

A Computational Approach for the Discovery of Novel DNA Methyltransferase Inhibitors




Abstract
:1. Introduction
2. Materials and Methods
2.1. Pharmacophore Model Generation and Validation
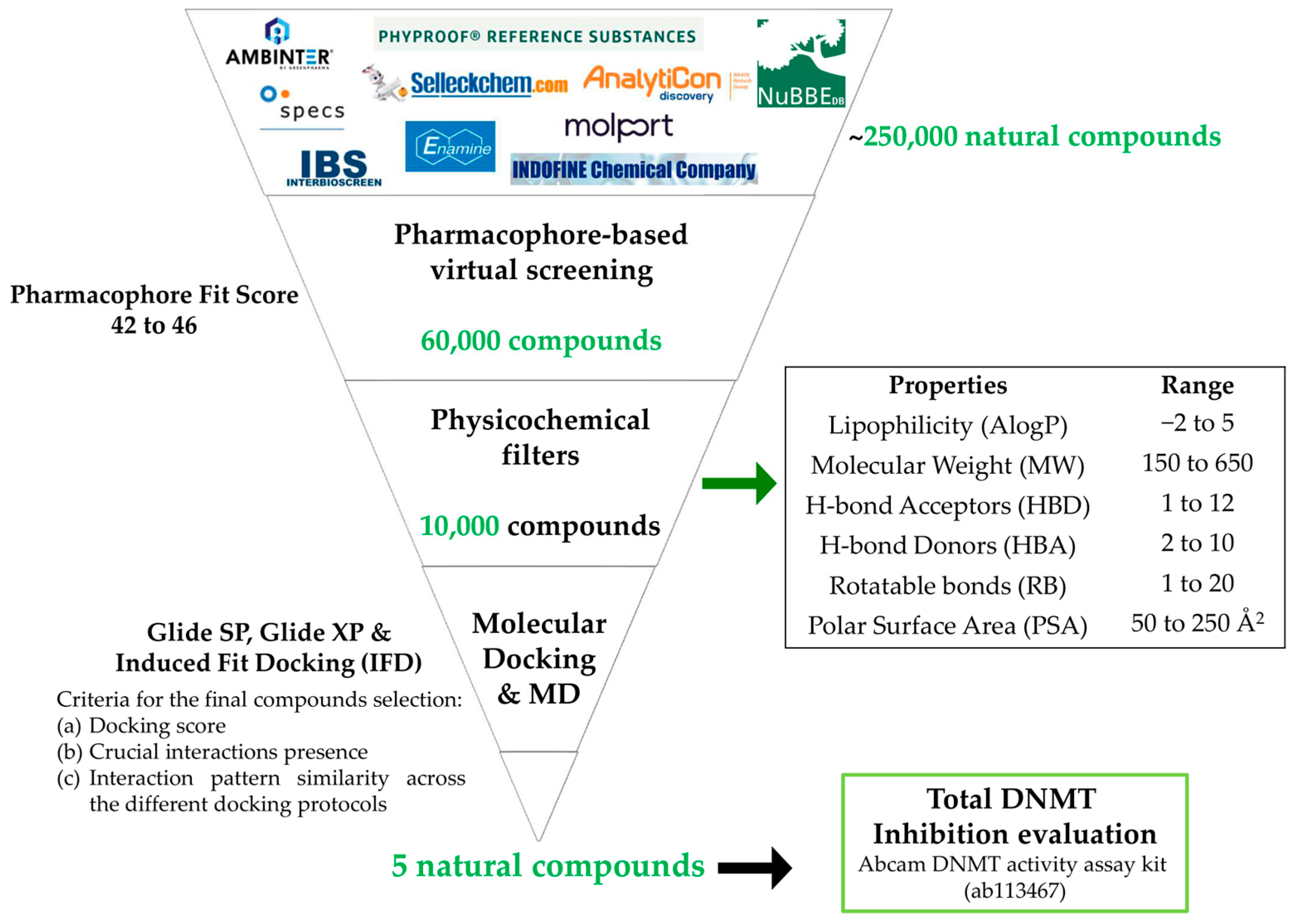
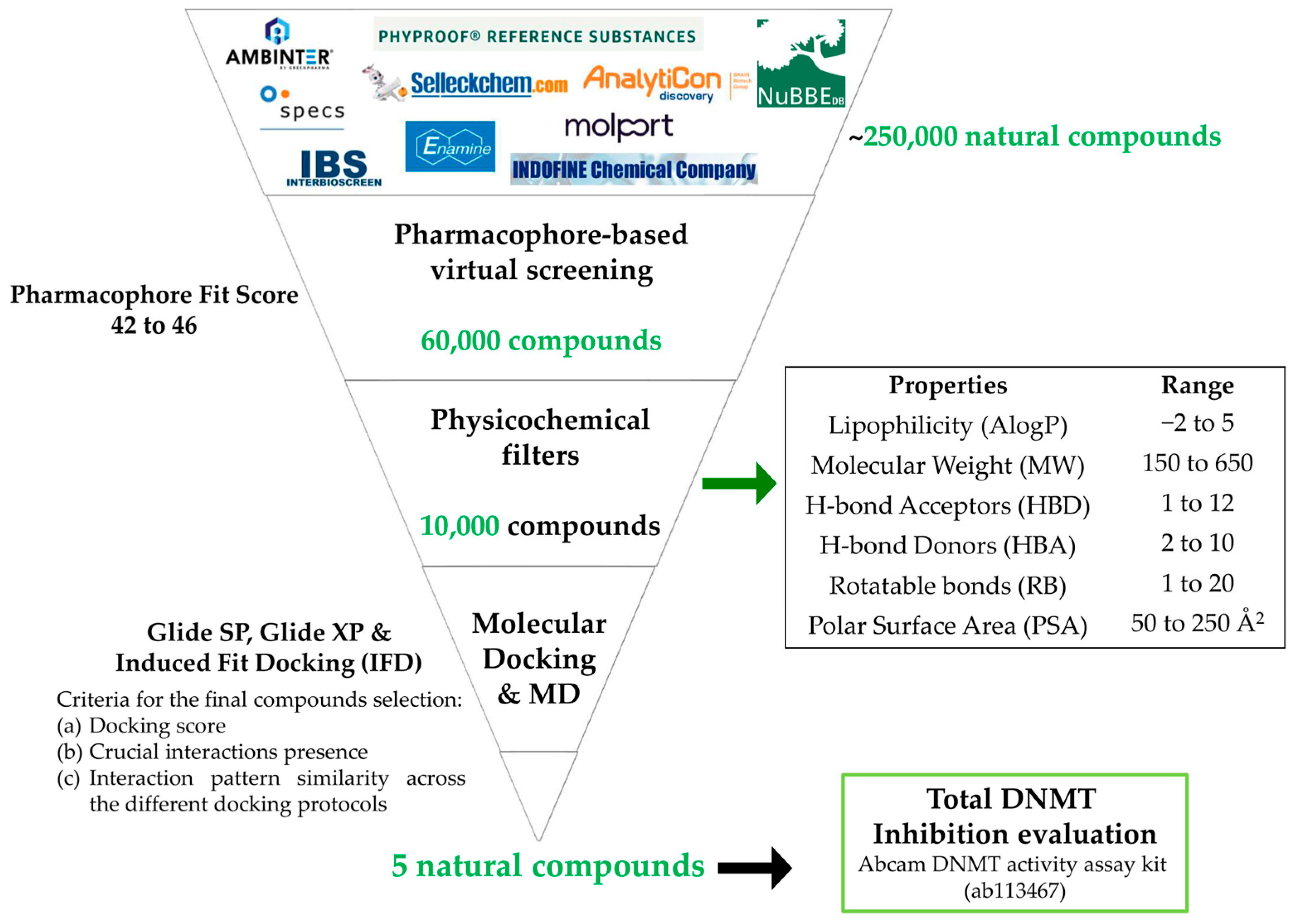
2.2. Virtual Screening
2.2.1. Pharmacophore-Based Virtual Screening
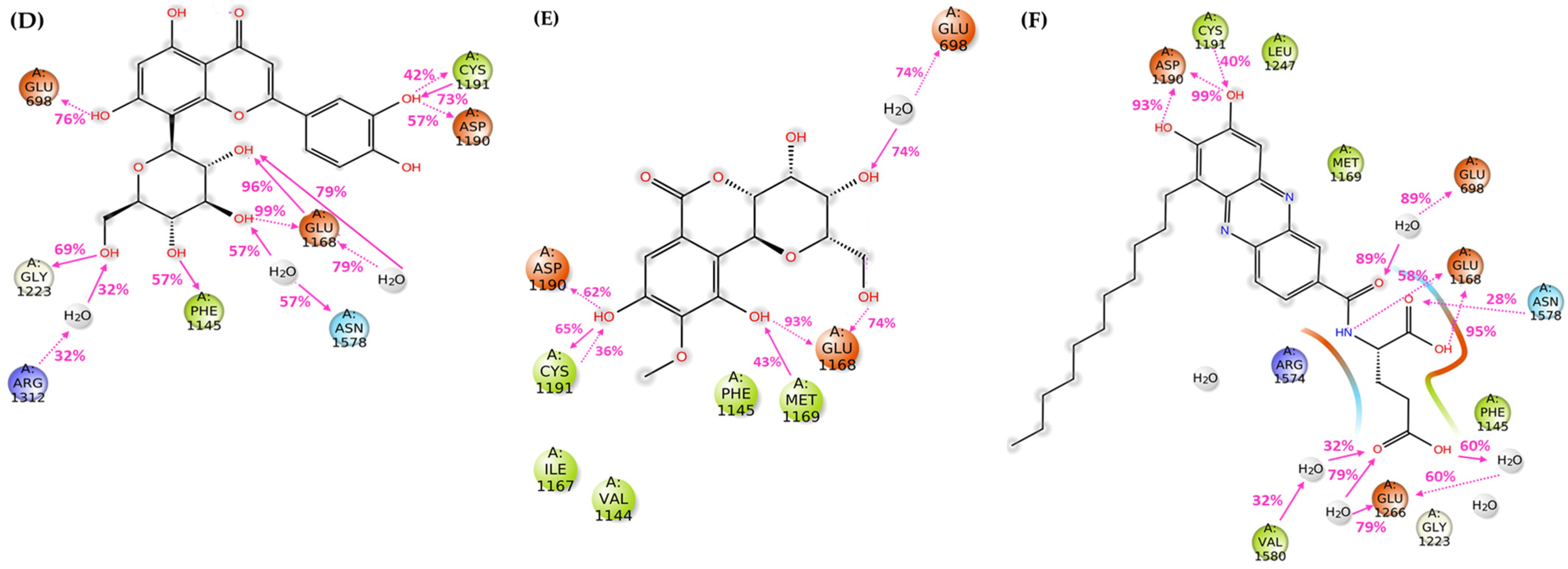
2.2.2. Molecular Docking Studies
2.2.3. Molecular Dynamics Simulations
2.2.4. ADMET Properties
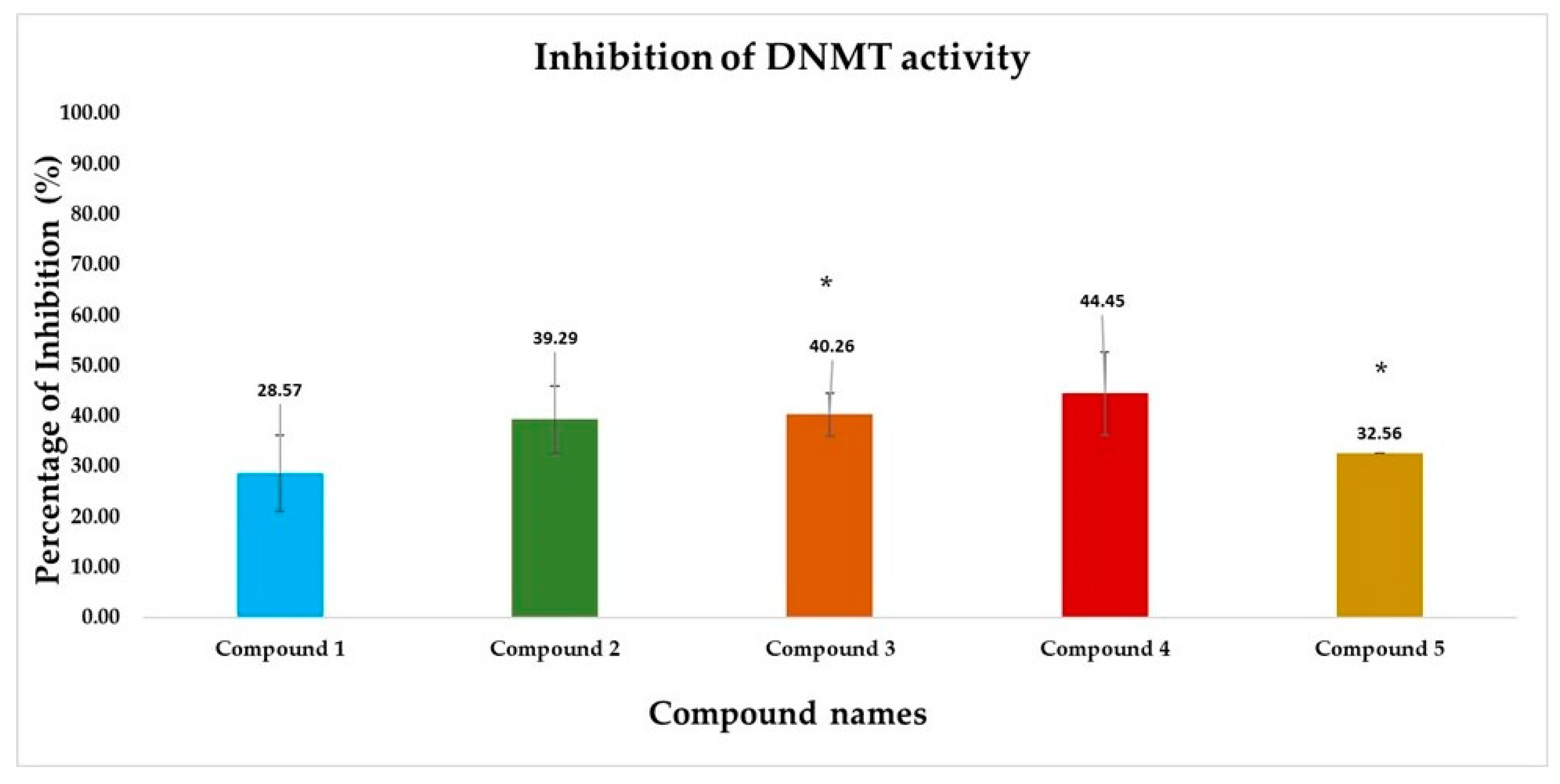
2.3. Total DNMT Inhibitory Activity Evaluation
DNMT Inhibition Measurements
3. Results
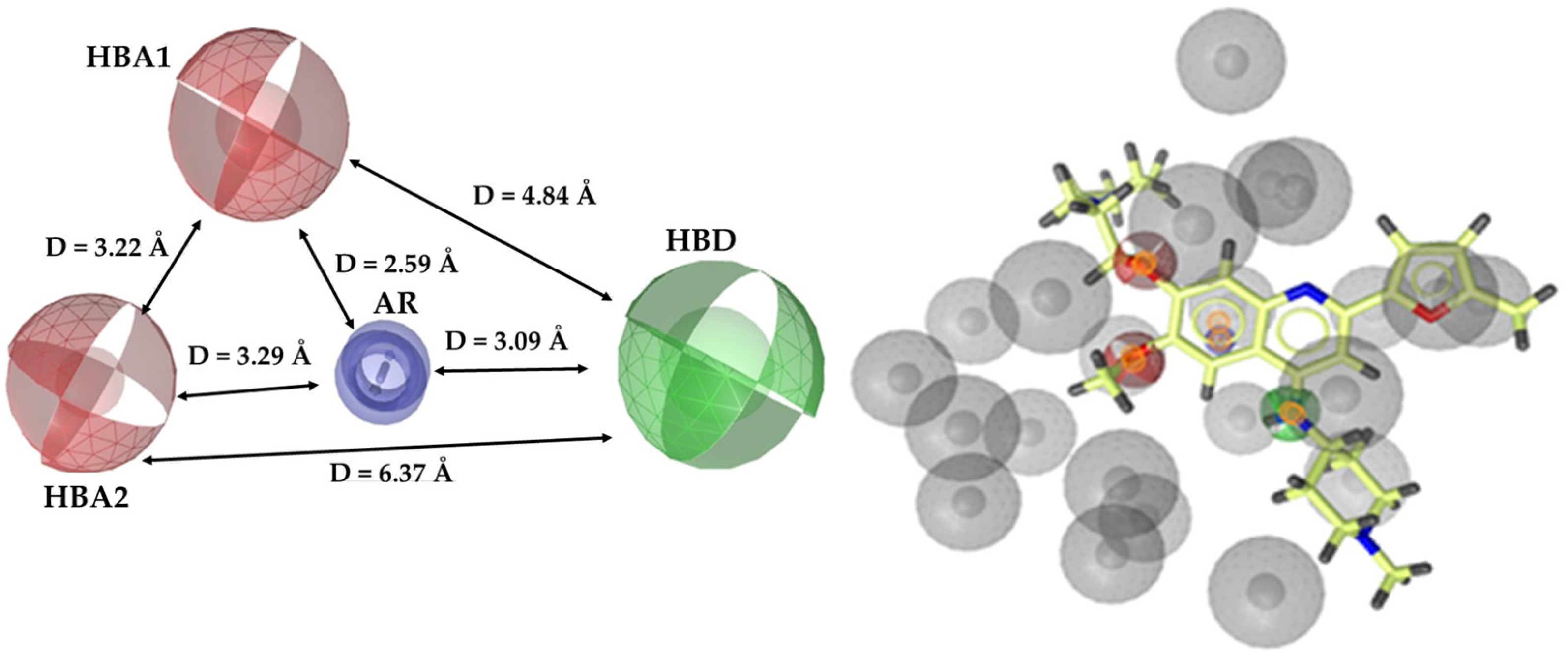
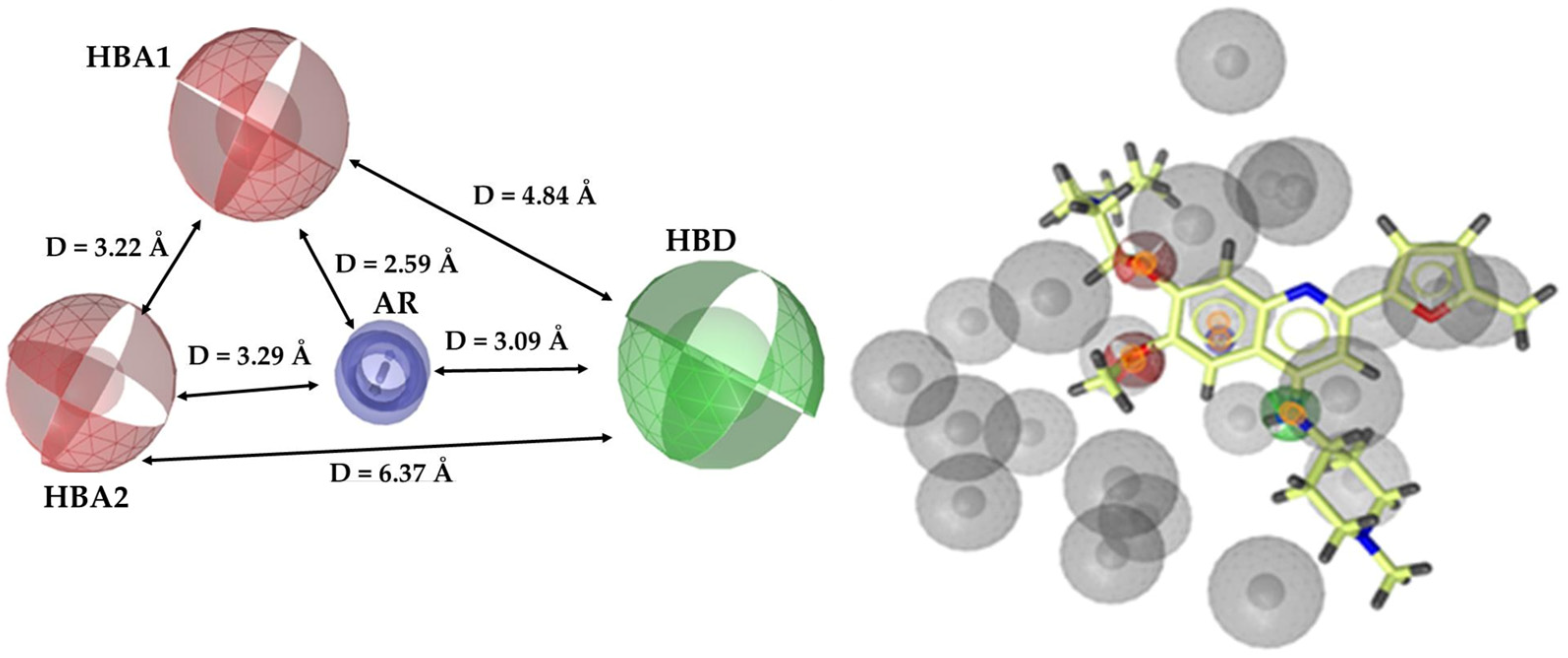
3.1. Pharmacophore Model
3.1.1. Ligand-Based Pharmacophore Model Generation
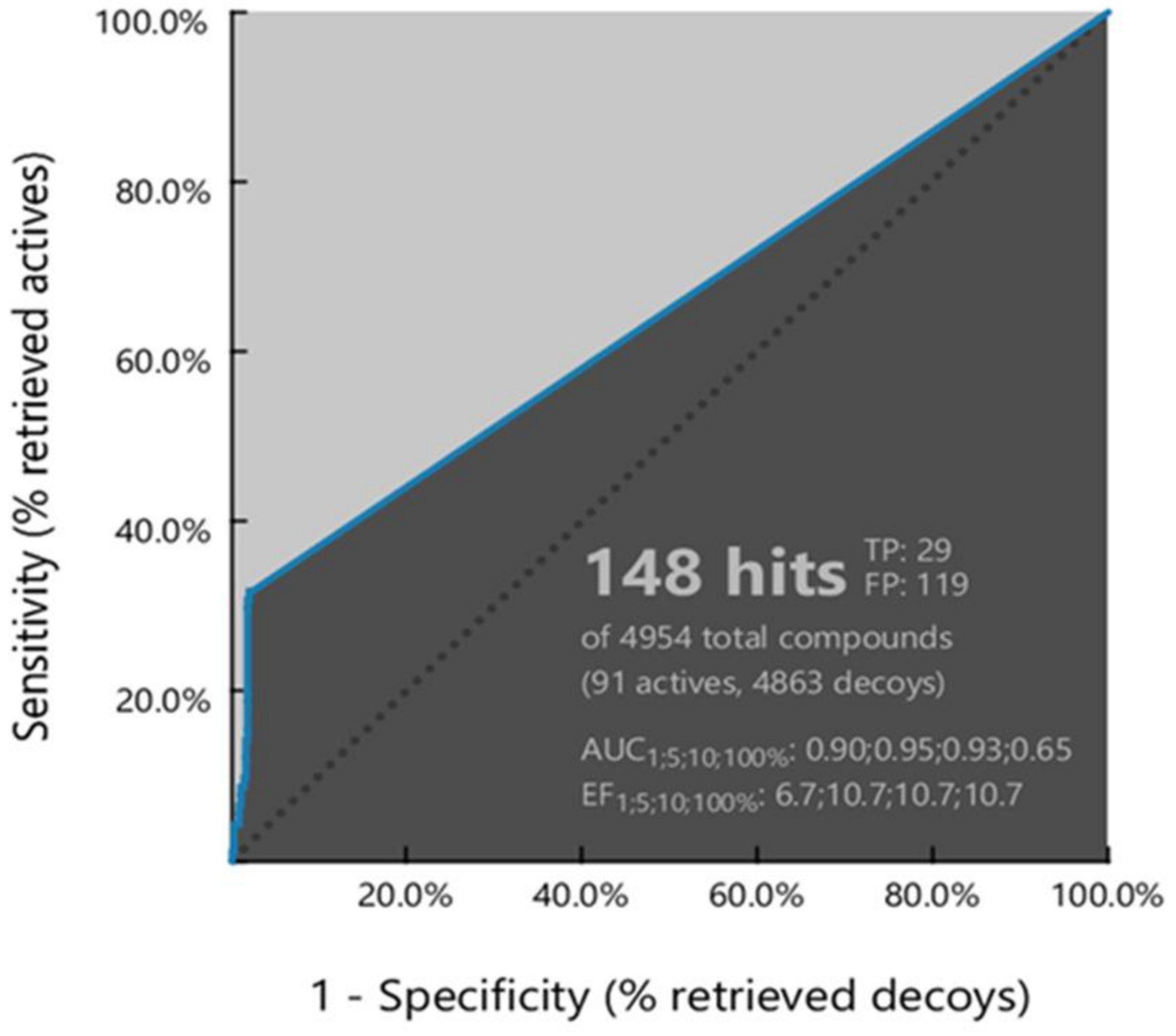
3.1.2. Ligand-Based Pharmacophore Model Validation
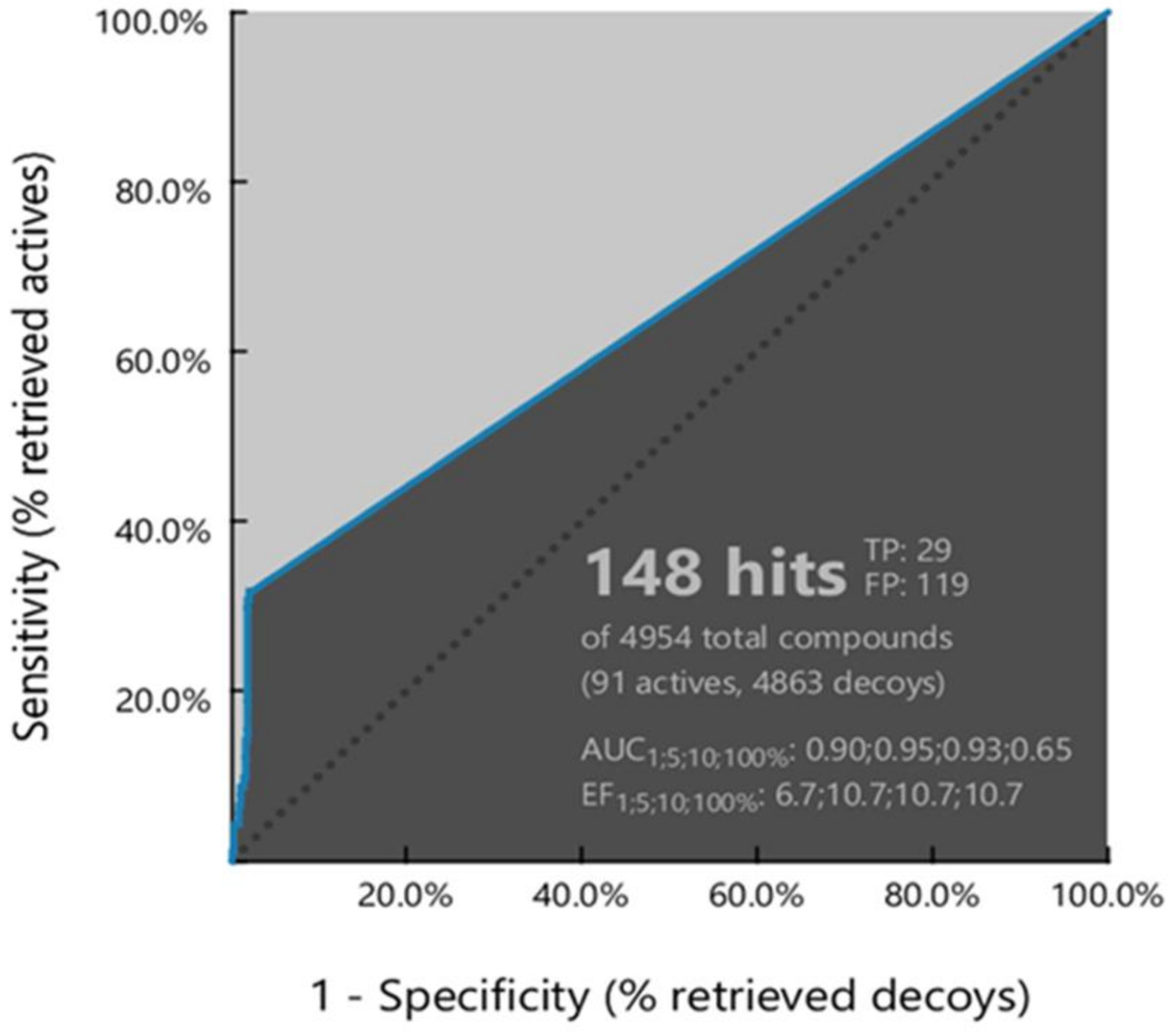
3.2. Virtual Screening (VS) Results
3.2.1. Pharmacophore-Based Virtual Screening
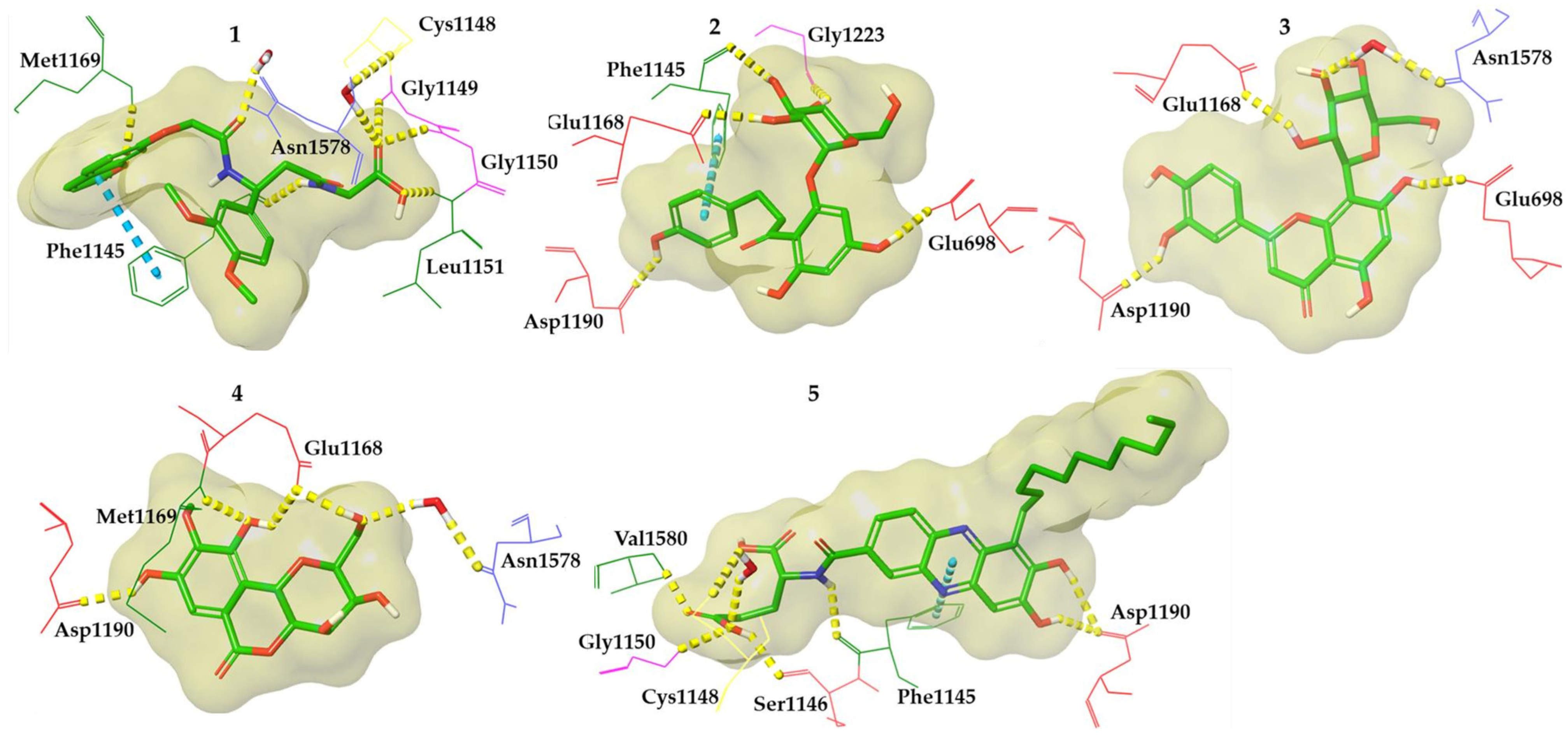
