
Unravelling the Mysteries of the Sonic Hedgehog Pathway in Cancer Stem Cells: Activity, Crosstalk and Regulation

Abstract
:1. Introduction
1.1. The Sonic Hedgehog (Shh) Pathway and Its Significance in Normal Development and Tissue Homeostasis
1.2. Canonical and Non-Canonical Pathways
1.3. Abnormalities in Shh Signalling
1.3.1. Birth Defects
1.3.2. Cancer
1.4. Introduction to CSCs and Their Pivotal Role in Tumorigenesis, Recurrence, and Resistance to Therapy
1.4.1. Overview
1.4.2. Metastatic Stem Cells
1.4.3. CSC Markers
1.4.4. CSC Markers and Shh
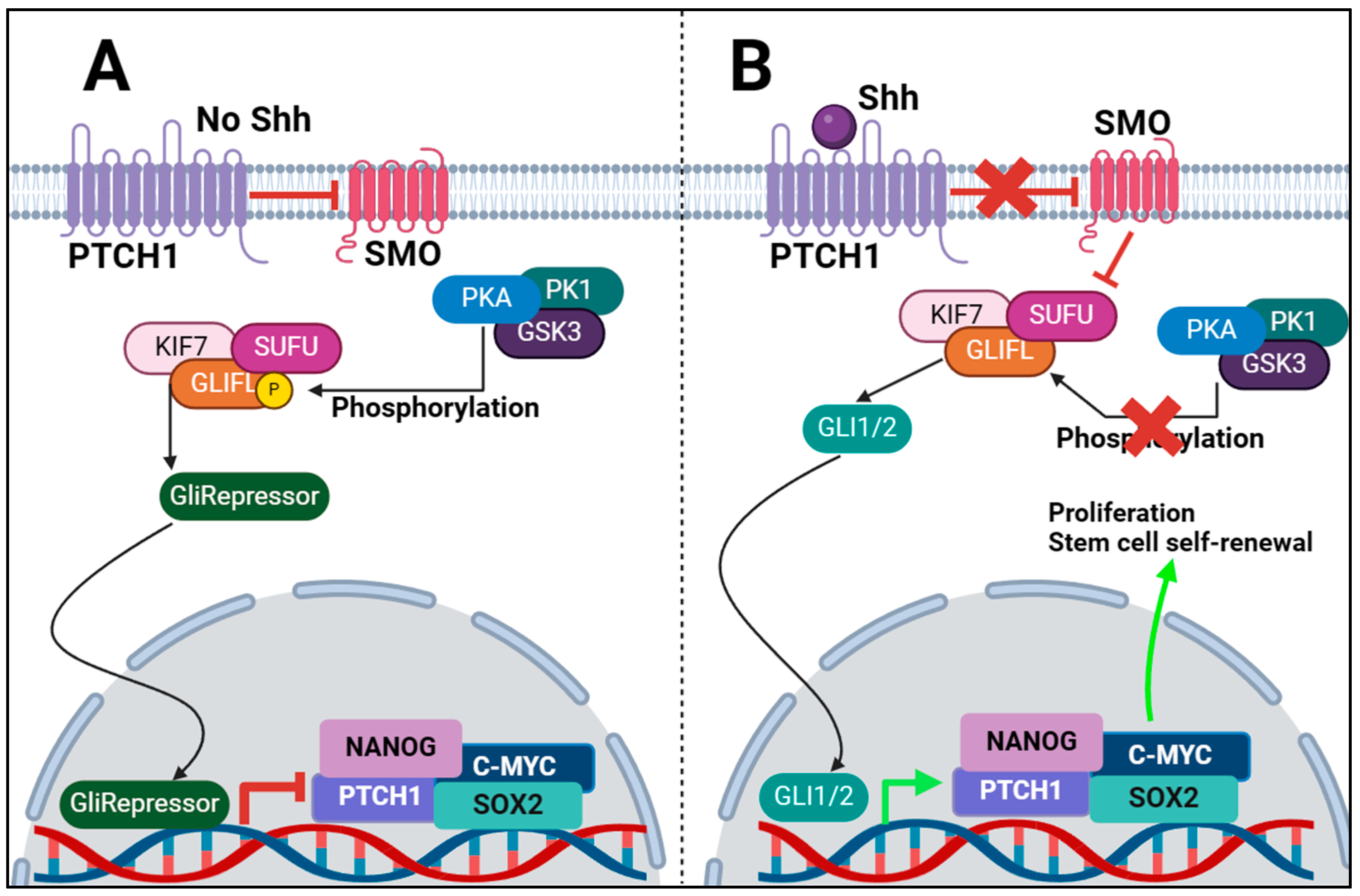
2. Shh Pathway in CSCs: Key Molecular Components of the Shh Pathway and Their Interactions within the Context of CSCs
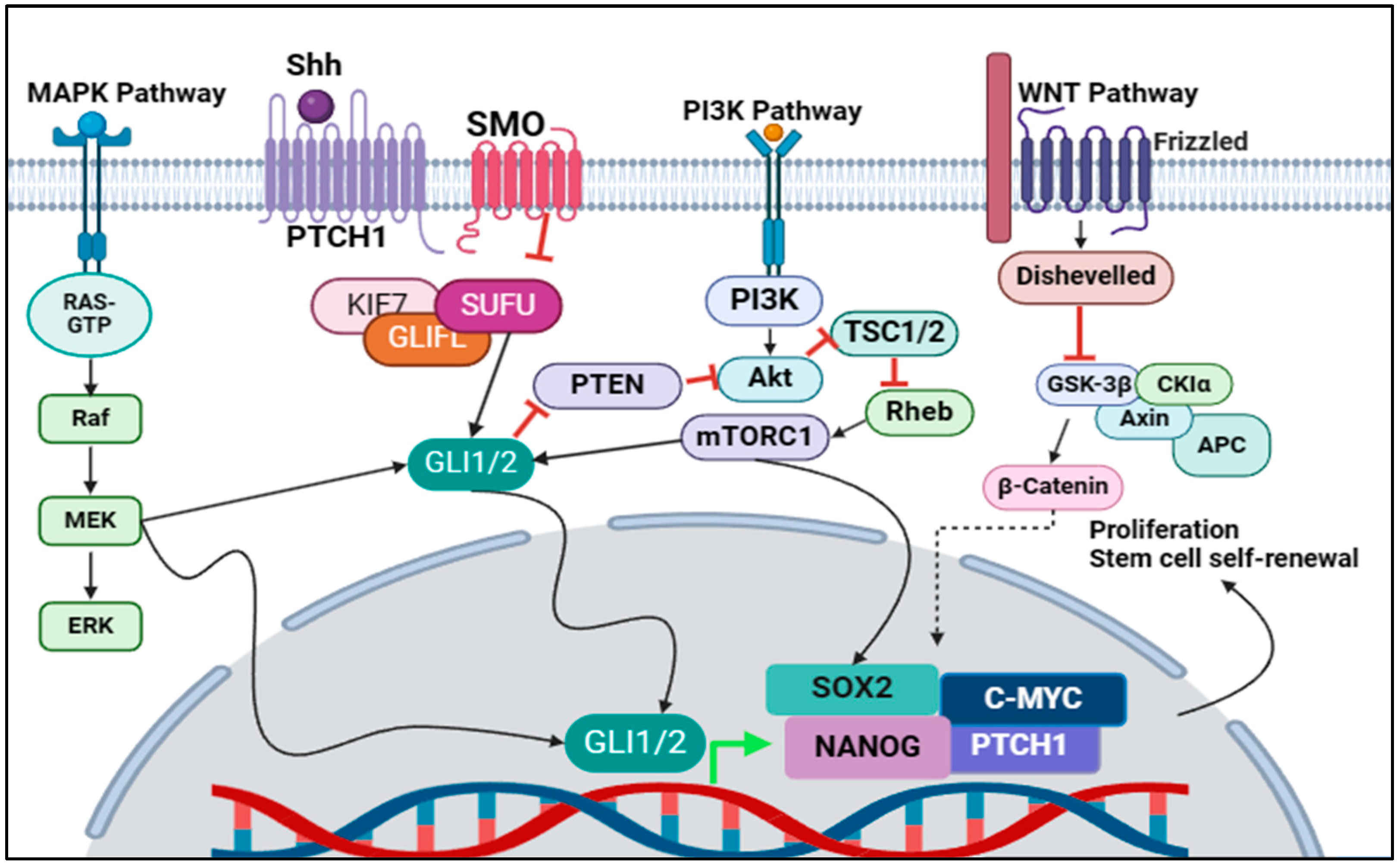
3. Crosstalk between the Shh Pathway and Other Signalling Pathways in the Regulation of CSC Properties
3.1. MAPK/Shh Crosstalk
3.2. PI3K/Shh Crosstalk
3.3. Wnt/Shh Crosstalk
3.4. Targeting Crosstalk of Signalling Pathways in Cancer Stem Cells
4. Regulation of the Shh Pathway in CSCs
4.1. Targeting Shh Ligand
4.2. Targeting Messenger Protein SMO
4.3. Targeting GLI1/2
4.4. miRNAs Targeting CD133
5. Small Molecule Inhibitors
6. Future Prospective
7. Conclusions
Funding
Conflicts of Interest
References
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Li, S.; Li, H.; Yang, C.; Lin, J. The role of Shh signalling pathway in central nervous system development and related diseases. Cell Biochem. Funct. 2021, 39, 180–189. [Google Scholar] [CrossRef]
- Yang, C.; Qi, Y.; Sun, Z. The Role of Sonic Hedgehog Pathway in the Development of the Central Nervous System and Aging-Related Neurodegenerative Diseases. Front. Mol. Biosci. 2021, 8, 711710. [Google Scholar] [CrossRef]
- Adelian, S.; Ahadi, A.M.; Ayat, H.; Teimori, H. Enhanced recombinant C-terminal domain of gli2 gene expression can improve wound healing through promoting cdc25b and N-Myc genes expression. Gene Rep. 2020, 20, 100754. [Google Scholar] [CrossRef]
- Petrova, E.; Rios-Esteves, J.; Ouerfelli, O.; Glickman, J.F.; Resh, M.D. Inhibitors of Hedgehog acyltransferase block Sonic Hedgehog signaling. Nat. Chem. Biol. 2013, 9, 247–249. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Unden, A.B.; Sandstedt, B.; Toftgard, R.; Zaphiropoulos, P.G. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat. Cell Biol. 1999, 1, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signalling: Kif7 is not that fishy after all. Curr. Biol. 2009, 19, R729–R731. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Huntzicker, E.G.; Estay, I.S.; Zhen, H.; Lokteva, L.A.; Jackson, P.K.; Oro, A.E. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006, 20, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.S.; Neill, G.W.; Regl, G.; Eichberger, T.; Frischauf, A.M.; Aberger, F.; Quinn, A.; Philpott, M. GLI2 is expressed in normal human epidermis and BCC and induces GLI1 expression by binding to its promoter. J. Investig. Dermatol. 2004, 122, 1503–1509. [Google Scholar] [CrossRef]
- Dai, P.; Akimaru, H.; Tanaka, Y.; Maekawa, T.; Nakafuku, M.; Ishii, S. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem. 1999, 274, 8143–8152. [Google Scholar] [CrossRef]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Noncanonical Hedgehog signaling. Vitam. Horm. 2012, 88, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D. Hedgehog signalling: Emerging evidence for non-canonical pathways. Cell Signal. 2009, 21, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Copp, A.J.; Greene, N.D. Genetics and development of neural tube defects. J. Pathol. 2010, 220, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.N.; Copp, A.J.; Teratology, M. The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 633–652. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Krauss, R.S. Modeling the complex etiology of holoprosencephaly in mice. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Hammond, N.L.; Brookes, K.J.; Dixon, M.J. Ectopic Hedgehog Signaling Causes Cleft Palate and Defective Osteogenesis. J. Dent. Res. 2018, 97, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Ehlen, H.W.; Buelens, L.A.; Vortkamp, A. Hedgehog signaling in skeletal development. Birth Defects Res. C Embryo Today 2006, 78, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, M.; Isohata, N.; Ohta, H.; Aoyagi, K.; Ochiya, T.; Saeki, N.; Yanagihara, K.; Nakanishi, Y.; Taniguchi, H.; Sakamoto, H.; et al. Hedgehog signal activation in gastric pit cell and in diffuse-type gastric cancer. Gastroenterology 2006, 131, 14–29. [Google Scholar] [CrossRef]
