
Sudden Cardiac Death in the Young: State-of-the-Art Review in Molecular Autopsy


Abstract
1. Introduction
2. Sudden Cardiac Death in the Young
2.1. Epidemiology
2.2. Causes Hereditary Heart Diseases
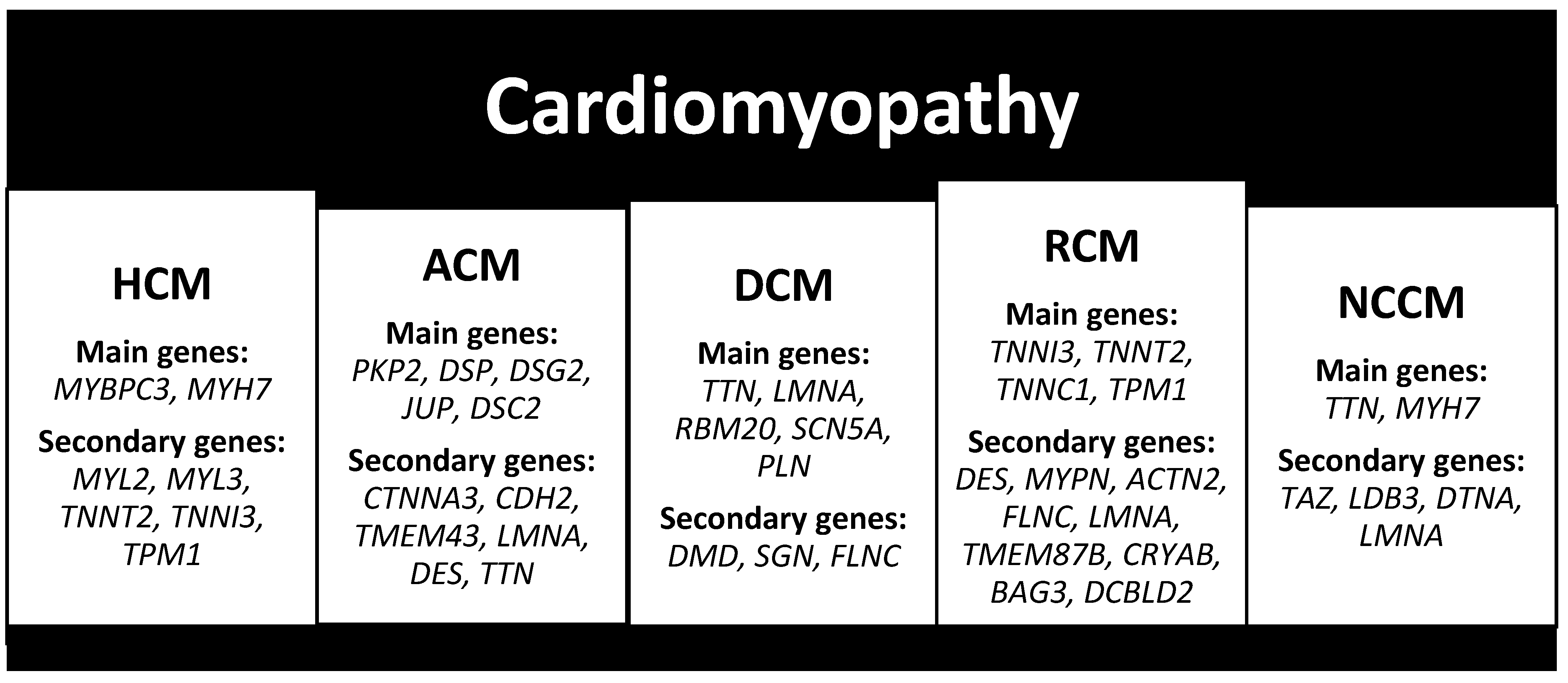
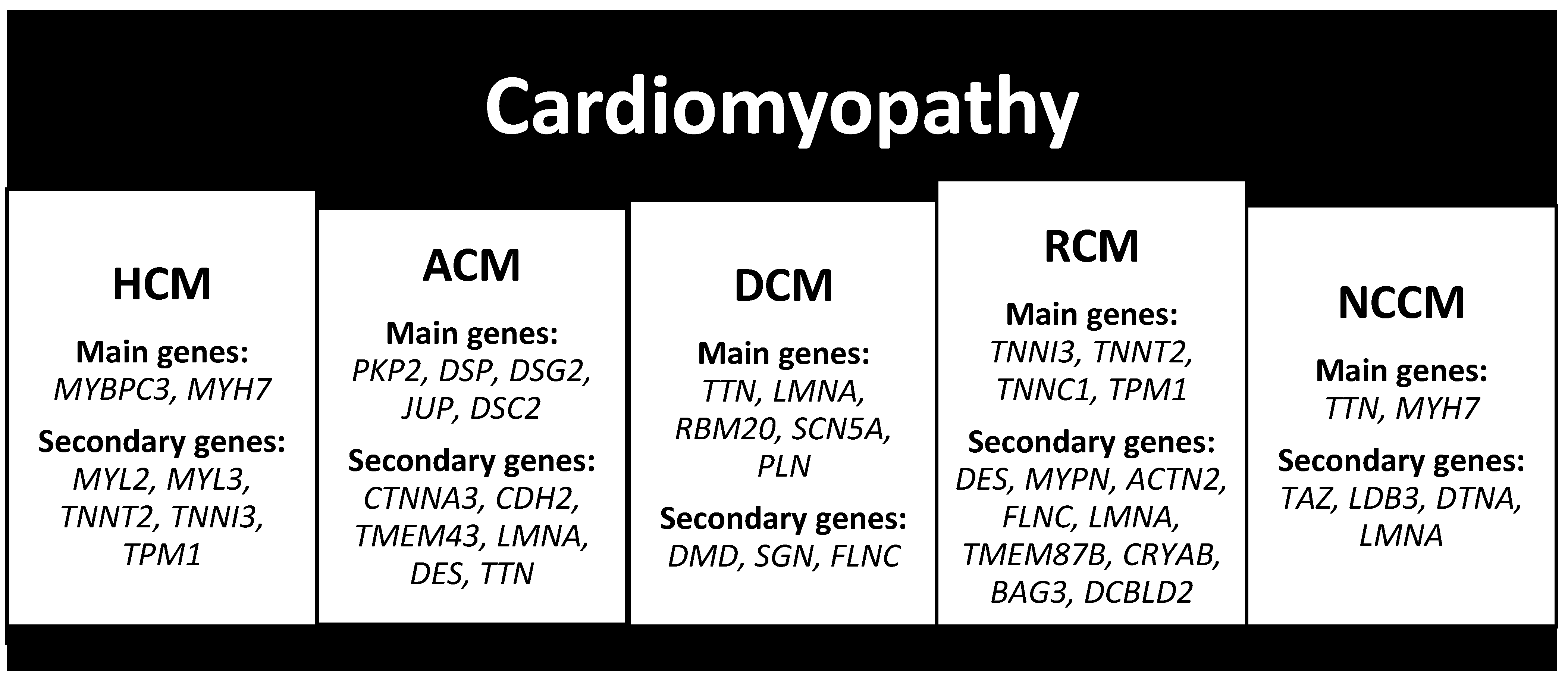
2.3. Cardiomyopathy
2.3.1. Hypertrophic Cardiomyopathy (HCM)
2.3.2. Arrhythmogenic Cardiomyopathy (ACM)
2.3.3. Dilated Cardiomyopathy (DCM)
2.3.4. Restrictive Cardiomyopathy (RCM)
2.3.5. Non-Compaction Cardiomyopathy (NCCM)
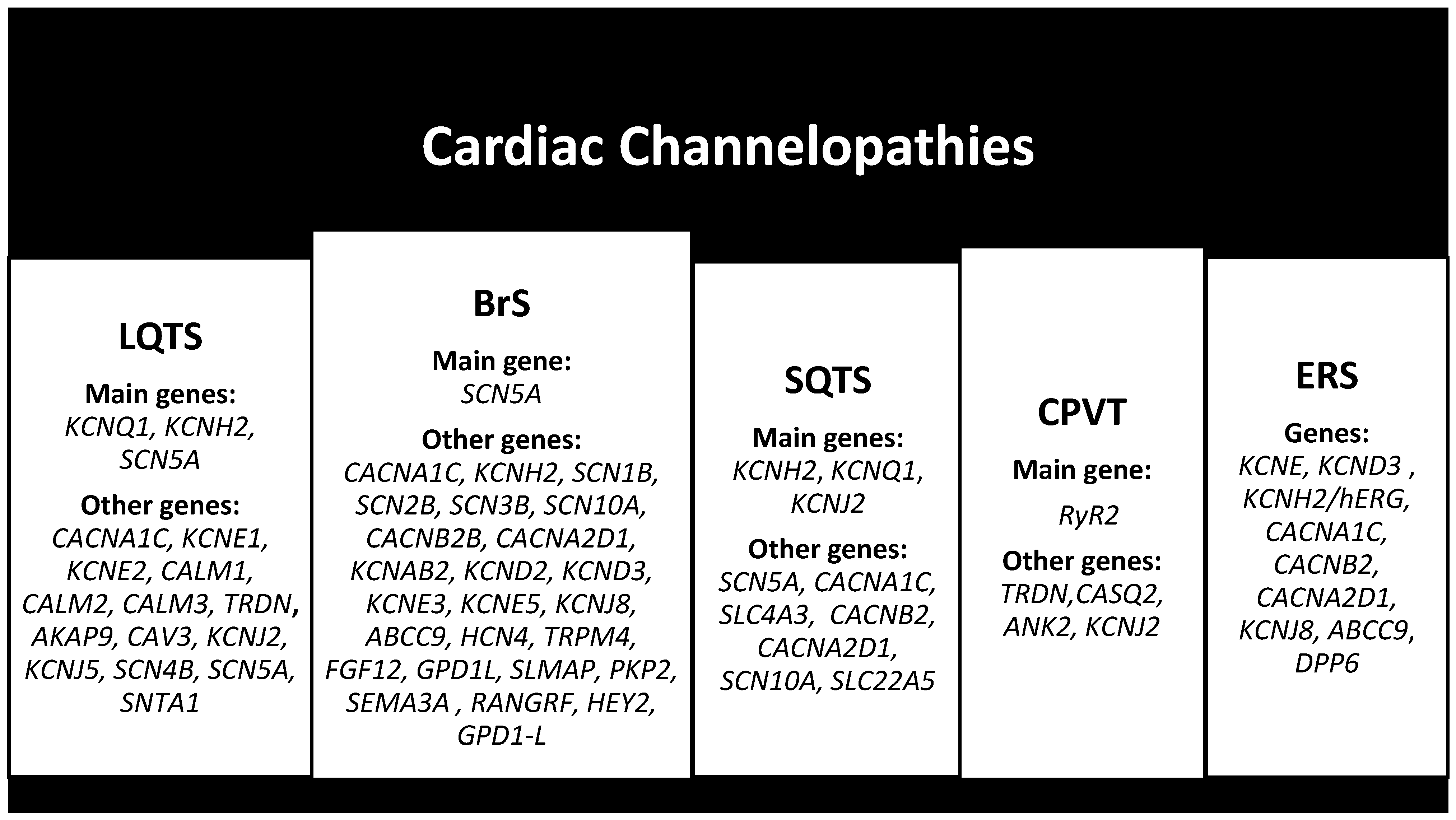
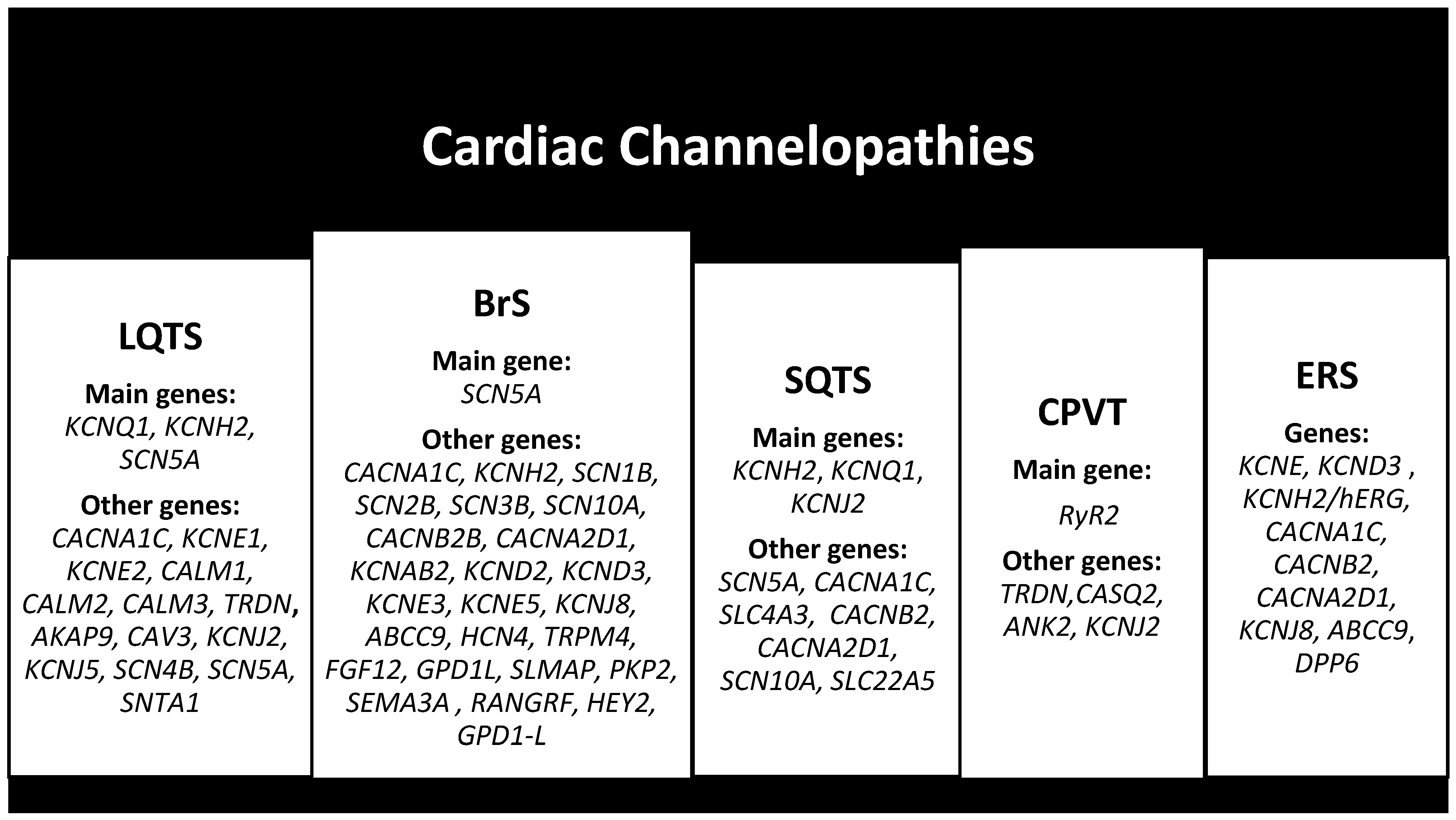
2.4. Cardiac Channelopathies
2.4.1. Long QT Syndrome (LQTS)
2.4.2. Brugada Syndrome (BrS)
2.4.3. Short QT Syndrome (SQTS)
2.4.4. Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)
2.4.5. Early Repolarization Syndrome (ERS)
3. Molecular Autopsy (MA)
4. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- De Gaspari, M.; Rizzo, S.; Thiene, G.; Basso, C. Causes of sudden death. Eur. Heart J. Suppl. 2023, 25 (Suppl. B), B16–B20. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Aguilera, B.; Banner, J.; Cohle, S.; d’Amati, G.; de Gouveia, R.H.; di Gioia, C.; Fabre, A.; Gallagher, P.J.; Leone, O.; et al. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows. Arch. 2017, 471, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G. Autopsy and sudden death. Eur. Heart J. Suppl. 2023, 25 (Suppl. C), C118–C129. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.L.; Lin, P.T.; Basso, C.; Bois, M.; Buja, L.M.; Cohle, S.D.; D’Amati, G.; Duncanson, E.; Fallon, J.T.; Firchau, D.; et al. Sudden cardiac death in the young: A consensus statement on recommended practices for cardiac examination by pathologists from the Society for Cardiovascular Pathology. Cardiovasc. Pathol. 2023, 63, 107497. [Google Scholar] [CrossRef] [PubMed]
- Abbas, R.; Abbas, A.; Khan, T.K.; Sharjeel, S.; Amanullah, K.; Irshad, Y. Sudden Cardiac Death in Young Individuals: A Current Review of Evaluation, Screening and Prevention. J. Clin. Med. Res. 2023, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.A.; Malhotra, A.; Mpondo, F.H.M.; Gupta, V.; Jain, R.; Gupta, S.; Jain, R. Sudden cardiac death in the adolescent population: A narrative review. Egypt. J. Intern. Med. 2023, 35, 36. [Google Scholar] [CrossRef]