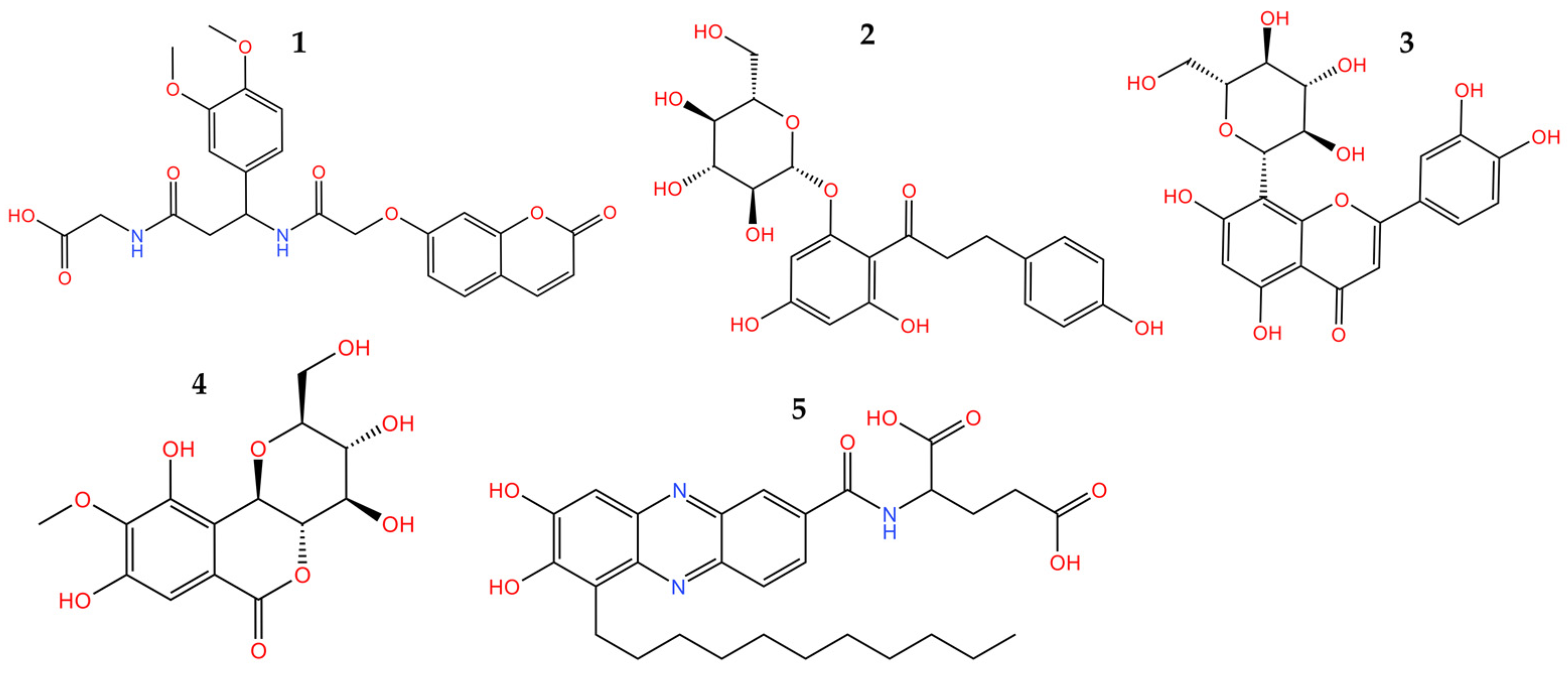
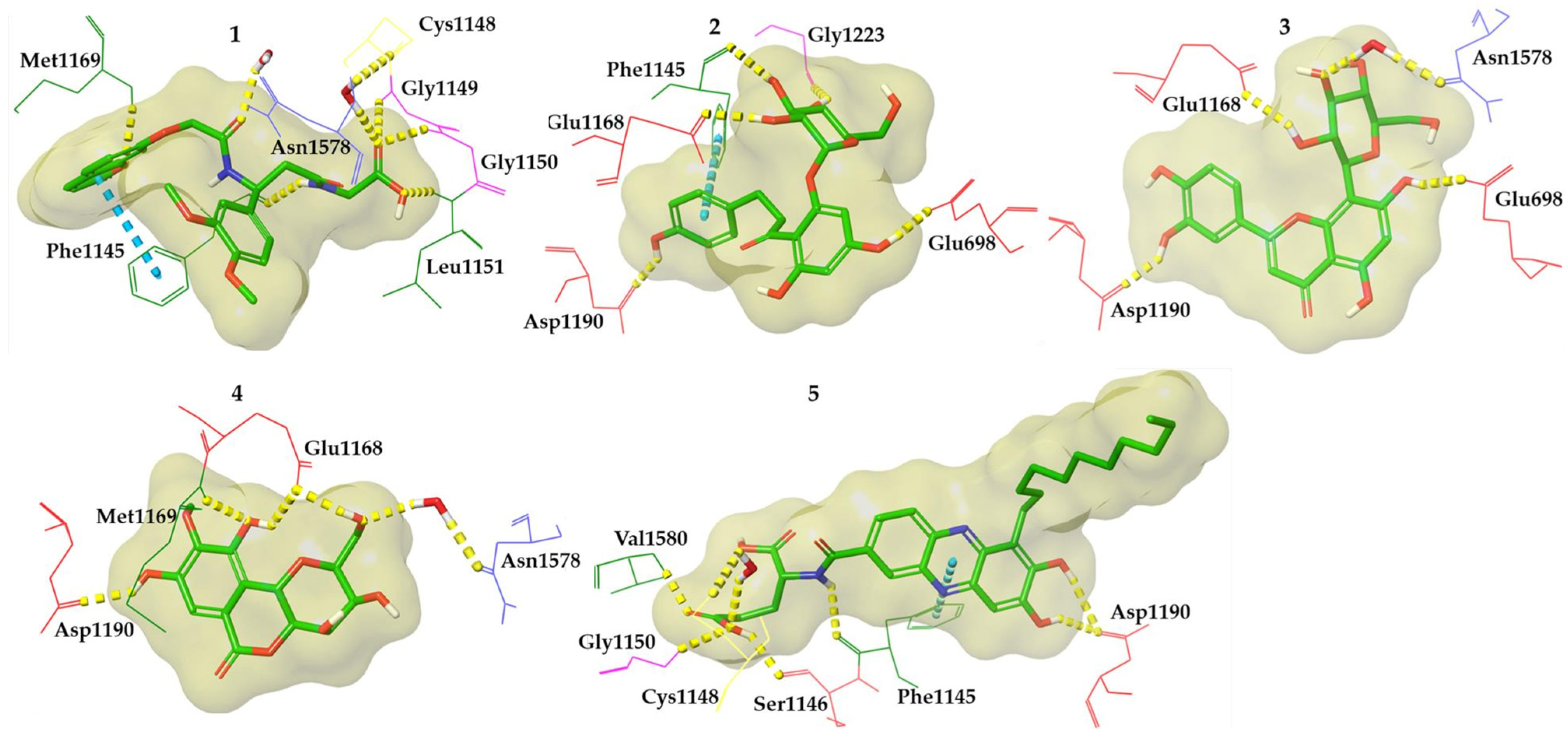
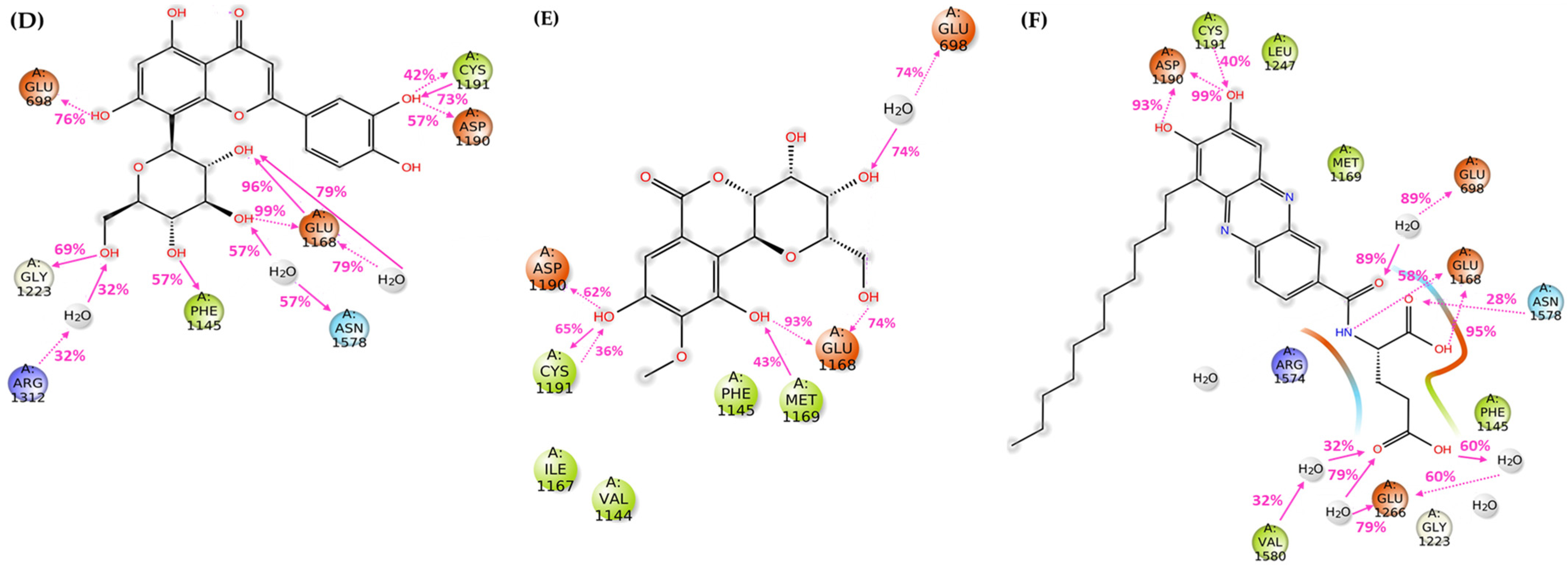
3.2.2. Molecular Docking
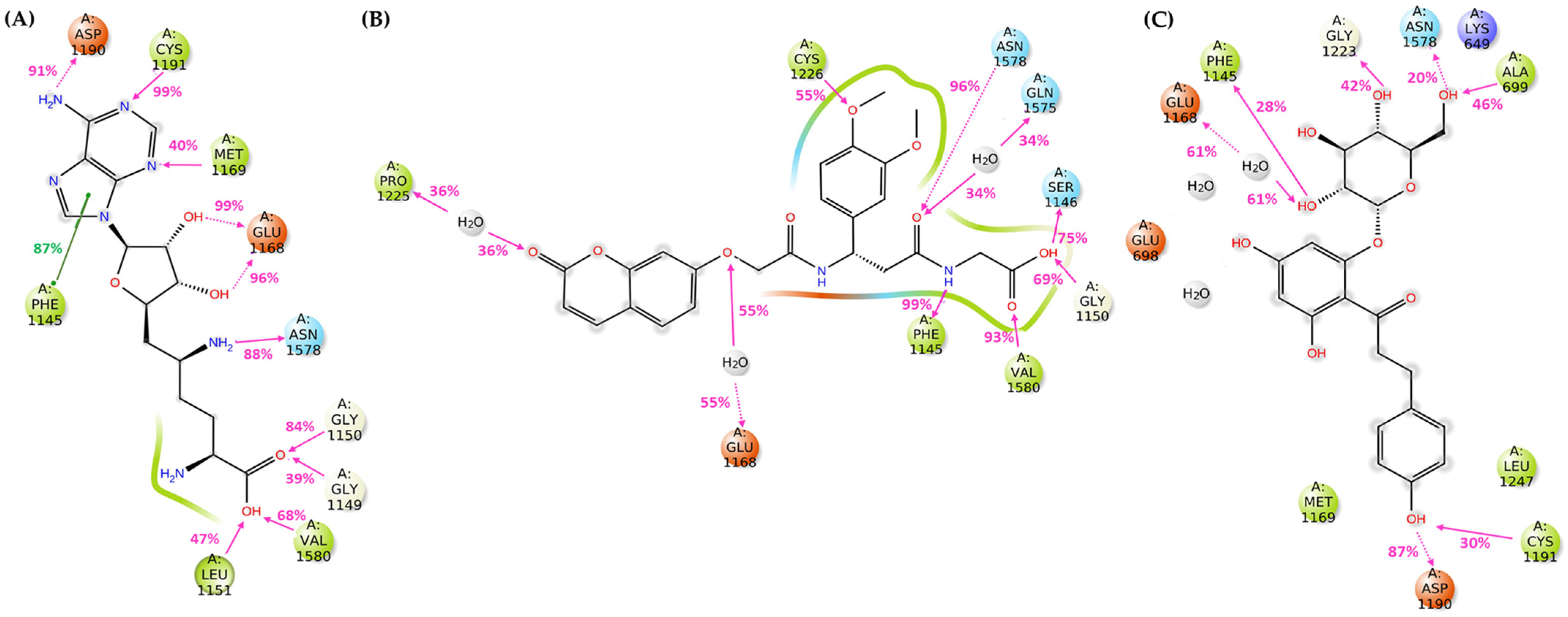
3.2.3. Molecular Dynamics Simulations
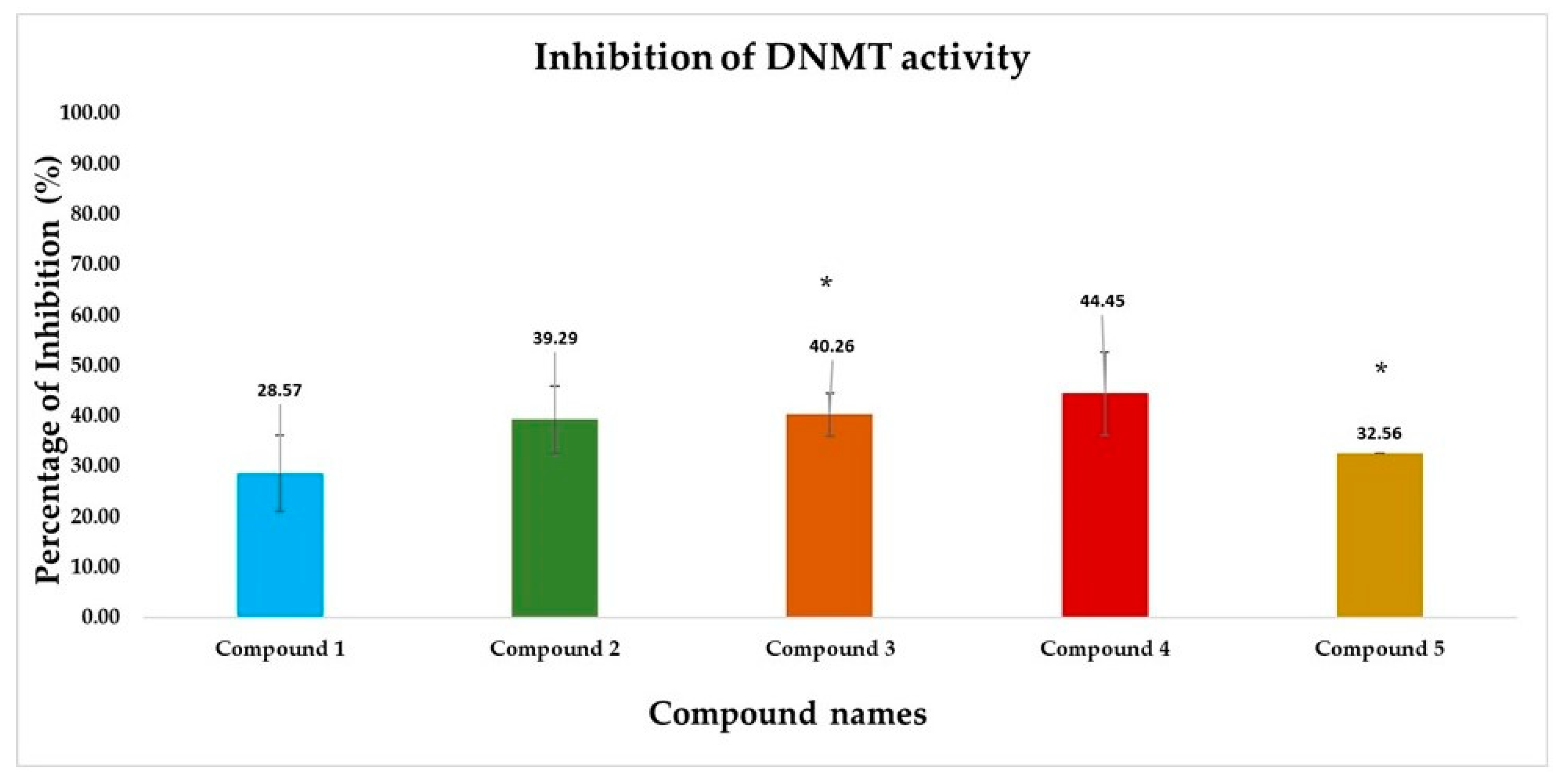
3.2.4. DNMT Inhibition Assay Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Sig Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-F.; Zhou, Y.-N.; Zhang, J.; Su, S.; Liu, Y.; Peng, G.-H.; Zang, W.; Cao, J. The Role of Epigenetic Methylation/Demethylation in the Regulation of Retinal Photoreceptors. Front. Cell Dev. Biol. 2023, 11, 1149132. [Google Scholar] [CrossRef] [PubMed]
- Farsetti, A.; Illi, B.; Gaetano, C. How Epigenetics Impacts on Human Diseases. Eur. J. Intern. Med. 2023, 114, 15–22. [Google Scholar] [CrossRef] [PubMed]
- La Torre, A.; Lo Vecchio, F.; Greco, A. Epigenetic Mechanisms of Aging and Aging-Associated Diseases. Cells 2023, 12, 1163. [Google Scholar] [CrossRef] [PubMed]