- Pasca di Magliano, M.; Sekine, S.; Ermilov, A.; Ferris, J.; Dlugosz, A.A.; Hebrok, M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006, 20, 3161–3173. [Google Scholar] [CrossRef]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Shevde, L.A. Hedgehog signaling: Modulation of cancer properies and tumor mircroenvironment. Mol. Cancer 2016, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Coletta, R.D.; Yeudall, W.A.; Salo, T. Grand Challenges in Oral Cancers. Front. Oral Health 2020, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Baniebrahimi, G.; Mir, F.; Khanmohammadi, R. Cancer stem cells and oral cancer: Insights into molecular mechanisms and therapeutic approaches. Cancer Cell Int. 2020, 20, 113. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H. Cancer stem cells and self-renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef]
- Chen, K.; Huang, Y.H.; Chen, J.L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef]
- Chen, D.; Wang, C.Y. Targeting cancer stem cells in squamous cell carcinoma. Precis. Clin. Med. 2019, 2, 152–165. [Google Scholar] [CrossRef]
- Zhang, X.; Powell, K.; Li, L. Breast Cancer Stem Cells: Biomarkers, Identification and Isolation Methods, Regulating Mechanisms, Cellular Origin, and Beyond. Cancers 2020, 12, 3765. [Google Scholar] [CrossRef]
- Celia-Terrassa, T.; Jolly, M.K. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition in Cancer Metastasis. Cold Spring Harb. Perspect. Med. 2020, 10, a036905. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Celia-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Mondal, N.; Greco, T.M.; Wei, Y.; Spadazzi, C.; Lin, S.C.; Zheng, H.; Cheung, C.; Magnani, J.L.; Lin, S.H.; et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 2019, 21, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, S.; Zivanovic, N.; Sharma, R.; Pe’er, D.; Bodenmiller, B. Learning time-varying information flow from single-cell epithelial to mesenchymal transition data. PLoS ONE 2018, 13, e0203389. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, D.; Sharma, R.; Nainys, J.; Yim, K.; Kathail, P.; Carr, A.J.; Burdziak, C.; Moon, K.R.; Chaffer, C.L.; Pattabiraman, D.; et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 2018, 174, 716–729 e727. [Google Scholar] [CrossRef] [PubMed]
- Karacosta, L.G.; Anchang, B.; Ignatiadis, N.; Kimmey, S.C.; Benson, J.A.; Shrager, J.B.; Tibshirani, R.; Bendall, S.C.; Plevritis, S.K. Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nat. Commun. 2019, 10, 5587. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354 e315. [Google Scholar] [CrossRef]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef]
- Ko, Y.C.; Choi, H.S.; Liu, R.; Lee, D.S. Physalin A, 13,14-Seco-16, 24-Cyclo-Steroid, Inhibits Stemness of Breast Cancer Cells by Regulation of Hedgehog Signaling Pathway and Yes-Associated Protein 1 (YAP1). Int. J. Mol. Sci. 2021, 22, 8718. [Google Scholar] [CrossRef] [PubMed]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112 e114. [Google Scholar] [CrossRef] [PubMed]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massague, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zurrer-Hardi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Celia-Terrassa, T.; Liu, D.D.; Choudhury, A.; Hang, X.; Wei, Y.; Zamalloa, J.; Alfaro-Aco, R.; Chakrabarti, R.; Jiang, Y.Z.; Koh, B.I.; et al. Normal and cancerous mammary stem cells evade interferon-induced constraint through the miR-199a-LCOR axis. Nat. Cell Biol. 2017, 19, 711–723. [Google Scholar] [CrossRef]
- Ma, C.; Zhao, J.Z.; Lin, R.T.; Zhou, L.; Chen, Y.N.; Yu, L.J.; Shi, T.Y.; Wang, M.; Liu, M.M.; Liu, Y.R.; et al. Combined overexpression of cadherin 6, cadherin 11 and cluster of differentiation 44 is associated with lymph node metastasis and poor prognosis in oral squamous cell carcinoma. Oncol. Lett. 2018, 15, 9498–9506. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.H.; Kim, R.H. An Updated Review of Oral Cancer Stem Cells and Their Stemness Regulation. Crit. Rev. Oncog. 2018, 23, 189–200. [Google Scholar] [CrossRef]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef]
- Wang, L.; Zuo, X.; Xie, K.; Wei, D. The Role of CD44 and Cancer Stem Cells. Methods Mol. Biol. 2018, 1692, 31–42. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, W.; He, R.; Zhang, F.; Wang, H.; Li, P.; Shao, R.G.; Xu, X. Lidamycin decreases CD133 expression in hepatocellular carcinoma via the Notch signaling pathway. Oncol. Lett. 2017, 14, 7889–7895. [Google Scholar] [CrossRef]
- Aghajani, M.; Mansoori, B.; Mohammadi, A.; Asadzadeh, Z.; Baradaran, B. New emerging roles of CD133 in cancer stem cell: Signaling pathway and miRNA regulation. J. Cell. Physiol. 2019, 234, 21642–21661. [Google Scholar] [CrossRef] [PubMed]