- DeWitt, E.; Etheridge, S.P. Sudden Cardiac Death in Adolescents: Allowing the Dead to Speak. J. Am. Coll. Cardiol. 2023, 81, 1018–1020. [Google Scholar] [CrossRef]
- D’Ascenzi, F.; Valentini, F.; Pistoresi, S.; Frascaro, F.; Piu, P.; Cavigli, L.; Valente, S.; Focardi, M.; Cameli, M.; Bonifazi, M.; et al. Causes of sudden cardiac death in young athletes and non-athletes: Systematic review and meta-analysis: Sudden cardiac death in the young. Trends. Cardiovasc. Med. 2022, 32, 299–308. [Google Scholar] [CrossRef]
- Finocchiaro, G.; Westaby, J.; Sheppard, M.N.; Papadakis, M.; Sharma, S. Sudden Cardiac Death in Young Athletes: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2024, 83, 350–370. [Google Scholar] [CrossRef]
- Basso, C.; Fornes, P.; Gallagher, P.J.; de Gouveia, R.H.; Sheppard, M.; Thiene, G.; van der Wal, A.; on behalf of the Association for European Cardiovascular Pathology. Guidelines for autopsy investigation of sudden cardiac death. Virchows. Arch. 2008, 452, 11–18. [Google Scholar] [CrossRef]
- Markwerth, P.; Bajanowski, T.; Tzimas, I.; Dettmeyer, R. Sudden cardiac death-update. Int. J. Legal. Med. 2021, 135, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Shimizu, W.; Albert, C.M. The spectrum of epidemiology underlying sudden cardiac death. Circ. Res. 2015, 116, 1887–1906. [Google Scholar] [CrossRef] [PubMed]
- Banner, J.; Basso, C.; Tolkien, Z.; Kholova, I.; Michaud, K.; Gallagher, P.J. Autopsy examination in sudden cardiac death: A current perspective on behalf of the Association for European Cardiovascular Pathology. Virchows Arch. 2021, 478, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Isbister, J.; Semsarian, C. Sudden cardiac death: An update. Intern. Med. J. 2019, 49, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G. Sudden cardiac death in young people with apparently normal heart. Cardiovasc. Res. 2001, 50, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Risgaard, B. Sudden cardiac death: A nationwide cohort study among the young. Dan. Med. J. 2016, 63, B5321. [Google Scholar] [PubMed]
- Virmani, R.; Burke, A.P.; Farb, A. Sudden cardiac death. Cardiovasc. Pathol. 2001, 10, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N. Engl. J. Med. 2016, 374, 2441–2452. [Google Scholar] [CrossRef]
- Srinivasan, N.T.; Schilling, R.J. Sudden Cardiac Death and Arrhythmias. Arrhythm. Electrophysiol. Rev. 2018, 7, 111–117. [Google Scholar] [CrossRef]