- Sahafnejad, Z.; Ramazi, S.; Allahverdi, A. An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review. Genes 2023, 14, 873. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Liou, J.-P. Recent Developments in Epigenetic Cancer Therapeutics: Clinical Advancement and Emerging Trends. J. Biomed. Sci. 2021, 28, 27. [Google Scholar] [CrossRef] [PubMed]
- Babar, Q.; Saeed, A.; Tabish, T.A.; Pricl, S.; Townley, H.; Thorat, N. Novel Epigenetic Therapeutic Strategies and Targets in Cancer. Biochim. Et Biophys. Acta BBA Mol. Basis Dis. 2022, 1868, 166552. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Zhou, Q.; Wen, X.; Wang, R.; Liu, R.; Wang, T.; Shi, J.; Hu, Y.; Hou, J. Small-Molecule Inhibitors Overcome Epigenetic Reprogramming for Cancer Therapy. Front. Pharmacol. 2021, 12, 702360. [Google Scholar] [CrossRef] [PubMed]
- Rabal, O.; San José-Enériz, E.; Agirre, X.; Sánchez-Arias, J.A.; De Miguel, I.; Ordoñez, R.; Garate, L.; Miranda, E.; Sáez, E.; Vilas-Zornoza, A.; et al. Design and Synthesis