

Changing the Landscape of Solid Tumor Therapy from Apoptosis-Promoting to Apoptosis-Inhibiting Strategies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Expert Opinions on Solid Tumor Therapy
1.2. Objectives
2. Danger of Information-Generating Approaches to Cancer Biology: The Current State of Confusion
2.1. Off-Target Effects of Drugs Designed for Targeted Therapies
2.2. Precision Oncology for Treating Patients with Solid Tumors: Ever-Increasing Information and More (Empty) Promises
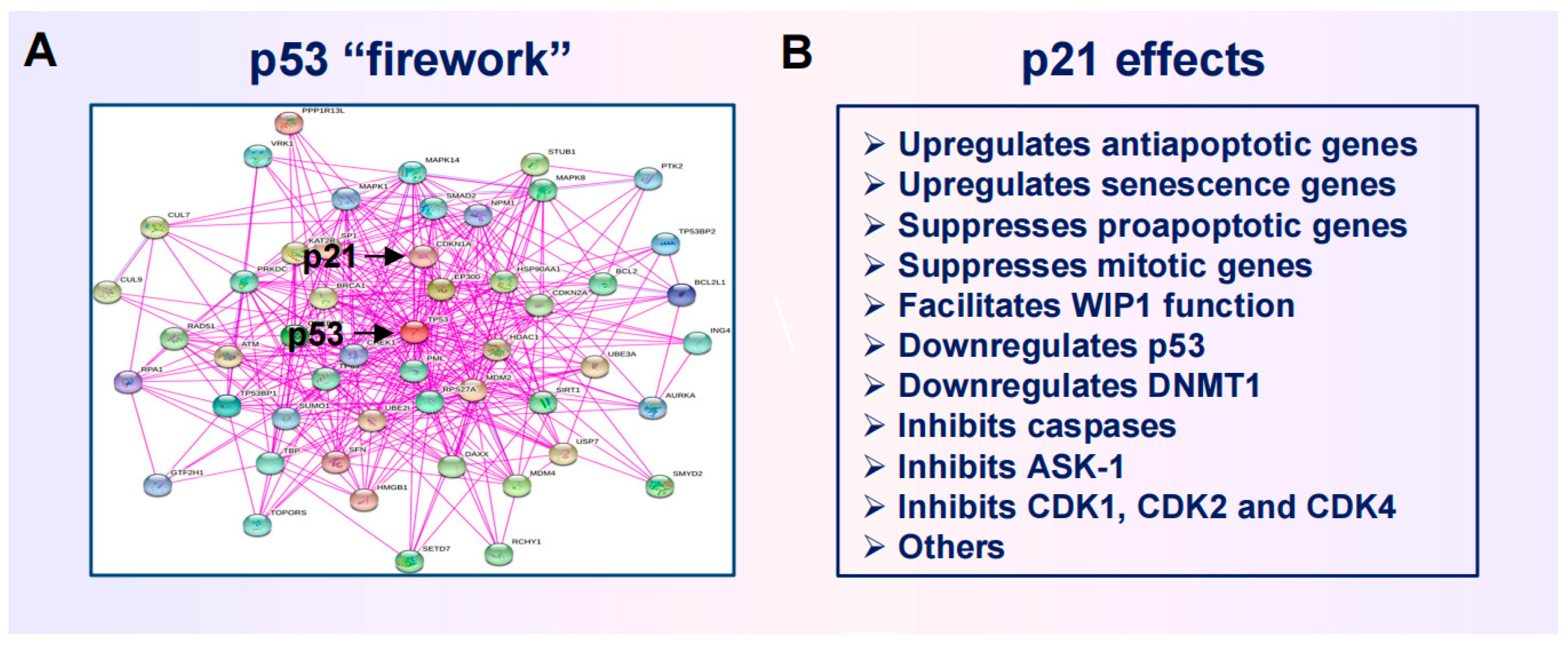
2.3. Anticancer Strategies Targeting the p53 “Firework”
3. Impact of Genome Chaos on Cancer Cell Resistance to Therapeutic Agents
3.1. Therapy-Induced Dormancy via Polyploidy/Multinucleation
3.2. The Creation of PGCCs Complicates the Interpretation of Chemosensitivity Data
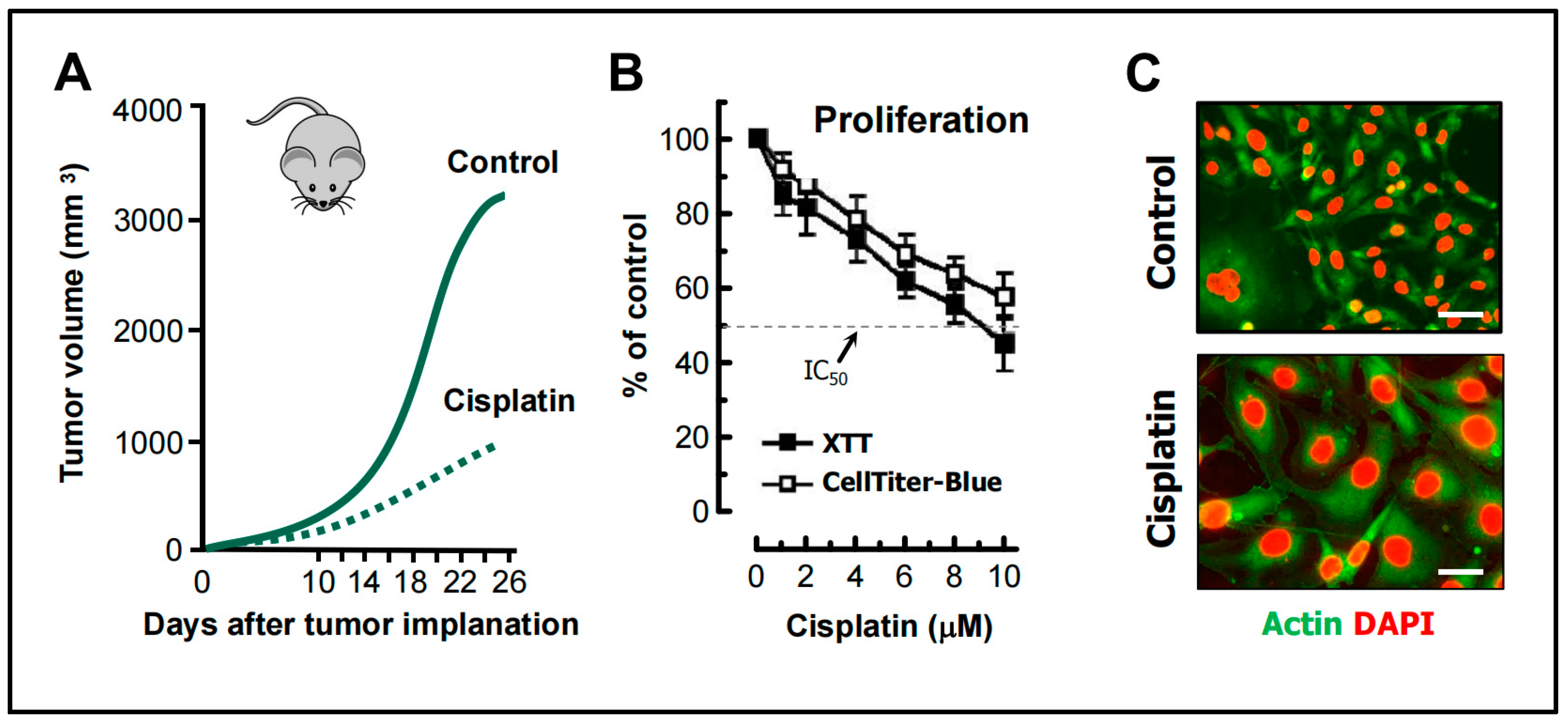
- Weng et al. [68] studied the mechanism of cisplatin resistance in B16-F10 (mouse melanoma) cells grown in C57BL (immune-proficient) mice. As expected, cisplatin treatment resulted in a significant reduction (by ~75%) of tumor volume compared to the control group (Figure 5A). Tumors in cisplatin-treated mice were highly enriched with PGCCs, which were shown to be