- Papadakis, M.; Raju, H.; Behr, E.R.; De Noronha, S.V.; Spath, N.; Kouloubinis, A.; Sheppard, M.N.; Sharma, S. Sudden cardiac death with autopsy findings of uncertain significance: Potential for erroneous interpretation. Circ. Arrhythm. Electrophysiol. 2013, 6, 588–596. [Google Scholar] [CrossRef]
- Siontis, K.C.; Ommen, S.R.; Geske, J.B. Art and science of risk stratification of sudden cardiac death in hypertrophic cardiomyopathy: Current state, unknowns, and future directions. Prog. Cardiovasc. Dis. 2023, 80, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Basit, H.; Brito, D.; Sharma, S. Hypertrophic Cardiomyopathy. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Thakkar, K.; Karajgi, A.R.; Kallamvalappil, A.M.; Avanthika, C.; Jhaveri, S.; Shandilya, A.; Anusheel; Al-Masri, R. Sudden cardiac death in childhood hypertrophic cardiomyopathy. Dis. Mon. 2023, 69, 101548. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Q.; Guo, F.; Fu, L.; Dong, Y.; Xie, S.; Ding, X.; Hu, S.; Zhou, X.D.; Jiang, Y.; Zhou, H.; et al. 1-Deoxynojirimycin promotes cardiac function and rescues mitochondrial cristae in mitochondrial hypertrophic cardiomyopathy. J. Clin. Investig. 2023, 133, e164660. [Google Scholar] [CrossRef]
- De la Guía-Galipienso, F.; Ugedo-Alzaga, K.; Grazioli, G.; Quesada-Ocete, F.J.; Feliu-Rey, E.; Perez, M.V.; Quesada-Dorador, A.; Sanchis-Gomar, F. Arrhythmogenic Cardiomyopathy and Athletes: A Dangerous Relationship. Curr. Probl. Cardiol. 2023, 48, 101799. [Google Scholar] [CrossRef] [PubMed]
- Alblaihed, L.; Kositz, C.; Brady, W.J.; Al-Salamah, T.; Mattu, A. Diagnosis and management of arrhythmogenic right ventricular cardiomyopathy. Am. J. Emerg. Med. 2023, 65, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.J.; Morrissette-McAlmon, J.; Tung, L.; Boheler, K.R. Understanding Arrhythmogenic Cardiomyopathy: Advances through the Use of Human Pluripotent Stem Cell Models. Genes 2023, 14, 1864. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Marra, M.P.; Zorzi, A.; Beffagna, G.; Cipriani, A.; De Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Hustings, N.; Van Ballaer, V.; Barrios, L. Cardiac MRI after Sudden Cardiac Arrest in a Young Woman Prompts Diagnosis of Familial Dilated Cardiomyopathy. J. Belg. Soc. Radiol. 2023, 107, 21. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Eldemire, R.; Mestroni, L.; Taylor, M.R.G. Genetics of Dilated Cardiomyopathy. Annu. Rev. Med. 2024, 75, 417–426. [Google Scholar] [CrossRef]