of Novel Epigenetic Inhibitors Targeting Histone Deacetylases, DNA Methyltransferase 1, and Lysine Methyltransferase G9a with In Vivo Efficacy in Multiple Myeloma. J. Med. Chem. 2021, 64, 3392–3426. [Google Scholar] [CrossRef] [PubMed]
- Gul, S. Epigenetic Assays for Chemical Biology and Drug Discovery. Clin. Epigenet 2017, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Sadybekov, A.V.; Katritch, V. Computational Approaches Streamlining Drug Discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef]
- Sessions, Z.; Sánchez-Cruz, N.; Prieto-Martínez, F.D.; Alves, V.M.; Santos, H.P.; Muratov, E.; Tropsha, A.; Medina-Franco, J.L. Recent Progress on Cheminformatics Approaches to Epigenetic Drug Discovery. Drug Discov. Today 2020, 25, 2268–2276. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, G.; Li, Y.; Lei, D.; Xiang, J.; Ouyang, L.; Wang, Y.; Yang, J. Recent Progress in DNA Methyltransferase Inhibitors as Anticancer Agents. Front. Pharmacol. 2022, 13, 1072651. [Google Scholar] [CrossRef]
- Prado-Romero, D.L.; Gómez-García, A.; Cedillo-González, R.; Villegas-Quintero, H.; Avellaneda-Tamayo, J.F.; López-López, E.; Saldívar-González, F.I.; Chávez-Hernández, A.L.; Medina-Franco, J.L. Consensus Docking Aid to Model the Activity of an Inhibitor of DNA Methyltransferase 1 Inspired by de Novo Design. Front. Drug Discov. 2023, 3, 1261094. [Google Scholar] [CrossRef]
- Oliveira, T.; Silva, M.; Maia, E.; Silva, A.; Taranto, A. Virtual Screening Algorithms in Drug Discovery: A Review Focused on Machine and Deep Learning Methods. DDC 2023, 2, 311–334. [Google Scholar] [CrossRef]
- Biala, G.; Kedzierska, E.; Kruk-Slomka, M.; Orzelska-Gorka, J.; Hmaidan, S.; Skrok, A.; Kaminski, J.; Havrankova, E.; Nadaska, D.; Malik, I. Research in the Field of Drug Design and Development. Pharmaceuticals 2023, 16, 1283. [Google Scholar] [CrossRef]
- Holdgate, G.A.; Bardelle, C.; Lanne, A.; Read, J.; O’Donovan, D.H.; Smith, J.M.; Selmi, N.; Sheppard, R. Drug Discovery for Epigenetics Targets. Drug Discov. Today 2022, 27, 1088–1098. [Google Scholar] [CrossRef]