- Cierpikowski, P.; Lis-Nawara, A.; Bar, J. SHH Expression Is Significantly Associated With Cancer Stem Cell Markers in Oral Squamous Cell Carcinoma. Anticancer Res. 2021, 41, 5405–5413. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-H.; Zhai, E.-T.; Chen, S.-L.; Wu, H.; Wu, K.-M.; Zhang, X.-H.; Chen, C.-Q.; Cai, S.-R.; He, Y.-L. CD44, Sonic Hedgehog, and Gli1 Expression Are Prognostic Biomarkers in Gastric Cancer Patients after Radical Resection. Gastroenterol. Res. Pract. 2016, 2016, 1013045. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Sheen, I.S.; Jeng, W.J.; Yu, M.C.; Hsiau, H.I.; Chang, F.Y.; Tsai, H.H. Activation of the sonic hedgehog signaling pathway occurs in the CD133 positive cells of mouse liver cancer Hepa 1-6 cells. Onco Targets Ther. 2013, 6, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Nandi, S.; Dey, M.; Sonabend, A.M.; Lesniak, M.S. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133(+) glioma stem cells to temozolomide therapy. Mol. Med. 2011, 17, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Kohler, B.; Schonicke, A.; Scharwachter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef]
- Szkandera, J.; Kiesslich, T.; Haybaeck, J.; Gerger, A.; Pichler, M. Hedgehog signaling pathway in ovarian cancer. Int. J. Mol. Sci. 2013, 14, 1179–1196. [Google Scholar] [CrossRef]
- Etheridge, L.A.; Crawford, T.Q.; Zhang, S.; Roelink, H. Evidence for a role of vertebrate Disp1 in long-range Shh signaling. Development 2010, 137, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Li, Y.J.; Kawakami, T.; Xu, S.M.; Chuang, P.T. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 2004, 18, 641–659. [Google Scholar] [CrossRef] [PubMed]
- Buglino, J.A.; Resh, M.D. Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog. J. Biol. Chem. 2008, 283, 22076–22088. [Google Scholar] [CrossRef] [PubMed]
- Nicolis, S.K. Cancer stem cells and “stemness” genes in neuro-oncology. Neurobiol. Dis. 2007, 25, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lu, Y.; Li, Y.; Prinz, R.A. Sonic Hedgehog Signaling in Thyroid Cancer. Front. Endocrinol. 2017, 8, 284. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhu, Y.; Deng, S.; Chen, Y.; Li, W.; Sun, J.; Xu, X. Targeting the Sonic Hedgehog Pathway to Suppress the Expression of the Cancer Stem Cell (CSC)-Related Transcription Factors and CSC-Driven Thyroid Tumor Growth. Cancers 2021, 13, 418. [Google Scholar] [CrossRef] [PubMed]
- Heiden, K.B.; Williamson, A.J.; Doscas, M.E.; Ye, J.; Wang, Y.; Liu, D.; Xing, M.; Prinz, R.A.; Xu, X. The sonic hedgehog signaling pathway maintains the cancer stem cell self-renewal of anaplastic thyroid cancer by inducing snail expression. J. Clin. Endocrinol. Metab. 2014, 99, E2178–E2187. [Google Scholar] [CrossRef] [PubMed]
- Carina, V.; Zito, G.; Pizzolanti, G.; Richiusa, P.; Criscimanna, A.; Rodolico, V.; Tomasello, L.; Pitrone, M.; Arancio, W.; Giordano, C. Multiple pluripotent stem cell markers in human anaplastic thyroid cancer: The putative upstream role of SOX2. Thyroid 2013, 23, 829–837. [Google Scholar] [CrossRef]
- Satheesha, S.; Manzella, G.; Bovay, A.; Casanova, E.A.; Bode, P.K.; Belle, R.; Feuchtgruber, S.; Jaaks, P.; Dogan, N.; Koscielniak, E.; et al. Targeting hedgehog signaling reduces self-renewal in embryonal rhabdomyosarcoma. Oncogene 2016, 35, 2020–2030. [Google Scholar] [CrossRef]
- Zhu, R.; Gires, O.; Zhu, L.; Liu, J.; Li, J.; Yang, H.; Ju, G.; Huang, J.; Ge, W.; Chen, Y.; et al. TSPAN8 promotes cancer cell stemness via activation of sonic Hedgehog signaling. Nat. Commun. 2019, 10, 2863. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yin, S.; Zeng, H.; Li, H.; Wan, X. Regulation of Embryonic Stem Cell Self-Renewal. Life 2022, 12, 1151. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, M.R.; Kim, T.; Kim, Y.W.; Cho, M.Y. Activated STAT3 may participate in tumor progression through increasing CD133/survivin expression in early stage of colon cancer. Biochem. Biophys. Res. Commun. 2018, 497, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.W.; Song, Y.; Kim, S.H.; Kim, J.S.; Kim, K.M.; Choi, E.K.; Kim, J.; Seo, H.R. CD133 confers cancer stem-like cell properties by stabilizing EGFR-AKT signaling in hepatocellular carcinoma. Cancer Lett. 2017, 389, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Batlle, E.; Massague, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Geyer, N.; Gerling, M. Hedgehog Signaling in Colorectal Cancer: All in the Stroma? Int. J. Mol. Sci. 2021, 22, 1025. [Google Scholar] [CrossRef] [PubMed]