- Tsatsopoulou, A.; Protonotarios, I.; Xylouri, Z.; Papagiannis, I.; Anastasakis, A.; Germanakis, I.; Patrianakos, A.; Nyktari, E.; Gavras, C.; Papadopoulos, G.; et al. Cardiomyopathies in children: An overview. Hellenic. J. Cardiol. 2023, 72, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Ditaranto, R.; Caponetti, A.G.; Ferrara, V.; Parisi, V.; Minnucci, M.; Chiti, C.; Baldassarre, R.; Di Nicola, F.; Bonetti, S.; Hasan, T.; et al. Pediatric Restrictive Cardiomyopathies. Front. Pediatr. 2022, 9, 745365. [Google Scholar] [CrossRef]
- Brodehl, A.; Gerull, B. Genetic Insights into Primary Restrictive Cardiomyopathy. J. Clin. Med. 2022, 11, 2094. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Yavari, M.; Al-Abcha, A.; Banga, S.; Abela, G. Ventricular non-compaction review. Heart Fail. Rev. 2022, 27, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Ader, F.; Roux, M.; Donal, E.; Eicher, J.; Aoutil, N.; Huttin, O.; Selton-Suty, C.; Coisne, D.; Jondeau, G.; et al. Targeted panel sequencing in adult patients with left ventricular non-compaction reveals a large genetic heterogeneity. Clin. Genet. 2019, 95, 356–367. [Google Scholar] [CrossRef]
- Sebastian, S.A.; Panthangi, V.; Sethi, Y.; Padda, I.; Khan, U.; Affas, Z.R.; Mareddy, C.; Dolack, L.; Johal, G. Precision Medicine and Cardiac Channelopathies: Human iPSCs Take the Lead. Curr. Probl. Cardiol. 2023, 48, 101990. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, J.; Song, M.; Yu, Y.; Geng, J.; Lin, D.; Yang, J.; Wu, J.; Li, K.; Yu, Y.; et al. Whole-exome sequencing and electrophysiological study reveal a novel loss-of-function mutation of KCNA10 in epinephrine provoked long QT syndrome with familial history of sudden cardiac death. Leg. Med. 2023, 62, 102245. [Google Scholar] [CrossRef]
- Pappone, C.; Ciconte, G.; Anastasia, L.; Gaita, F.; Grant, E.; Micaglio, E.; Locati, E.T.; Calovic, Z.; Vicedomini, G.; Santinelli, V. Right ventricular epicardial arrhythmogenic substrate in long-QT syndrome patients at risk of sudden death. Europace 2023, 25, 948–955. [Google Scholar] [CrossRef]
- Mahendran, S.; Gupta, I.; Davis, J.; Davis, A.J.; Orchard, J.W.; Orchard, J.J. Comparison of methods for correcting QT interval in athletes and young people: A systematic review. Clin. Cardiol. 2023, 46, 1106–1115. [Google Scholar] [CrossRef]