- Juárez-Mercado, K.E.; Prieto-Martínez, F.D.; Sánchez-Cruz, N.; Peña-Castillo, A.; Prada-Gracia, D.; Medina-Franco, J.L. Expanding the Structural Diversity of DNA Methyltransferase Inhibitors. Pharmaceuticals 2020, 14, 17. [Google Scholar] [CrossRef]
- Barba-Ostria, C.; Carrera-Pacheco, S.E.; Gonzalez-Pastor, R.; Heredia-Moya, J.; Mayorga-Ramos, A.; Rodríguez-Pólit, C.; Zúñiga-Miranda, J.; Arias-Almeida, B.; Guamán, L.P. Evaluation of Biological Activity of Natural Compounds: Current Trends and Methods. Molecules 2022, 27, 4490. [Google Scholar] [CrossRef]
- Ratovitski, E. Anticancer Natural Compounds as Epigenetic Modulators of Gene Expression. CG 2017, 18, 175–205. [Google Scholar] [CrossRef]
- Borsoi, F.T.; Neri-Numa, I.A.; De Oliveira, W.Q.; De Araújo, F.F.; Pastore, G.M. Dietary Polyphenols and Their Relationship to the Modulation of Non-Communicable Chronic Diseases and Epigenetic Mechanisms: A Mini-Review. Food Chem. Mol. Sci. 2023, 6, 100155. [Google Scholar] [CrossRef]
- Rajendran, P.; Abdelsalam, S.A.; Renu, K.; Veeraraghavan, V.; Ben Ammar, R.; Ahmed, E.A. Polyphenols as Potent Epigenetics Agents for Cancer. Int. J. Mol. Sci. 2022, 23, 11712. [Google Scholar] [CrossRef]
- Li, F.; Qasim, S.; Li, D.; Dou, Q.P. Updated Review on Green Tea Polyphenol Epigallocatechin-3-Gallate as a Cancer Epigenetic Regulator. Semin. Cancer Biol. 2022, 83, 335–352. [Google Scholar] [CrossRef]
- El Omari, N.; Bakrim, S.; Bakha, M.; Lorenzo, J.M.; Rebezov, M.; Shariati, M.A.; Aboulaghras, S.; Balahbib, A.; Khayrullin, M.; Bouyahya, A. Natural Bioactive Compounds Targeting Epigenetic Pathways in Cancer: A Review on Alkaloids, Terpenoids, Quinones, and Isothiocyanates. Nutrients 2021, 13, 3714. [Google Scholar] [CrossRef]
- LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters|Journal of Chemical Information and Modeling. Available online: https://pubs.acs.org/doi/10.1021/ci049885e (accessed on 14 February 2024).