more malignant than the parental cancer cells. A similar observation was reported by Puig et al. [70] with rat colon carcinoma isografts established in immunocompetent rats. In the latter study, tumor repopulation resulting from PGCCs was observed late (>40 days) after cisplatin treatment (for details, see [25]).
- Figure 5B shows the cisplatin sensitivity of MDA-MB-231 breast carcinoma cells evaluated by the 96-well plate XTT and CellTiter-Blue assays. The IC50 (half-maximum inhibitory concentration) values were ~10 µM.
- Further studies involving single cell assays demonstrated that cisplatin sensitivity reflected proliferation arrest via the creation of PGCCs (predominantly mononucleated giants; Figure 5C) and that virtually all emerging PGCCs remained adherent to the culture dish and remained viable and metabolically active [69].
3.3. Common Features of PGCCs and Senescent Cancer Cells
4. Rethinking Pro-Apoptotic Strategies to Combat Solid Tumors
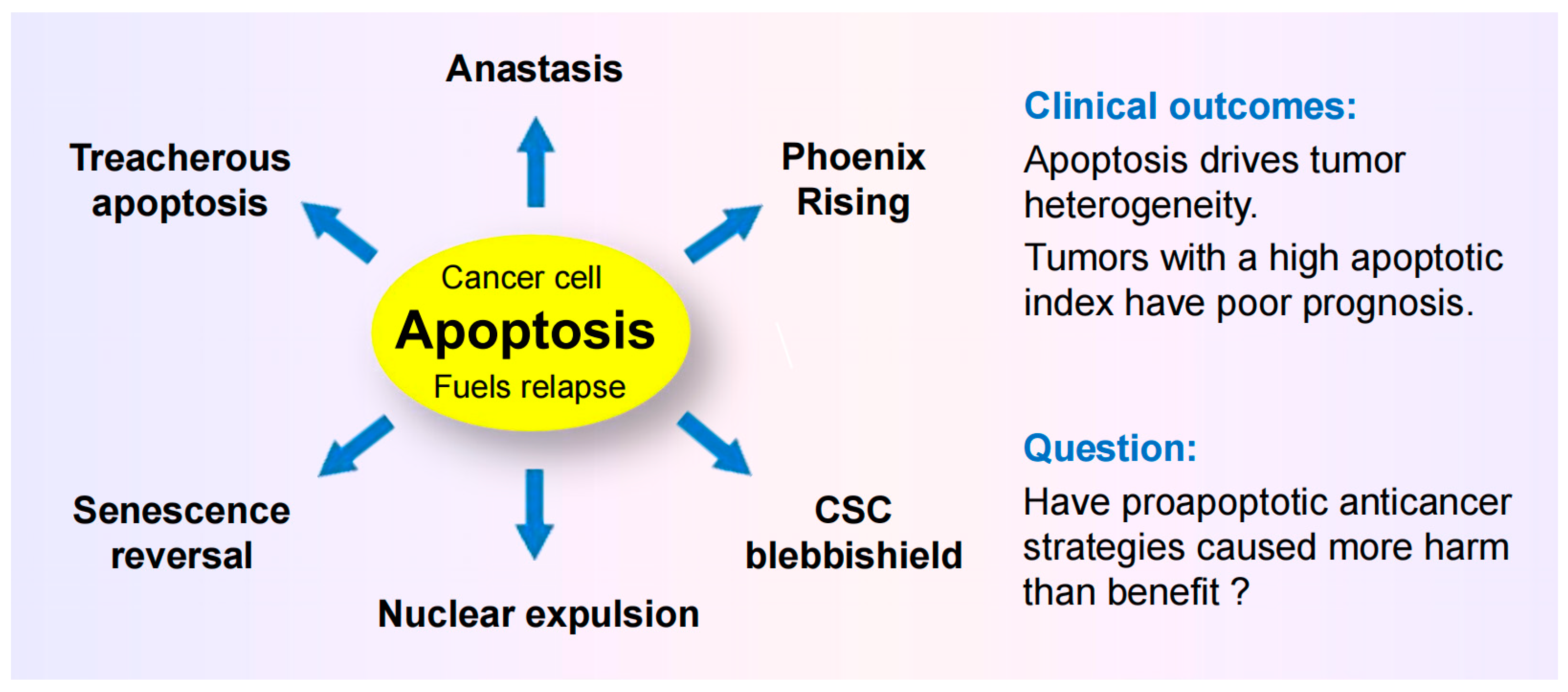
4.1. Dark Side of Apoptosis in Solid Tumor Therapy
4.1.1. Pro-Survival Function of Caspase 3
4.1.2. Anastasis
4.1.3. Treacherous Apoptosis
4.1.4. Nuclear Expulsion
4.1.5. Relationship between Apoptosis and Cancer Aggressiveness
4.2. Cancer Stem Cell Survival after Engaging Apoptosis
4.3. Questions
5. Exploiting the Apoptotic Threshold for Managing Solid Tumors
5.1. Apoptosis-Promoting Preclinical Chemosensitivity Studies Often Generate Clinically Irrelevant Information
- Cisplatin concentrations between 5 and 10 µM used in cell-based studies are determined to be comparable to concentrations that may be achieved in tumor/tissues of treated patients and tumor-bearing laboratory animals [70]. Higher concentrations result in severe side effects in animal studies [70]. Thus, ~10 µM cisplatin is denoted as the maximum tolerated dose (MTD) in Figure 6.
- The basis for this discrepancy is known [109]. Relatively low concentrations of cisplatin induce sufficient amounts of DNA lesions that inhibit cell proliferation, whereas very high drug concentrations are needed to damage the cytoplasmic compartments to engage apoptosis.
- Such a discrepancy in IC50 values for inhibition of cell proliferation and induction of apoptosis has been observed for various solid tumor-derived cell lines after exposure to cisplatin or other chemotherapeutic drugs (e.g., oxaliplatin, paclitaxel, and doxorubicin) (for details, see [108,109,110]).
