr. Issues Mol. Biol. 2024, 46, 3193-3208. https://doi.org/10.3390/cimb46040200
Peggion S, Najem S, Kolman JP, Reinshagen K, Pagerols Raluy L. Revisiting Neuroblastoma: Nrf2, NF-κB and Phox2B as a Promising Network in Neuroblastoma. Current Issues in Molecular Biology. 2024; 46(4):3193-3208. https://doi.org/10.3390/cimb46040200
Chicago/Turabian StylePeggion, Sara, Safiullah Najem, Jan Philipp Kolman, Konrad Reinshagen, and Laia Pagerols Raluy. 2024. "Revisiting Neuroblastoma: Nrf2, NF-κB and Phox2B as a Promising Network in Neuroblastoma" Current Issues in Molecular Biology 46, no. 4: 3193-3208. https://doi.org/10.3390/cimb46040200
APA StylePeggion, S., Najem, S., Kolman, J. P., Reinshagen, K., & Pagerols Raluy, L. (2024). Revisiting Neuroblastoma: Nrf2, NF-κB and Phox2B as a Promising Network in Neuroblastoma. Current Issues in Molecular Biology, 46(4), 3193-3208. https://doi.org/10.3390/cimb46040200