- Qi, Y.; Wang, D.; Wang, D.; Jin, T.; Yang, L.; Wu, H.; Li, Y.; Zhao, J.; Du, F.; Song, M.; et al. HEDD: The Human Epigenetic Drug Database. Database 2016, 2016, baw159. [Google Scholar] [CrossRef]
- Huang, Z.; Jiang, H.; Liu, X.; Chen, Y.; Wong, J.; Wang, Q.; Huang, W.; Shi, T.; Zhang, J. HEMD: An Integrated Tool of Human Epigenetic Enzymes and Chemical Modulators for Therapeutics. PLoS ONE 2012, 7, e39917. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-3: Maestro; Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-3: LigPrep; Schrödinger, LLC: New York, NY, USA, 2020.
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Langer, T.; Wolber, G. Pharmacophore Definition and 3D Searches. Drug Discov. Today Technol. 2004, 1, 203–207. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-3: QikProp; Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 202-3: Canvas; Schrödinger, LLC: New York, NY, USA, 2020.
- Schluckebier, G.; Kozak, M.; Bleimling, N.; Weinhold, E.; Saenger, W. Differential Binding of S-Adenosylmethionine S-Adenosylhomocysteine and Sinefungin to the Adenine-Specific DNA Methyltransferase M.TaqI. J. Mol. Biol. 1997, 265, 56–67. [Google Scholar] [CrossRef]
- Xie, T.; Yu, J.; Fu, W.; Wang, Z.; Xu, L.; Chang, S.; Wang, E.; Zhu, F.; Zeng, S.; Kang, Y.; et al. Insight into the Selective Binding Mechanism of DNMT1 and DNMT3A Inhibitors: A Molecular Simulation Study. Phys. Chem. Chem. Phys. 2019, 21, 12931–12947. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-3: Protein Preparation Wizard; Epik; Schrödinger, LLC: New York, NY, USA; Impact, Schrödinger, LLC: New York, NY, USA; Prime, Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-3: Glide; Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-3: Induced Fit Docking Protocol; Glide, Schrödinger, LLC: New York, NY, USA; Prime, Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-3: Desmond Molecular Dynamics System; D.E. Shaw Research: New York, NY, USA; Maestro-Desmond Interoperability Tools, Schrödinger: New York, NY, USA, 2020.
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; Biancaniello, C.; Argenio, M.A.; Facchiano, A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals 2022, 15, 646. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, A.; Schuster, D. Methods for Generating and Applying Pharmacophore Models as Virtual Screening Filters and for Bioactivity Profiling. Methods 2015, 71, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Furlan, V.; Bren, U. Insight into Inhibitory Mechanism of PDE4D by Dietary Polyphenols Using Molecular Dynamics Simulations and Free Energy Calculations. Biomolecules 2021, 11, 479. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef]
- Guinan, M.; Benckendorff, C.; Smith, M.; Miller, J.D. Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues. Molecules 2020, 25, 2050. https://doi.org/10.3390/molecules25092050.
- Medina-Franco, J.L.; López-Vallejo, F.; Kuck, D.; Lyko, F. Natural Products as DNA Methyltransferase Inhibitors: A Computer-Aided Discovery Approach. Mol. Divers. 2011, 15, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Majid, S.; Dar, A.A.; Shahryari, V.; Hirata, H.; Ahmad, A.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Dahiya, R. Genistein Reverses Hypermethylation and Induces Active Histone Modifications in Tumor Suppressor Gene B-Cell Translocation Gene 3 in Prostate Cancer. Cancer 2010, 116, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Sun, Q.; Li, D.; Miao, S.; Chen, S.; Song, L.; Gao, C.; Chen, Y.; Tan, C.; Jiang, Y. Design, Synthesis and Anticancer Potential of NSC-319745 Hydroxamic Acid Derivatives as DNMT and HDAC Inhibitors. Eur. J. Med. Chem. 2017, 134, 281–292. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ROC Curve Performance (100% of the Screening) | |||
| Sensitivity (Se) | 0.32 | Enrichment Factor (EF) | 10.7 |
| False Positive Rate (1-Sp) | 0.02 | Area Under the Curve (AUC) | 0.65 |
| Calculated Statistical Significance Variables Values | |||
| 1T | 4954 | 5HT | 148 |
| 2A | 91 | 6HA | 29 |
| 3I | 14 | 7HI | 1 |
| 4D | 4849 | 8HD | 118 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kritsi, E.; Christodoulou, P.; Tsiaka, T.; Georgiadis, P.; Zervou, M. A Computational Approach for the Discovery of Novel DNA Methyltransferase Inhibitors. Curr. Issues Mol. Biol. 2024, 46, 3394-3407. https://doi.org/10.3390/cimb46040213
Kritsi E, Christodoulou P, Tsiaka T, Georgiadis P, Zervou M. A Computational Approach for the Discovery of Novel DNA Methyltransferase Inhibitors. Current Issues in Molecular Biology. 2024; 46(4):3394-3407. https://doi.org/10.3390/cimb46040213
Chicago/Turabian StyleKritsi, Eftichia, Paris Christodoulou, Thalia Tsiaka, Panagiotis Georgiadis, and Maria Zervou. 2024. "A Computational Approach for the Discovery of Novel DNA Methyltransferase Inhibitors" Current Issues in Molecular Biology 46, no. 4: 3394-3407. https://doi.org/10.3390/cimb46040213
APA StyleKritsi, E., Christodoulou, P., Tsiaka, T., Georgiadis, P., & Zervou, M. (2024). A Computational Approach for the Discovery of Novel DNA Methyltransferase Inhibitors. Current Issues in Molecular Biology, 46(4), 3394-3407. https://doi.org/10.3390/cimb46040213