- Abdelmaksoud, N.M.; Abulsoud, A.I.; Doghish, A.S.; Abdelghany, T.M. From resistance to resilience: Uncovering chemotherapeutic resistance mechanisms; insights from established models. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188993. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Wang, R.; Boulbes, D.R.; Zhang, H.; Watowich, S.S.; Xia, L.; Ye, X.; Bhattacharya, R.; Ellis, L.M. Macrophage conditioned medium promotes colorectal cancer stem cell phenotype via the hedgehog signaling pathway. PLoS ONE 2018, 13, e0190070. [Google Scholar] [CrossRef]
- Jinushi, M.; Komohara, Y. Tumor-associated macrophages as an emerging target against tumors: Creating a new path from bench to bedside. Biochim. Biophys. Acta 2015, 1855, 123–130. [Google Scholar] [CrossRef]
- Yang, S.H.; Sharrocks, A.D.; Whitmarsh, A.J. MAP kinase signalling cascades and transcriptional regulation. Gene 2013, 513, 1–13. [Google Scholar] [CrossRef]
- Park, J.I. MAPK-ERK Pathway. Int. J. Mol. Sci. 2023, 24, 9666. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Rovida, E.; Stecca, B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: A crosstalk providing therapeutic opportunities? Semin. Cancer Biol. 2015, 35, 154–167. [Google Scholar] [CrossRef]
- Riobo, N.A.; Haines, G.M.; Emerson, C.P., Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006, 66, 839–845. [Google Scholar] [CrossRef]
- Whisenant, T.C.; Ho, D.T.; Benz, R.W.; Rogers, J.S.; Kaake, R.M.; Gordon, E.A.; Huang, L.; Baldi, P.; Bardwell, L. Computational prediction and experimental verification of new MAP kinase docking sites and substrates including Gli transcription factors. PLoS Comput. Biol. 2010, 6, e1000908. [Google Scholar] [CrossRef]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef]
- Mazumdar, T.; Devecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. Blocking Hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res. 2011, 71, 5904–5914. [Google Scholar] [CrossRef]
- Mazumdar, T.; DeVecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. The GLI genes as the molecular switch in disrupting Hedgehog signaling in colon cancer. Oncotarget 2011, 2, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xu, Z. Cross-signaling among phosphinositide-3 kinase, mitogen-activated protein kinase and sonic hedgehog pathways exists in esophageal cancer. Int. J. Cancer 2011, 129, 275–284. [Google Scholar] [CrossRef]
- Ebrahimi, N.; Afshinpour, M.; Fakhr, S.S.; Kalkhoran, P.G.; Shadman-Manesh, V.; Adelian, S.; Beiranvand, S.; Rezaei-Tazangi, F.; Khorram, R.; Hamblin, M.R.; et al. Cancer stem cells in colorectal cancer: Signaling pathways involved in stemness and therapy resistance. Crit. Rev. Oncol. Hematol. 2023, 182, 103920. [Google Scholar] [CrossRef]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Douville, J.; Beaulieu, R.; Balicki, D. ALDH1 as a functional marker of cancer stem and progenitor cells. Stem Cells Dev. 2009, 18, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.Y.; Kaf, R.M.; Ahmed, M.M.; Elwan, A.; Ashour, H.R.; Ibrahim, A. The Prognostic Value of Cancer Stem Cell Markers (Notch1, ALDH1, and CD44) in Primary Colorectal Carcinoma. J. Gastrointest. Cancer 2019, 50, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, K.; Chauhan, S.; Kumar, D. Expression analysis and regulation of GLI and its correlation with stemness and metabolic alteration in human brain tumor. 3 Biotech 2023, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, Y.H.; Shim, S.; Kim, A.; Jang, H.; Lee, S.J.; Park, S.; Seo, S.; Jang, W.I.; Lee, S.B.; et al. Radiation-Activated PI3K/AKT Pathway Promotes the Induction of Cancer Stem-Like Cells via the Upregulation of SOX2 in Colorectal Cancer. Cells 2021, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhu, G.; Huang, J.; Li, L.; Du, Y.; Gao, Y.; Wu, D.; Wang, X.; Hsieh, J.T.; He, D.; et al. Non-canonical GLI1/2 activation by PI3K/AKT signaling in renal cell carcinoma: A novel potential therapeutic target. Cancer Lett. 2016, 370, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Du, J.; Zhao, L.; Liu, W.; Zhao, T.; Liang, H.; Fang, P.; Zhang, K.; Zeng, H. GLI1 reduces drug sensitivity by regulating cell cycle through PI3K/AKT/GSK3/CDK pathway in acute myeloid leukemia. Cell Death Dis. 2021, 12, 231. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef]
- Valenti, G.; Quinn, H.M.; Heynen, G.; Lan, L.; Holland, J.D.; Vogel, R.; Wulf-Goldenberg, A.; Birchmeier, W. Cancer Stem Cells Regulate Cancer-Associated Fibroblasts via Activation of Hedgehog Signaling in Mammary Gland Tumors. Cancer Res. 2017, 77, 2134–2147. [Google Scholar] [CrossRef]