- Adler, A.; Novelli, V.; Amin, A.S.; Abiusi, E.; Care, M.; Nannenberg, E.A.; Feilotter, H.; Amenta, S.; Mazza, D.; Bikker, H.; et al. An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome. Circulation 2020, 141, 418–428. [Google Scholar] [CrossRef]
- Bauer, R.; Timothy, K.W.; Golden, A. Update on the Molecular Genetics of Timothy Syndrome. Front. Pediatr. 2021, 9, 668546. [Google Scholar] [CrossRef] [PubMed]
- Li, K.H.C.; Lee, S.; Yin, C.; Liu, T.; Ngarmukos, T.; Conte, G.; Yan, G.X.; Sy, R.W.; Letsas, K.P.; Tse, G.; et al. Brugada syndrome: A comprehensive review of pathophysiological mechanisms and risk stratification strategies. Int. J. Cardiol. Heart Vasc. 2020, 26, 100468. [Google Scholar] [CrossRef] [PubMed]
- Popa, I.P.; Șerban, D.N.; Mărănducă, M.A.; Șerban, I.L.; Tamba, B.I.; Tudorancea, I. Brugada Syndrome: From Molecular Mechanisms and Genetics to Risk Stratification. Int. J. Mol. Sci. 2023, 24, 3328. [Google Scholar] [CrossRef] [PubMed]
- Gaita, F.; Cerrato, N.; Giustetto, C.; Martino, A.; Bergamasco, L.; Millesimo, M.; Barbonaglia, L.; Carvalho, P.; Caponi, D.; Saglietto, A.; et al. Asymptomatic Patients with Brugada ECG Pattern: Long-Term Prognosis From a Large Prospective Study. Circulation 2023, 148, 1543–1555. [Google Scholar] [CrossRef] [PubMed]
- Moras, E.; Gandhi, K.; Narasimhan, B.; Brugada, R.; Brugada, J.; Brugada, P.; Krittanawong, C. Genetic and Molecular Mechanisms in Brugada Syndrome. Cells 2023, 12, 1791. [Google Scholar] [CrossRef] [PubMed]
- Pasero, E.; Gaita, F.; Randazzo, V.; Meynet, P.; Cannata, S.; Maury, P.; Giustetto, C. Artificial Intelligence ECG Analysis in Patients with Short QT Syndrome to Predict Life-Threatening Arrhythmic Events. Sensors 2023, 23, 8900. [Google Scholar] [CrossRef]
- Badura, K.; Buławska, D.; Dąbek, B.; Witkowska, A.; Lisińska, W.; Radzioch, E.; Skwira, S.; Młynarska, E.; Rysz, J.; Franczyk, B. Primary Electrical Heart Disease-Principles of Pathophysiology and Genetics. Int. J. Mol. Sci. 2024, 25, 1826. [Google Scholar] [CrossRef] [PubMed]
- Hancox, J.C.; Du, C.Y.; Butler, A.; Zhang, Y.; Dempsey, C.E.; Harmer, S.C.; Zhang, H. Pro-arrhythmic effects of gain-of-function potassium channel mutations in the short QT syndrome. Philos. Trans. R Soc. Lond. B Biol. Sci. 2023, 378, 20220165. [Google Scholar] [CrossRef] [PubMed]