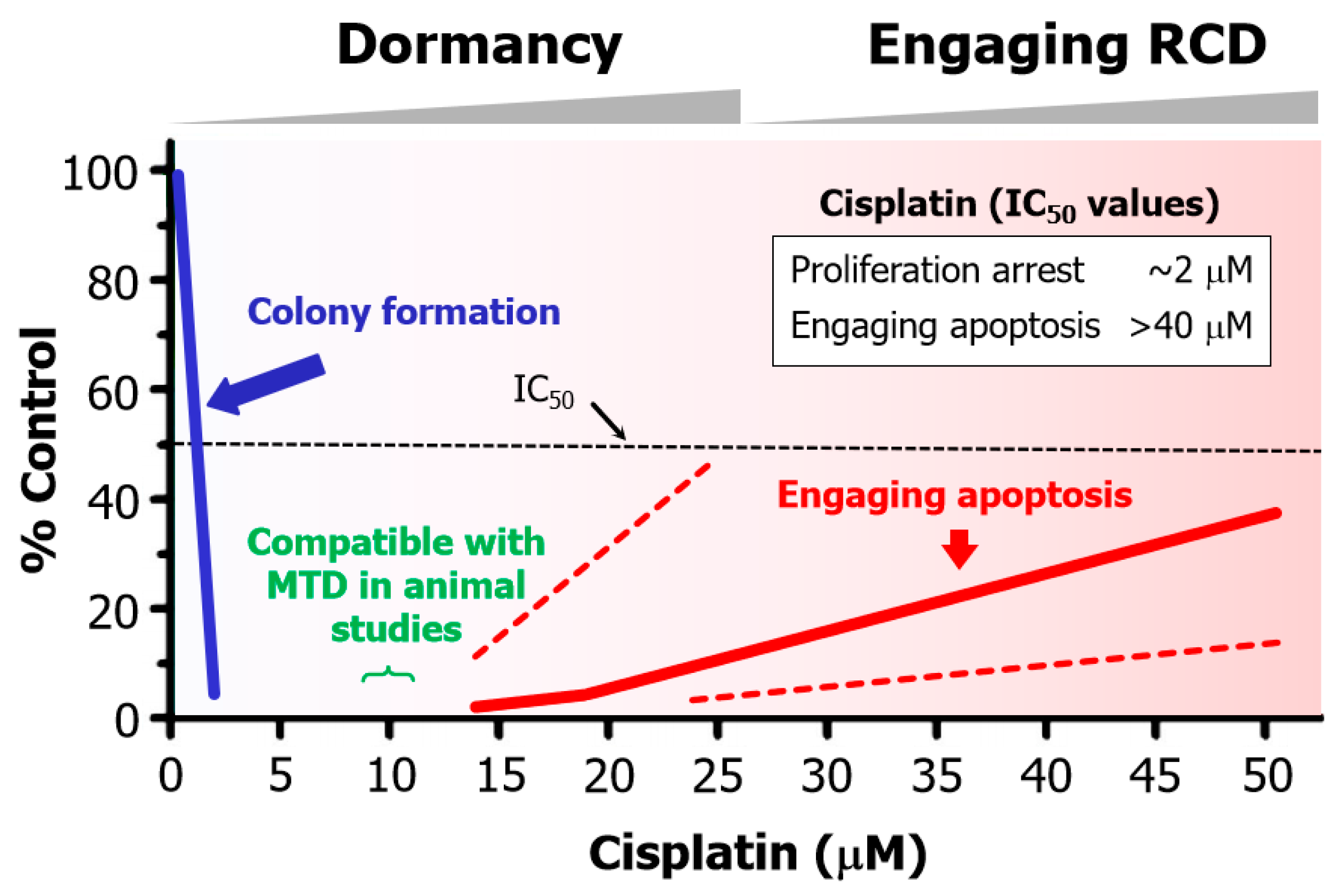
5.2. Chemotherapy-Induced Cancer Cell Dormancy: Lesser Evil Than Apoptosis?
6. Conclusions
6.1. Preclinical “Down” Assays have Caused More Harm Than Benefit
6.2. Conflicting Reports on Cancer Cell Fate after Engaging Apoptosis
6.3. Intratumor Heterogeneity: How Complex Does It Get?
6.4. Effective Anticancer Strategies Need to Target More Than One Therapy-Resistant Cancer Sub-Population within a Tumor
6.5. Where Will All This Lead?
6.6. Further Reading
Funding
Conflicts of Interest
Appendix A

References
- Prasad, V. Perspective: The precision-oncology illusion. Nature 2016, 537, S63. [Google Scholar] [CrossRef] [PubMed]
- Szabo, L. Cancer Treatment Hype Gives False Hope to Many Patients. Kaiser Health News. 2017. Available online: https://www.usatoday.com/story/news/2017/04/27/cancer-treatment-hype-gives-false-hope-many-patients/100972794/ (accessed on 15 April 2024).
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Joyner, M.J.; Paneth, N. Promises, promises, and precision medicine. J. Clin. Investig. 2019, 129, 946–948. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.-C.; Dawson, S.-J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Pich, O.; Bailey, C.; Watkins, T.B.K.; Zaccaria, S.; Jamal-Hanjani, M.; Swanton, C. The translational challenges of precision oncology. Cancer Cell 2022, 40, 458–478. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2022, 81, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Lohse, S. Mapping uncertainty in precision medicine: A systematic scoping review. J. Eval. Clin. Pract. 2023, 29, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Fojo, T. Journeys to failure that litter the path to developing new cancer therapeutics. JAMA Netw. Open 2023, 6, e2324949. [Google Scholar] [CrossRef] [PubMed]
- Suehnholz, S.P.; Nissan, M.H.; Zhang, H.; Kundra, R.; Nandakumar, S.; Lu, C.; Carrero, S.; Dhaneshwar, A.; Fernandez, N.; Xu, B.W.; et al. Quantifying the expanding landscape of clinical actionability for patients with cancer. Cancer Discov. 2024, 14, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Kailen, W.G. Preclinical Cancer Target Validation: How Not to Be Wrong. NIH Wednesday Afternoon Lectures (WELS) Series. 24 January 2018. Available online: https://videocast.nih.gov/watch=27066 (accessed on 15 April 2024).
- Belluz, J. Most Cancer Drugs Fail in Testing. This Might Be a Big Reason Why. Science—VOX Blog. 2019. Available online: https://www.vox.com/2019/9/16/20864066/cancer-studies-fail (accessed on 15 April 2024).
- Horgan, J. The Cancer Industry: Hype vs. Reality. Cancer Medicine Generates Enormous Revenues but Marginal Benefits for Patients. 2020. Available online: https://www.scientificamerican.com/blog/cross-check/the-cancer-industry-hype-vs-reality/ (accessed on 15 April 2024).
- Horgan, J. The Cancer Industry: Hype Versus Reality. 2023. Available online: https://johnhorgan.org/cross-check/the-cancer-industry-hype-versus-reality (accessed on 15 April 2024).