- Luo, W.; Rodriguez, M.; Valdez, J.M.; Zhu, X.; Tan, K.; Li, D.; Siwko, S.; Xin, L.; Liu, M. Lgr4 is a key regulator of prostate development and prostate stem cell differentiation. Stem Cells 2013, 31, 2492–2505. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, J.; Li, D.; Lai, L.; Siwko, S.; Li, Y.; Liu, M. Lgr4 regulates mammary gland development and stem cell activity through the pluripotency transcription factor Sox2. Stem Cells 2013, 31, 1921–1931. [Google Scholar] [CrossRef]
- Bhal, S.; Kundu, C.N. Targeting crosstalk of signaling pathways in cancer stem cells: A promising approach for development of novel anti-cancer therapeutics. Med. Oncol. 2023, 40, 82. [Google Scholar] [CrossRef] [PubMed]
- Chaudary, N.; Pintilie, M.; Hedley, D.; Hill, R.P.; Milosevic, M.; Mackay, H. Hedgehog inhibition enhances efficacy of radiation and cisplatin in orthotopic cervical cancer xenografts. Br. J. Cancer 2017, 116, 50–57. [Google Scholar] [CrossRef]
- Chatterjee, S.; Sil, P.C. Targeting the crosstalks of Wnt pathway with Hedgehog and Notch for cancer therapy. Pharmacol. Res. 2019, 142, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, R.; Pelullo, M.; Zema, S.; Nardozza, F.; Checquolo, S.; Lauer, D.M.; Bufalieri, F.; Palermo, R.; Felli, M.P.; Vacca, A.; et al. Maml1 acts cooperatively with Gli proteins to regulate sonic hedgehog signaling pathway. Cell Death Dis. 2017, 8, e2942. [Google Scholar] [CrossRef] [PubMed]
- Paluszczak, J.; Wisniewska, D.; Kostrzewska-Poczekaj, M.; Kiwerska, K.; Grenman, R.; Mielcarek-Kuchta, D.; Jarmuz-Szymczak, M. Prognostic significance of the methylation of Wnt pathway antagonists-CXXC4, DACT2, and the inhibitors of sonic hedgehog signaling-ZIC1, ZIC4, and HHIP in head and neck squamous cell carcinomas. Clin. Oral Investig. 2017, 21, 1777–1788. [Google Scholar] [CrossRef]
- Ge, Q.; Hu, Y.; He, J.; Chen, F.; Wu, L.; Tu, X.; Qi, Y.; Zhang, Z.; Xue, M.; Chen, S.; et al. Zic1 suppresses gastric cancer metastasis by regulating Wnt/beta-catenin signaling and epithelial-mesenchymal transition. FASEB J. 2020, 34, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wang, Y.; Qian, M.; Qiao, Y.; Zou, S.; Chen, C.; Zhang, X.; Chen, Y.; Zhao, Y.; Zhu, G.; et al. Sirt1 suppresses Wnt/betaCatenin signaling in liver cancer cells by targeting betaCatenin in a PKAalpha-dependent manner. Cell Signal. 2017, 37, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Leung, H.W.; Lau, E.Y.T.; Leung, C.O.N.; Lei, M.M.L.; Mok, E.H.K.; Ma, V.W.S.; Cho, W.C.S.; Ng, I.O.L.; Yun, J.P.; Cai, S.H.; et al. NRF2/SHH signaling cascade promotes tumor-initiating cell lineage and drug resistance in hepatocellular carcinoma. Cancer Lett. 2020, 476, 48–56. [Google Scholar] [CrossRef]
- Fu, S.; Wang, Y.; Li, H.; Chen, L.; Liu, Q. Regulatory Networks of LncRNA MALAT-1 in Cancer. Cancer Manag. Res. 2020, 12, 10181–10198. [Google Scholar] [CrossRef]
- Guo, F.; Cao, Z.; Guo, H.; Li, S. The action mechanism of lncRNA-HOTAIR on the drug resistance of non-small cell lung cancer by regulating Wnt signaling pathway. Exp. Ther. Med. 2018, 15, 4885–4889. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.S.; Li, L.J.; Huang, H.W.; Yang, H.F.; Wu, D.P. MYC-regulated lncRNA NEAT1 promotes B cell proliferation and lymphomagenesis via the miR-34b-5p-GLI1 pathway in diffuse large B-cell lymphoma. Cancer Cell Int. 2020, 20, 87. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Li, X. Application of CRISPR-Cas9 for Long Noncoding RNA Genes in Cancer Research. Hum. Gene Ther. 2019, 30, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ericson, J.; Morton, S.; Kawakami, A.; Roelink, H.; Jessell, T.M. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell 1996, 87, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Maun, H.R.; Wen, X.; Lingel, A.; de Sauvage, F.J.; Lazarus, R.A.; Scales, S.J.; Hymowitz, S.G. Hedgehog pathway antagonist 5E1 binds hedgehog at the pseudo-active site. J. Biol. Chem. 2010, 285, 26570–26580. [Google Scholar] [CrossRef] [PubMed]
- Teichman, J.; Dodbiba, L.; Thai, H.; Fleet, A.; Morey, T.; Liu, L.; McGregor, M.; Cheng, D.; Chen, Z.; Darling, G.; et al. Hedgehog inhibition mediates radiation sensitivity in mouse xenograft models of human esophageal adenocarcinoma. PLoS ONE 2018, 13, e0194809. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Yue, W.; Wei, B.; Wang, N.; Li, T.; Guan, L.; Shi, S.; Zeng, Q.; Pei, X.; Chen, L. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS ONE 2011, 6, e17687. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A.; et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef] [PubMed]