- Henriquez, E.; A Hernandez, E.; Mundla, S.R.; Wankhade, D.H.; Saad, M.; Ketha, S.S.; Martinez, G.C.; Ahmed, F.S.; Hussain, M.S.; Hernandez, E.A.; et al. Catecholaminergic Polymorphic Ventricular Tachycardia and Gene Therapy: A Comprehensive Review of the Literature. Cureus 2023, 15, e47974. [Google Scholar] [CrossRef]
- Jurisic, S.; Medeiros-Domingo, A.; Berger, F.; Balmer, C.; Brunckhorst, C.; Ruschitzka, F.; Saguner, A.M.; Duru, F. Catecholaminergic Polymorphic Ventricular Tachycardia: Multiple Clinical Presentations of a Genetically Determined Disease. J. Clin. Med. 2023, 13, 47. [Google Scholar] [CrossRef]
- Zeppenfeld, K.; Tfelt-Hansen, J.; De Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar]
- Ji, H.Y.; Hu, N.; Liu, R.; Zhou, H.R.; Gao, W.L.; Quan, X.Q. Worldwide prevalence of early repolarization pattern in general population and physically active individuals: A meta-analysis. Medicine 2021, 100, e25978. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G. Molecular autopsy in sudden cardiac death. Glob. Cardiol. Sci. Pract. 2023, 2023, e202308. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Barrios, E.; Grassi, S.; Brión, M.; Toro, R.; Cesar, S.; Cruzalegui, J.; Coll, M.; Alcalde, M.; Brugada, R.; Greco, A.; et al. Molecular autopsy: Twenty years of post-mortem diagnosis in sudden cardiac death. Front. Med. 2023, 10, 1118585. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Barrios, E.; Cesar, S.; Cruzalegui, J.; Hernandez, C.; Arbelo, E.; Fiol, V.; Brugada, J.; Brugada, R.; Campuzano, O.; Sarquella-Brugada, G. Clinical Genetics of Inherited Arrhythmogenic Disease in the Pediatric Population. Biomedicines 2022, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.; Pérez-Serra, A.; Mates, J.; Del Olmo, B.; Puigmulé, M.; Fernandez-Falgueras, A.; Iglesias, A.; Picó, F.; Lopez, L.; Brugada, R.; et al. Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death. Biology 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.; Puckelwartz, M.J.; Pesce, L.L.; Dellefave-Castillo, L.M.; Vanoye, C.G.; Potet, F.; Page, P.; Kearns, S.D.; Pottinger, T.; White, S.; et al. Genomic Autopsy of Sudden Deaths in Young Individuals. JAMA Cardiol. 2021, 6, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Goudal, A.; Karakachoff, M.; Lindenbaum, P.; Baron, E.; Bonnaud, S.; Kyndt, F.; Arnaud, M.; Minois, D.; Bourcereau, E.; Thollet, A.; et al. Burden of rare variants in arrhythmogenic cardiomyopathy with right dominant form-associated genes provides new insights for molecular diagnosis and clinical management. Hum. Mutat. 