- Azra, R. The First Cell: And the Human Costs of Pursuing Cancer to the Last; Basic Books: New York, NY, USA, 2019. [Google Scholar]
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11, eaaw8412. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta. Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Sadri, A. Is target-based drug discovery efficient? Discovery and “off-target” mechanisms of all drugs. J. Med. Chem. 2023, 66, 12651–12677. [Google Scholar] [CrossRef] [PubMed]
- Bruin, M.A.C.; Sonke, G.S.; Beijnen, J.H.; Huitema, A.D.R. Pharmacokinetics and harmacodynamics of PARP inhibitors in oncology. Clin. Pharmacokinet. 2022, 61, 1649–1675. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Murray, D. Intratumor heterogeneity and therapy resistance: Contributions of dormancy, apoptosis reversal (anastasis) and cell fusion to disease recurrence. Int. J. Mol. Sci. 2020, 21, 1308. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of polyploid/multinucleated giant cancer cells in metastasis and disease relapse following anticancer treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. Coming full circle-from endless complexity to simplicity and back again. Cell 2014, 157, 267–271. [Google Scholar] [CrossRef]
- Kailen, W.G. Publish Houses of Brick, not Mansions of Straw. Nature 2017, 5454, 387. [Google Scholar]
- Mirzayans, R.; Murray, D. What are the reasons for continuing failures in cancer therapy? Are misleading/inappropriate preclinical assays to be blamed? Might some modern therapies cause more harm than benefit? Int. J. Mol. Sci. 2022, 23, 13217. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Murray, D. Intratumor heterogeneity and treatment resistance of solid tumors with a focus on polyploid/senescent giant cancer cells (PGCCs). Int. J. Mol. Sci. 2023, 24, 11534. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Uversky, V.N. p53 proteoforms and intrinsic disorder: An illustration of the protein structure-function continuum concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Iwakuma, T. Emerging non-canonical functions and regulation of p53. Int. J. Mol. Sci. 2018, 19, 1015. [Google Scholar] [CrossRef] [PubMed]
- Horvat, A.; Tadijan, A.; Vlaši’c, I.; Slade, N. p53/p73 Protein network in colorectal cancer and other human malignancies. Cancers 2021, 13, 2885. [Google Scholar] [CrossRef] [PubMed]
- Montero-Calle, A.; Garranzo-Asensio, M.; Torrente-Rodríguez, R.M.; Ruiz-Valdepeñas Montiel, V.; Poves, C.; Dziaková, J.; Sanz, R.; Díaz del Arco, C.; Pingarrón, J.M.; Fernández-Aceñero, M.J.; et al. p53 and p63 Proteoforms Derived from Alternative Splicing Possess Differential Seroreactivity in Colorectal Cancer with Distinct Diagnostic Ability from the Canonical Proteins. Cancers 2023, 15, 2102. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Sai, Y.; Zhou, L.; Liu, Z.; Du, P.; Wu, J.; Zhang, J. Current insights into the regulation of programmed cell death by TP53 mutation in cancer. Front. Oncol. 2022, 12, 1023427. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Berger, S.L. The interplay between epigenetic changes and the p53 protein in stem cells. Genes Dev. 2017, 31, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.E.; Reich, N.O. p53 and TDG are dominant in regulating the activity of the human de novo DNA methyltransferase DNMT3A on nucleosomes. J. Biol. Chem. 2021, 296, 100058. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.; Mirzayans, R.; McBride, W.H. Defenses against pro-oxidant forces—Maintenance of cellular and genomic integrity and longevity. Radiat. Res. 2018, 190, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Markowska, M.; Budzinska, M.A.; Coenen-Stass, A.; Kang, S.; Kizling, E.; Kolmus, K.; Koras, K.; Staub, E.; Szczurek, E. Synthetic lethality prediction in DNA damage repair, chromatin remodeling and the cell cycle using multi-omics data from cell lines and patients. Sci Rep. 2023, 13, 7049. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Pietrocola, F.; Guilbaud, E.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostini, M.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; et al. Apoptotic cell death in disease-Current understanding of the NCCD 2023. Cell Death Differ. 2023, 30, 1097–1154. [Google Scholar] [PubMed]
- Biswas, U.; Roy, R.; Ghosh, S.; Chakrabarti, G. The interplay between autophagy and apoptosis: Its implication in lung cancer and therapeutics. Cancer Lett. 2024, 585, 21666. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Kin, T.; Beck, W.T. Impact of complex apoptotic signaling pathways on cancer cell sensitivity to therapy. Cancers 2024, 16, 984. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Strasser, A.; Kayagaki, N.; Dixit, V.M. Cell death. Cell 2024, 187, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Webster, J.D.; Newton, K. Control of cell death in health and disease. Annu. Rev. Pathol. 2024, 19, 157–180. [Google Scholar] [CrossRef] [PubMed]
- Kulbay, M.; Paimboeuf, A.; Ozdemir, D.; Bernier, J. Review of cancer cell resistance mechanisms to apoptosis and actual targeted therapies. J. Cell. Biochem. 2022, 123, 1736–1761. [Google Scholar] [CrossRef] [PubMed]