- Coon, V.; Laukert, T.; Pedone, C.A.; Laterra, J.; Kim, K.J.; Fults, D.W. Molecular therapy targeting Sonic hedgehog and hepatocyte growth factor signaling in a mouse model of medulloblastoma. Mol. Cancer Ther. 2010, 9, 2627–2636. [Google Scholar] [CrossRef]
- Stanton, B.Z.; Peng, L.F.; Maloof, N.; Nakai, K.; Wang, X.; Duffner, J.L.; Taveras, K.M.; Hyman, J.M.; Lee, S.W.; Koehler, A.N.; et al. A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat. Chem. Biol. 2009, 5, 154–156. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Ray, H. Safety and Tolerability of Sonic Hedgehog Pathway Inhibitors in Cancer. Drug Saf. 2019, 42, 263–279. [Google Scholar] [CrossRef]
- Matevossian, A.; Resh, M.D. Hedgehog Acyltransferase as a target in estrogen receptor positive, HER2 amplified, and tamoxifen resistant breast cancer cells. Mol. Cancer 2015, 14, 72. [Google Scholar] [CrossRef]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Chang, C.F.; Lin, S.S. Sonic Hedgehog Signaling in Organogenesis, Tumors, and Tumor Microenvironments. Int. J. Mol. Sci. 2020, 21, 758. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Chang, C.F.; Sheen, I.S.; Jeng, C.J.; Wang, C.H. Cellular and Molecular Biology of Cancer Stem Cells of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 1417. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, L.; Zhang, Z.; Liu, X.; Gao, H.; Zhuang, Y.; Yang, P.; Kornmann, M.; Tian, X.; Yang, Y. Hedgehog Signaling Regulates Epithelial-Mesenchymal Transition in Pancreatic Cancer Stem-Like Cells. J. Cancer 2016, 7, 408–417. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, S.L.; Kim, J.H.; Lee, D.S. The FDA-Approved Anti-Asthma Medicine Ciclesonide Inhibits Lung Cancer Stem Cells through Hedgehog Signaling-Mediated SOX2 Regulation. Int. J. Mol. Sci. 2020, 21, 1014. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Jiao, M.; Wu, D.; Wu, K.; Li, X.; Zhu, G.; Yang, L.; Wang, X.; Hsieh, J.T.; et al. Genistein inhibits the stemness properties of prostate cancer cells through targeting Hedgehog-Gli1 pathway. Cancer Lett. 2012, 323, 48–57. [Google Scholar] [CrossRef]
- Fan, P.; Fan, S.; Wang, H.; Mao, J.; Shi, Y.; Ibrahim, M.M.; Ma, W.; Yu, X.; Hou, Z.; Wang, B.; et al. Genistein decreases the breast cancer stem-like cell population through Hedgehog pathway. Stem Cell Res. Ther. 2013, 4, 146. [Google Scholar] [CrossRef]
- Zhang, Q.; Cao, W.S.; Wang, X.Q.; Zhang, M.; Lu, X.M.; Chen, J.Q.; Chen, Y.; Ge, M.M.; Zhong, C.Y.; Han, H.Y. Genistein inhibits nasopharyngeal cancer stem cells through sonic hedgehog signaling. Phytother. Res. 2019, 33, 2783–2791. [Google Scholar] [CrossRef]
- Li, E.; Zhang, T.; Sun, X.; Li, Y.; Geng, H.; Yu, D.; Zhong, C. Sonic hedgehog pathway mediates genistein inhibition of renal cancer stem cells. Oncol. Lett. 2019, 18, 3081–3091. [Google Scholar] [CrossRef]
- Yu, D.; Shin, H.S.; Lee, Y.S.; Lee, D.; Kim, S.; Lee, Y.C. Genistein attenuates cancer stem cell characteristics in gastric cancer through the downregulation of Gli1. Oncol. Rep. 2014, 31, 673–678. [Google Scholar] [CrossRef]
- El-Rayes, B.F.; Philip, P.A.; Sarkar, F.H.; Shields, A.F.; Ferris, A.M.; Hess, K.; Kaseb, A.O.; Javle, M.M.; Varadhachary, G.R.; Wolff, R.A.; et al. A phase II study of isoflavones, erlotinib, and gemcitabine in advanced pancreatic cancer. Investig. New Drugs 2011, 29, 694–699. [Google Scholar] [CrossRef]
- Takimoto, C.H.; Glover, K.; Huang, X.; Hayes, S.A.; Gallot, L.; Quinn, M.; Jovanovic, B.D.; Shapiro, A.; Hernandez, L.; Goetz, A.; et al. Phase I pharmacokinetic and pharmacodynamic analysis of unconjugated soy isoflavones administered to individuals with cancer. Cancer Epidemiol. Biomark. Prev. 2003, 12, 1213–1221. [Google Scholar]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef]
- Srivastava, R.K.; Kaylani, S.Z.; Edrees, N.; Li, C.; Talwelkar, S.S.; Xu, J.; Palle, K.; Pressey, J.G.; Athar, M. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget 2014, 5, 12151–12165. [Google Scholar] [CrossRef]
- Tong, W.; Qiu, L.; Qi, M.; Liu, J.; Hu, K.; Lin, W.; Huang, Y.; Fu, J. GANT-61 and GDC-0449 induce apoptosis of prostate cancer stem cells through a GLI-dependent mechanism. J. Cell. Biochem. 2018, 119, 3641–3652. [Google Scholar] [CrossRef]