2022, 43, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Agustín, A.; Pinsach-Abuin, M.L.; Pagans, S. Role of Non-Coding Variants in Brugada Syndrome. Int. J. Mol. Sci. 2020, 21, 8556. [Google Scholar] [CrossRef]
- Mont Girbau, J.L.; Wilde, A.A.; Semsarian, C.; Márquez, M.F.; Deneke, T.; Kaufman, E.S. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace 2022, 24, 1307–1367. [Google Scholar]
- Isbister, J.C.; Nowak, N.; Yeates, L.; Singer, E.S.; Sy, R.W.; Ingles, J.; Raju, H.; Bagnall, R.D.; Semsarian, C. Concealed Cardiomyopathy in Autopsy-Inconclusive Cases of Sudden Cardiac Death and Implications for Families. J. Am. Coll. Cardiol. 2022, 80, 2057–2068. [Google Scholar] [CrossRef] [PubMed]
- Karra, R.; Nafissi, N.A. Expanding the Molecular Autopsy to Uncover Occult Cardiomyopathy: Concealed to Revealed. J. Am. Coll. Cardiol. 2022, 80, 2069–2071. [Google Scholar] [CrossRef] [PubMed]
- Stiles, M.K.; Wilde, A.A.; Abrams, D.J.; Ackerman, M.J.; Albert, C.M.; Behr, E.R.; Chugh, S.S.; Cornel, M.C.; Gardner, K.; Ingles, J.; et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. Heart Rhythm 2021, 18, e1–e50. [Google Scholar] [CrossRef]
- Behr, E.R.; Scrocco, C.; Wilde, A.A.M.; Marijon, E.; Crotti, L.; E Iliodromitis, K.; A Remme, C.; Kosiuk, J.; Rudaka, I.; Brugada, G.S.; et al. Investigation on Sudden Unexpected Death in the Young (SUDY) in Europe: Results of the European Heart Rhythm Association Survey. Europace 2022, 24, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Ingles, J.; Wilde, A.A. Sudden cardiac death in the young: The molecular autopsy and a practical approach to surviving relatives. Eur. Heart J. 2015, 36, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.; Fernandez-Falgueras, A.; Iglesias, A.; del Olmo, B.; Nogue-Navarro, L.; Simon, A.; Serra, A.P.; Puigmule, M.; Lopez, L.; Pico, F.; et al. Unpredicted Aberrant Splicing Products Identified in Postmortem Sudden Cardiac Death Samples. Int. J. Mol. Sci. 2022, 23, 12640. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Barrios, E.; Sarquella-Brugada, G.; Perez-Serra, A.; Fernandez-Falgueras, A.; Cesar, S.; Alcalde, M.; Coll, M.; Puigmulé, M.; Iglesias, A.; Ferrer-Costa, C.; et al. Reevaluation of ambiguous genetic variants in sudden unexplained deaths of a young cohort. Int. J. Legal. Med. 2023, 137, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G.; Fernandez-Falgueras, A.; Coll, M.; Iglesias, A.; Ferrer-Costa, C.; Cesar, S.; Arbelo, E.; García-Álvarez, A.; Jordà, P.; et al. Reanalysis and reclassification of rare genetic variants associated with inherited arrhythmogenic syndromes. EBioMedicine 2020, 54, 102732. [Google Scholar] [CrossRef]
- Sarquella-Brugada, G.; Fernandez-Falgueras, A.; Cesar, S.; Arbelo, E.; Coll, M.; Perez-Serra, A.; Puigmulé, M.; Iglesias, A.; Alcalde, M.; Vallverdú-Prats, M.; et al. Clinical impact of rare variants associated with inherited channelopathies: A 5-year update. Hum. Genet. 2022, 141, 1579–1589. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Cardiac Channelopathies | Recommended Gene Mutations | Suspected |
|---|---|---|
| LQTS | KCNQ1, KCNH2, SCN5A (80%) | Young died suddenly during physical exercise or swimming or sleep |
| BrS | SCN5A (30%) | Young died suddenly at night |
| SQTS | KCNQ1, KCNJ2, KCNH2 (45%) | Newborn died suddenly |
| CPVT | RyR2 (55%) | Young died suddenly during adrenergic stress. |
| Cardiomyopathies | Most Frequent Gene Mutations |
|---|---|
| HCM | MYBPC3 (40–45%), MYH7 (15–25%) |
| ACM | PKP2 (10–45%), DSP (10–15%), DSG2 (10%), JUP and DSC2 (1–2%) |
| DCM | TTN (20–25%), LMNA (8%), RBM20 (1–5%), SCN5A (2–3%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salzillo, C.; Sansone, V.; Napolitano, F. Sudden Cardiac Death in the Young: State-of-the-Art Review in Molecular Autopsy. Curr. Issues Mol. Biol. 2024, 46, 3313-3327. https://doi.org/10.3390/cimb46040207
Salzillo C, Sansone V, Napolitano F. Sudden Cardiac Death in the Young: State-of-the-Art Review in Molecular Autopsy. Current Issues in Molecular Biology. 2024; 46(4):3313-3327. https://doi.org/10.3390/cimb46040207
Chicago/Turabian StyleSalzillo, Cecilia, Vincenza Sansone, and Francesco Napolitano. 2024. "Sudden Cardiac Death in the Young: State-of-the-Art Review in Molecular Autopsy" Current Issues in Molecular Biology 46, no. 4: 3313-3327. https://doi.org/10.3390/cimb46040207
APA StyleSalzillo, C., Sansone, V., & Napolitano, F. (2024). Sudden Cardiac Death in the Young: State-of-the-Art Review in Molecular Autopsy. Current Issues in Molecular Biology, 46(4), 3313-3327. https://doi.org/10.3390/cimb46040207