- Tuval, A.; Strandgren, C.; Heldin, A.; Palomar-Siles, M.; Wiman, K.G. Pharmacological reactivation of p53 in the era of precision anticancer medicine. Nat. Rev. Clin. Oncol. 2024, 21, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. Targeting the p53 protein for cancer therapies: The translational impact of p53 research. Cancer Res. 2022, 82, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.W.; Beatty, P.H.; Lewis, J.D. Molecular targeting of the most functionally complex gene in precision oncology: p53. Cancers 2022, 14, 5176. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Pommier, Y. BRCAness, homologous recombination deficiencies, and synthetic lethality. Cancer Res. 2023, 83, 1173–1174. [Google Scholar] [CrossRef] [PubMed]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Cong, K.; Cantor, S.B. Exploiting replication gaps for cancer therapy. Mol. Cell 2022, 82, 2363–2369. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur. J. Med. Chem. 2022, 230, 114109. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: The first synthetic lethal targeted therapy. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.; Heng, H.H. Genome chaos, information creation, and cancer emergence: Searching for new frameworks on the 50th anniversary of the “war on cancer”. Genes 2022, 13, 101. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H. Genome Chaos: Rethinking Genetics, Evolution, and Molecular Medicine; Academic Press: Cambridge, MA, USA, 2019; p. 556. [Google Scholar]
- Ye, C.J.; Sharpe, Z.; Alemara, S.; Mackenzie, S.; Liu, G.; Abdallah, B.; Horne, S.; Regan, S.; Heng, H.H. Micronuclei and genome chaos: Changing the system inheritance. Genes 2019, 10, 366. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.A. How chaotic is genome chaos? Cancers 2021, 13, 1358. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Erenpreisa, J.; Sikora, E. Polyploid giant cancer cells: An emerging new field of cancer biology. Semin. Cancer Biol. 2022, 81, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Coward, J.; Harding, A. Size does matter: Why polyploid tumor cells are critical drug targets in the war on cancer. Front. Oncol. 2014, 4, 123. [Google Scholar] [CrossRef] [PubMed]
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef]
- Chen, J.; Niu, N.; Zhang, J.; Qi, L.; Shen, W.; Donkena, K.V.; Feng, Z.; Liu, J. Polyploid giant cancer cells (PGCCs): The evil roots of cancer. Curr. Cancer Drug Targets 2019, 19, 360–367. [Google Scholar] [CrossRef]
- Dudkowska, M.; Staniak, K.; Bojko, A.; Sikora, E. Chapter Five—The role of autophagy in escaping therapy-induced polyploidy/senescence Author links open overlay panel. Adv. Cancer Res. 2021, 150, 209–247. [Google Scholar] [PubMed]
- Zhang, J.; Qiao, Q.; Xu, H.; Zhou, R.; Liu, X. Human cell polyploidization: The good and the evil. Semin. Cancer Biol. 2022, 81, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Niu, N.; Li, X.; Zhang, X.; Sood, A.K. The life cycle of polyploid giant cancer cells and dormancy in cancer: Opportunities for novel therapeutic interventions. Semin. Cancer Biol. 2022, 81, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Trabzonlu, L.; Pienta, K.J.; Trock, B.J.; De Marzo, A.M.; Amend, S.R. Presence of cells in the polyaneuploid cancer cell (PACC) state predicts the risk of recurrence in prostate cancer. Prostate 2023, 83, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.H.; Wu, C.S.; Wu, J.C.; Kung, M.L.; Wu, M.H.; Tai, M.H. Cisplatin-induced giant cells formation is involved in chemoresistance of melanoma cells. Int. J. Mol. Sci. 2020, 21, 7892. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Murray, D. Impact of chemotherapeutic drugs on cancer cell proliferation, morphology and metabolic activity. J. Cancer Biol. Res. 2018, 6, 1118. [Google Scholar]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar] [PubMed]
- Barley, R.D.C.; Enns, L.; Paterson, M.C.; Mirzayans, R. Aberrant p21WAF1-dependent growth arrest as the possible mechanism of abnormal resistance to ultraviolet light cytotoxicity in Li-Fraumeni syndrome fifibroblast strains heterozygous for TP53 mutations. Oncogene 1998, 17, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wouters, B.G. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 1999, 59, 1391–1399. [Google Scholar] [PubMed]
- te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joe, S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar] [PubMed]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef] [PubMed]
- Tonnessen-Murray, C.A.; Frey, W.D.; Rao, S.G.; Shahbandi, A.; Ungerleider, N.A.; Olayiwola, J.O.; Murray, L.B.; Vinson, B.T.; Chrisey, D.B.; Lord, C.J.; et al. hemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J. Cell Biol. 2019, 218, 3827–3844. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Czarnecka, J.; Kominek, A.; Barszcz, K.; Bernas, T.; Piwocka, K.; Kaminska, B. Some chemotherapeutics-treated colon cancer cells display a specific phenotype being a combination of stem-like and senescent cell features. Cancer Biol. Ther. 2018, 19, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka-Herok, J.; Sliwinska, M.A.; Herok, M.; Targonska, A.; Strzeszewska-Potyrala, A.; Bojko, A.; Wolny, A.; Mosieniak, G.; Sikora, E. Therapy-induced senescent/polyploid cancer cells undergo atypical divisions associated with altered expression of meiosis, spermatogenesis and EMT genes. Int. J. Mol. Sci. 2022, 23, 8288. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Ichim, G.; Tait, S.W.G. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.; Tang, H.L. Anastasis: Recovery from the brink of cell death. R. Soc. Open Sci. 2018, 5, 180442. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Kaakati, R.; Lee, A.K.; Liu, X.; Li, F.; Li, C.-Y. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metastasis Rev. 2018, 37, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Berthenet, K.; Castillo Ferrer, C.; Fanfone, D.; Popgeorgiev, N.; Neves, D.; Bertolino, P.; Gibert, B.; Hernandez-Vargas, H.; Ichim, G. Failed apoptosis enhances melanoma cancer cell aggressiveness. Cell Rep. 2020, 31, 107731. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Murray, D. Do TUNEL and other apoptosis assays detect cell death in preclinical studies? Int. J. Mol. Sci. 2020, 21, 9090. [Google Scholar] [CrossRef] [PubMed]