- Kurebayashi, J.; Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T. Anti-cancer stem cell activity of a hedgehog inhibitor GANT61 in estrogen receptor-positive breast cancer cells. Cancer Sci. 2017, 108, 918–930. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Shirjang, S.; Baradaran, B. MicroRNAs in the Diagnosis and Treatment of Cancer. Immunol. Investig. 2017, 46, 880–897. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Mohammadi, A.; Mansoori, B.; Aghapour, M.; Shirjang, S.; Nami, S.; Baradaran, B. The Urtica dioica extract enhances sensitivity of paclitaxel drug to MDA-MB-468 breast cancer cells. Biomed. Pharmacother. 2016, 83, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Asadzadeh, Z.; Mansoori, B.; Mohammadi, A.; Aghajani, M.; Haji-Asgarzadeh, K.; Safarzadeh, E.; Mokhtarzadeh, A.; Duijf, P.H.G.; Baradaran, B. microRNAs in cancer stem cells: Biology, pathways, and therapeutic opportunities. J. Cell. Physiol. 2019, 234, 10002–10017. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.Y.; Liao, Y.J.; Cai, M.Y.; Ma, N.F.; Zhang, Q.; Chen, J.W.; Zhang, J.X.; Wang, F.W.; Wang, C.Y.; Chen, W.H.; et al. Eukaryotic Initiation Factor 5A2 Contributes to the Maintenance of CD133(+) Hepatocellular Carcinoma Cells via the c-Myc/microRNA-29b Axis. Stem Cells 2018, 36, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Mott, J.L.; Kurita, S.; Cazanave, S.C.; Bronk, S.F.; Werneburg, N.W.; Fernandez-Zapico, M.E. Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J. Cell. Biochem. 2010, 110, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Tsukasa, K.; Ding, Q.; Miyazaki, Y.; Matsubara, S.; Natsugoe, S.; Takao, S. miR-30 family promotes migratory and invasive abilities in CD133(+) pancreatic cancer stem-like cells. Hum. Cell 2016, 29, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Krantz, S.B.; Shields, M.A.; Dangi-Garimella, S.; Bentrem, D.J.; Munshi, H.G. Contribution of epithelial-mesenchymal transition to pancreatic cancer progression. Cancers 2010, 2, 2084–2097. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Rodriguez-Cruz, V.; Rameshwar, P. High expression of miR-9 in CD133(+) glioblastoma cells in chemoresistance to temozolomide. J. Cancer Stem Cell Res. 2015, 3, e1003. [Google Scholar] [CrossRef] [PubMed]
- Owens, A.E.; de Paola, I.; Hansen, W.A.; Liu, Y.W.; Khare, S.D.; Fasan, R. Design and Evolution of a Macrocyclic Peptide Inhibitor of the Sonic Hedgehog/Patched Interaction. J. Am. Chem. Soc. 2017, 139, 12559–12568. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.; Wang, J.; Yang, J.; Huang, W.; Lai, L.; Tan, W.; Liu, Y. Discovery of Small Molecule Inhibitors Targeting the Sonic Hedgehog. Front. Chem. 2020, 8, 498. [Google Scholar] [CrossRef]
- Tian, S.; Quan, H.; Xie, C.; Guo, H.; Lu, F.; Xu, Y.; Li, J.; Lou, L. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci. 2011, 102, 1374–1380. [Google Scholar] [CrossRef]
- Cao, W.; Li, Y.; Sun, H.; Yang, C.; Zhu, J.; Xie, C.; Li, X.; Wu, J.; Geng, S.; Wang, L.; et al. Apatinib Suppresses Gastric Cancer Stem Cells Properties by Inhibiting the Sonic Hedgehog Pathway. Front. Cell Dev. Biol. 2021, 9, 679806. [Google Scholar] [CrossRef] [PubMed]
- Sneha, S.; Nagare, R.P.; Sidhanth, C.; Krishnapriya, S.; Garg, M.; Ramachandran, B.; Murhekar, K.; Sundersingh, S.; Ganesan, T.S. The hedgehog pathway regulates cancer stem cells in serous adenocarcinoma of the ovary. Cell. Oncol. 2020, 43, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T.; Kurebayashi, J. Anti-cell growth and anti-cancer stem cell activities of the non-canonical hedgehog inhibitor GANT61 in triple-negative breast cancer cells. Breast Cancer 2017, 24, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Matsubara, S.; Ding, Q.; Tsukasa, K.; Yoshimitsu, M.; Kosai, K.; Takao, S. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol. Cancer 2016, 15, 49. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Shen, Y.; Chen, X.; He, J.; Liu, J.; Zu, X. Self-Renewal Signalling Pathway Inhibitors: Perspectives on Therapeutic Approaches for Cancer Stem Cells. Onco Targets Ther. 2020, 13, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Omar, A.; Ruff, P.; Penny, C. Inhibition of the Sonic Hedgehog Pathway using Small Molecule Inhibitors: Targeting Colon Cancer Stem Cells. Curr. Cancer Ther. Rev. 2023, 19, 138–155. [Google Scholar] [CrossRef]
- Yao, J.; An, Y.; Wie, J.S.; Ji, Z.L.; Lu, Z.P.; Wu, J.L.; Jiang, K.R.; Chen, P.; Xu, Z.K.; Miao, Y. Cyclopamine reverts acquired chemoresistance and down-regulates cancer stem cell markers in pancreatic cancer cell lines. Swiss. Med. Wkly. 2011, 141, w13208. [Google Scholar] [CrossRef] [PubMed]
- Meiss, F.; Andrlova, H.; Zeiser, R. Vismodegib. Recent Results Cancer Res. 2018, 211, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: Molecular mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef]