- Zaitceva, V.; Kopeina, G.S.; Zhivotovsky, B. Anastasis: Return journey from cell death. Cancers 2021, 13, 3671. [Google Scholar] [CrossRef] [PubMed]
- Castillo Ferrer, C.; Berthenet, K.; Ichim, G. Apoptosis—Fueling the oncogenic fire. FEBS J. 2021, 288, 4445–4463. [Google Scholar] [CrossRef]
- Mohammed, R.N.; Khosravi, M.; Rahman, H.S.; Adili, A.; Kamali, N.; Soloshenkov, P.P.; Thangavelu, L.; Saeedi, H.; Shomali, N.; Tamjidifar; et al. Anastasis: Cell recovery mechanisms and potential role in cancer. Cell Commun. Signal. 2022, 20, 81. [Google Scholar] [CrossRef] [PubMed]
- Corsi, F.; Capradossi, F.; Pelliccia, A.; Briganti, S.; Bruni, E.; Traversa, E.; Torino, F.; Reichle, A.; Ghibelli, L. Apoptosis as driver of therapy-induced cancer repopulation and acquired cell-resistance (CRAC): A simple in vitro model of Phoenix Rising in prostate cancer. Int. J. Mol. Sci. 2022, 23, 1152. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, E.; Eaves, C.J. Paradoxical roles of caspase-3 in regulating cell survival, proliferation, and tumorigenesis. J. Cell Biol. 2022, 221, e202201159. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R. Treacherous apoptosis—Cancer cells sacrifice themselves at the altar of heterogeneity. Hepatology 2022, 76, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Brohl, A.S. Classical epithelial-mesenchymal transition (EMT) and alternative cell death process-driven blebbishield metastatic-witch (BMW) pathways to cancer metastasis. Signal Transduct. Target. Ther. 2022, 7, 296. [Google Scholar] [CrossRef]
- Kalkavan, H.; Rühl, S.; Shaw, J.J.P.; Green, D.R. Non-lethal outcomes of engaging regulated cell death pathways in cancer. Nat. Cancer 2023, 4, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Nano, M.; Montell, D.J. Apoptotic signaling: Beyond cell death. Semin. Cell Dev. Biol. 2024, 156, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.T.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 2012, 23, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.; Talbot, C.C., Jr.; Fung, M.C.; Tang, H.L. Molecular signature of anastasis for reversal of apoptosis. F1000Research 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Khatib, S.A.; Ma, L.; Dang, H.; Forgues, M.; Chung, J.-Y.; Ylaya, K.; Hewitt, S.M.; Chaisaingmongkol, J.; Rucchirawat, M.; Wang, X.W. Single-cell biology uncovers apoptotic cell death and its spatial organization as a potential modifier of tumor diversity in HCC. Hepatology 2022, 76, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Park, W.Y.; Gray, J.M.; Holewinski, R.J.; Andresson, T.; So, J.S.; Carmona-Rivera, C.; Hollander, M.C.; Yang, H.H.; Lee, M.; Kaplan, M.J.; et al. Apoptosis-induced nuclear expulsion in tumor cells drives S100a4-mediated metastatic outgrowth through the RAGE pathway. Nat. Cancer 2023, 4, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, M.P.V. The dark side of apoptosis. In Molecular Mechanisms of Tumor Cell Resistance to Chemotherapy, Resistance to Targeted Anti-Cancer Therapeutics 1; Bonavida, B., Ed.; Springer: New York, NY, USA, 2013; pp. 245–258. [Google Scholar]
- Wang, R.A.; Li, Q.L.; Li, Z.S.; Zheng, P.J.; Zhang, H.Z.; Huang, X.F.; Chi, S.M.; Yang, A.G.; Cui, R. Apoptosis drives cancer cells proliferate and metastasize. J. Cell. Mol. Med. 2013, 17, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.A.; Li, Z.S.; Yan, Q.G.; Bian, X.W.; Ding, Y.Q.; Du, X.; Sun, B.C.; Zhang, X.H. Resistance to apoptosis should not be taken as a hallmark of cancer. Chin. J. Cancer 2014, 33, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, S.; Cheng, I.; Deng, M.; He, X.; Wang, Z.; Yang, C.H.; Zhao, X.Y.; Huang, J. High expression of anti-apoptotic protein Bcl-2 is a good prognostic factor in colorectal cancer: Result of a metaanalysis. World J. Gastroenterol. 2017, 23, 5018–5033. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, L.; Meyer, M.; Fay, J.; Curry, S.; Bacon, O.; Duessmann, H.; John, K.; Boland, K.C.; McNamara, D.A.; Kay, E.W.; et al. Low levels of Caspase-3 predict favourable response to 5FU-based chemotherapy in advanced colorectal cancer: Caspase-3 inhibition as a therapeutic approach. Cell Death Dis. 2016, 7, e2087. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.; Storr, S.J.; Zhang, Y.; Rakha, E.A.; Green, A.R.; Ellis, I.O.; Martin, S.G. Caspase-3 and caspase-8 expression in breast cancer: Caspase-3 is associated with survival. Apoptosis 2017, 22, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Lindner, A.U.; Lucantoni, F.; Varešlija, D.; Resler, A.; Murphy, B.M.; Gallagher, W.M.; Hill, A.D.K.; Young, L.S.; Prehn, J.H.M. Low cleaved caspase-7 levels indicate unfavourable outcome across all breast cancers. Mol. Med. 2018, 96, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhong, D.N.; Qin, H.; Wu, P.R.; Wei, K.L.; Chen, G.; He, R.Q.; Zhong, J.C. Caspase-3 over-expression is associated with poor overall survival and clinicopathological parameters in breast cancer: A meta-analysis of 3091 cases. Oncotarget 2018, 9, 8629–8641. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Kamat, A.M. Endocytosis and serpentine filopodia drive blebbishield-mediated resurrection of apoptotic cancer stem cells. Cell Death Disco. 2016, 2, 15069. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. The growing complexity of cancer cell response to DNA-damaging agents: Caspase 3 mediates cell death or survival? Int. J. Mol. Sci. 2016, 17, 708. [Google Scholar] [CrossRef]
- Berndtsson, M.; Hägg, M.; Panaretakis, T.; Havelka, A.M.; Shoshan, M.C.; Linder, S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int. J. Cancer 2007, 120, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.; Mirzayans, R. Cellular responses to platinum-based anticancer drugs and UVC: Role of p53 and implications for cancer therapy. Int. J. Mol. Sci. 2020, 21, 5766. [Google Scholar] [CrossRef] [PubMed]
- Li, F.L.; Liu, J.P.; Bao, R.X.; Yan, G.Q.; Feng, X.; Xu, Y.P.; Sun, Y.P.; Yan, W.; Ling, A.Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 508. [Google Scholar] [CrossRef]
- Barkinge, J.L.; Gudi, R.; Sarah, H.; Chu, F.; Borthakur, A.; Prabhakar, B.S.; Prasad, K.V.S. The p53 induced Siva-1 plays a signifi cant role in cisplatin-induced apoptosis. J. Carcinog. 2009, 8, 2. [Google Scholar] [PubMed]
- Zhang, H.; Sun, J.; Ma, R.; Zhao, S. Role of episamarcandin in promoting the apoptosis of human colon cancer HCT116 cells through the PI3K-Akt signaling pathway. Evid. Based Complement. Alternat. Med. 2021, 2021, 9663738. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Improving anticancer drug development begins with cell culture: Misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget 2017, 8, 8854–8866. [Google Scholar] [CrossRef] [PubMed]
- Nicoletto, R.E.; Ofner, C.M. Cytotoxic mechanisms of doxorubicin at clinically relevant concentrations in breast cancer cells. Cancer Chemother. Pharmacol. 2022, 89, 285–311. [Google Scholar] [CrossRef] [PubMed]
- Gourdier, I.; Crabbe, L.; Andreau, K.; Pau, B.; Kroemer, G. Oxaliplatin-induced mitochondrial apoptotic response of colon carcinoma cells does not require nuclear DNA. Oncogene 2004, 23, 7449–7457. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R. When therapy-induced cancer cell apoptosis fuels tumor relapse. Onco 2024, 4, 37–45. [Google Scholar] [CrossRef]
- Jänicke, R.U.; Sohn, D.; Schulze-Osthoff, K. The dark side of a tumor suppressor: Anti-apoptotic p53. Cell Death Differ. 2008, 15, 959–976. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Altschuler, S.J.; Wu, L.F. Patterns of early p21 dynamics determine proliferation-senescence cell fate after chemotherapy. Cell 2019, 178, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.S.; Johnson, D.B.; Balko, J.M. Corticosteroids and Cancer Immunotherapy. Clin. Cancer Res. 2023, 29, 2580–2587. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of wild-type p53 signaling in suppressing apoptosis in response to chemical genotoxic agents: Impact on chemotherapy outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, J.L.; Prendergast, G.C.; Messerschmidt, G.L. How cancers escape immune destruction and mechanisms of action for the new significantly active immune therapies: Helping nonimmunologists decipher recent advances. Oncologist 2016, 21, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Son, B.; Lee, S.; Youn, H.S.; Kim, E.G.; Kim, W.; Youn, B.H. The role of tumor microenvironment in therapeutic resistance. Oncotarget 2017, 8, 3933–3945. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular plasticity in cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.D.; Pang, K.; Wu, Z.X.; Dong, Y.; Hao, L.; Qin, J.X.; Wang, W.; Chen, Z.S.; Han, C.H. Tumor cell plasticity in targeted therapy-induced resistance: Mechanisms and new strategies. Signal Transduct. Target. Ther. 2023, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Piper, K. Who Fakes Cancer Research? Apparently, Lots of People. Future Perfect—VOX Blog. 2024. Available online: https://www.vox.com/future-perfect/24086809/fake-cancer-research-data-scientific-fraud (accessed on 20 May 2024).
- Bush, E. A Prestigious Cancer Institute Is Correcting Dozens of Papers and Retracting Others after a Blogger Cried Foul. 2024. Available online: https://www.nbcnews.com/science/science-news/allegations-research-misconduct-roil-dana-farber-cancer-institute-rcna135521 (accessed on 20 May 2024).


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirzayans, R. Changing the Landscape of Solid Tumor Therapy from Apoptosis-Promoting to Apoptosis-Inhibiting Strategies. Curr. Issues Mol. Biol. 2024, 46, 5379-5396. https://doi.org/10.3390/cimb46060322
Mirzayans R. Changing the Landscape of Solid Tumor Therapy from Apoptosis-Promoting to Apoptosis-Inhibiting Strategies. Current Issues in Molecular Biology. 2024; 46(6):5379-5396. https://doi.org/10.3390/cimb46060322
Chicago/Turabian StyleMirzayans, Razmik. 2024. "Changing the Landscape of Solid Tumor Therapy from Apoptosis-Promoting to Apoptosis-Inhibiting Strategies" Current Issues in Molecular Biology 46, no. 6: 5379-5396. https://doi.org/10.3390/cimb46060322
APA StyleMirzayans, R. (2024). Changing the Landscape of Solid Tumor Therapy from Apoptosis-Promoting to Apoptosis-Inhibiting Strategies. Current Issues in Molecular Biology, 46(6), 5379-5396. https://doi.org/10.3390/cimb46060322