- Li, W.; Yang, H.; Li, X.; Han, L.; Xu, N.; Shi, A. Signaling pathway inhibitors target breast cancer stem cells in triple-negative breast cancer. Oncol. Rep. 2019, 41, 437–446. [Google Scholar] [CrossRef]
- Pietrobono, S.; Stecca, B. Targeting the Oncoprotein Smoothened by Small Molecules: Focus on Novel Acylguanidine Derivatives as Potent Smoothened Inhibitors. Cells 2018, 7, 272. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, A.; Iovine, V.; Botta, B.; Quaglio, D.; D’Acquarica, I.; Ciogli, A.; Iazzetti, A.; Alfonsi, R.; Lospinoso Severini, L.; Infante, P.; et al. Chemical, computational and functional insights into the chemical stability of the Hedgehog pathway inhibitor GANT61. J. Enzyme Inhib. Med. Chem. 2018, 33, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Ghirga, F.; Mori, M.; Infante, P. Current trends in Hedgehog signaling pathway inhibition by small molecules. Bioorg. Med. Chem. Lett. 2018, 28, 3131–3140. [Google Scholar] [CrossRef] [PubMed]
- Galperin, I.; Dempwolff, L.; Diederich, W.E.; Lauth, M. Inhibiting Hedgehog: An Update on Pharmacological Compounds and Targeting Strategies. J. Med. Chem. 2019, 62, 8392–8411. [Google Scholar] [CrossRef]
- Schaffner, T.J.; Skoner, D.P. Ciclesonide: A safe and effective inhaled corticosteroid for the treatment of asthma. J. Asthma Allergy 2009, 2, 25–32. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Regulator | Role in Signalling |
|---|---|
| HHAT | Essential for Shh palmitoylation, stabilizing the ligand for secretion. |
| DISP1 | Required for long-range secretion of Shh to target cells. |
| Shh Ligand | Signal activator, initiates the Shh cascade by inhibiting PTCH1. |
| PTCH1 | In absence of Shh ligand, it prevents activation of Shh signalling by inhibiting SMO activity. |
| SMO | Inhibits SUFU activity when Shh ligand is present, which leads to GLI1/2 expression and signalling. |
| SUFU | When Shh is not present, prevents expression of GLI1/2 and produces GLIRepressor. When Shh is present, it gets inhibited, and GLI1/2 is produced. |
| GLI1/2 | Moves into nucleus and initiates transcription of target genes NANOG, SOX2. |
| GLIRepressor | Moves into nucleus and inhibits expression of GLI1/2 target genes. |
| Inhibitor | Target | Mechanism of Action | Clinical Trial Status | Trial Number (s) |
|---|---|---|---|---|
| Sonidegib | SMO | SMO receptor inhibitor | FDA-approved | |
| Vismodegib | SMO | SMO receptor inhibitor | FDA-approved | |
| ATO | GLI1 | Inhibits GLI1 activity | FDA-approved | |
| Glasdegib | SMO | SMO receptor inhibitor | FDA-approved | |
| Ciclesonide | SMO/GLI1/2 | Exact mechanism is unknown | FDA-approved | |
| ROBOTNIKININ | Shh | Shh ligand inhibitor | No trials to date * | N/A |
| 5E1 | Shh | Shh ligand inhibitor | No trials to date * | N/A |
| RU-SKI 43 | Shh | Shhat enzyme inhibitor, preventing Shh ligand synthesis | No trials to date * | N/A |
| Saridegib | SMO | SMO receptor inhibitor | Phase 2 | NCT01310816, NCT01130142 |
| Talidegib | SMO | SMO receptor inhibitor | Phase 2 | NCT05199584, NCT02530437, NCT01722292 |
| LEQ506 | SMO | SMO receptor inhibitor | Phase 1 | NCT01106508 |
| Cyclopamine | SMO | SMO receptor inhibitor | No trials to date * | N/A |
| Rivoceranib | SMO/GLI1/2 | Exact mechanism is unknown | Phase 3 | NCT03042611, NCT04639180 |
| Genistein | GLI1/2 | Decreases GLI1 activity, specifics unknown | Phase 2 and 3 | NCT01985763, NCT01126879, NCT00584532 |
| GANT61 | GLI1/2 | Inhibits GLI1/2 DNA binding ability | No trials to date * | N/A |
| Pirfenidone | GLI2 | Destabilises GLI2 protein to prevent activity | Phase 2 | NCT06142318, NCT05801133 |
| miRNAs | CD133 | Inhibits expression of CD133 | No trials to date * | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berrino, C.; Omar, A. Unravelling the Mysteries of the Sonic Hedgehog Pathway in Cancer Stem Cells: Activity, Crosstalk and Regulation. Curr. Issues Mol. Biol. 2024, 46, 5397-5419. https://doi.org/10.3390/cimb46060323
Berrino C, Omar A. Unravelling the Mysteries of the Sonic Hedgehog Pathway in Cancer Stem Cells: Activity, Crosstalk and Regulation. Current Issues in Molecular Biology. 2024; 46(6):5397-5419. https://doi.org/10.3390/cimb46060323
Chicago/Turabian StyleBerrino, Carlo, and Aadilah Omar. 2024. "Unravelling the Mysteries of the Sonic Hedgehog Pathway in Cancer Stem Cells: Activity, Crosstalk and Regulation" Current Issues in Molecular Biology 46, no. 6: 5397-5419. https://doi.org/10.3390/cimb46060323
APA StyleBerrino, C., & Omar, A. (2024). Unravelling the Mysteries of the Sonic Hedgehog Pathway in Cancer Stem Cells: Activity, Crosstalk and Regulation. Current Issues in Molecular Biology, 46(6), 5397-5419. https://doi.org/10.3390/cimb46060323