
Molecular Mechanisms in the Design of Novel Targeted Therapies for Neurodegenerative Diseases


Abstract
1. Introduction
2. Amyotrophic Lateral Sclerosis and Multiple Sclerosis
2.1. Clinical Symptoms and Course of Amyotrophic Lateral Sclerosis and Multiple Sclerosis
2.2. Molecular Mechanisms of Amyotrophic Lateral Sclerosis and Multiple Sclerosis Pathophysiology
2.3. Potential Treatment for Amyotrophic Lateral Sclerosis and Multiple Sclerosis
3. Alexander Disease
3.1. Clinical Symptoms and Course of Alexander Diseases
3.2. Molecular Mechanisms behind Alexander Disease
3.3. Potential Treatment for Alexander Disease
4. Huntington’s Disease
4.1. Clinical Symptoms and Course of Huntington’s Disease
4.2. Molecular Mechanisms of Huntington’s Disease
4.3. Potential Treatment for Huntington’s Disease
4.4. CRISPR-Cas9 in Huntington Disease Therapy
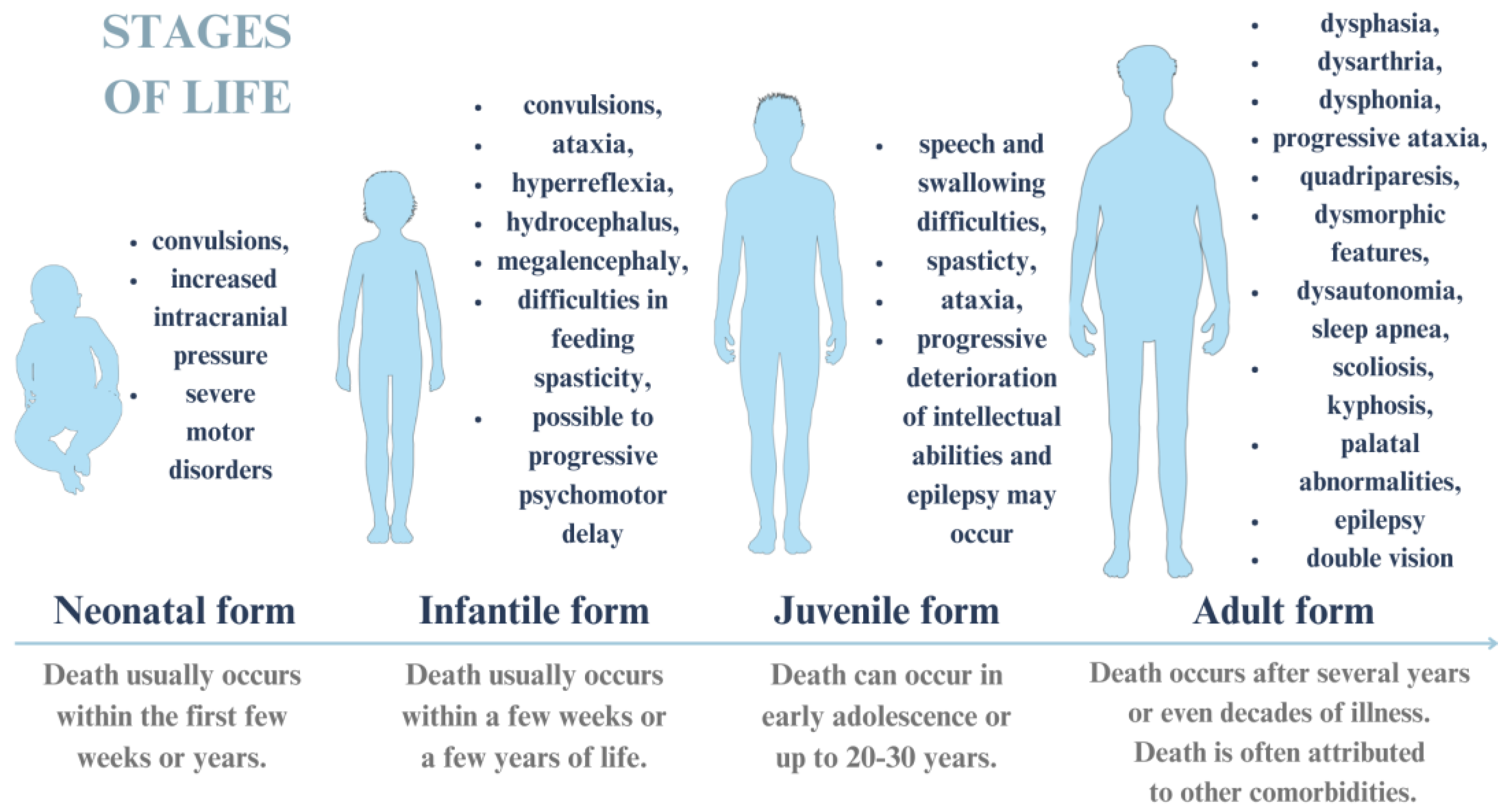
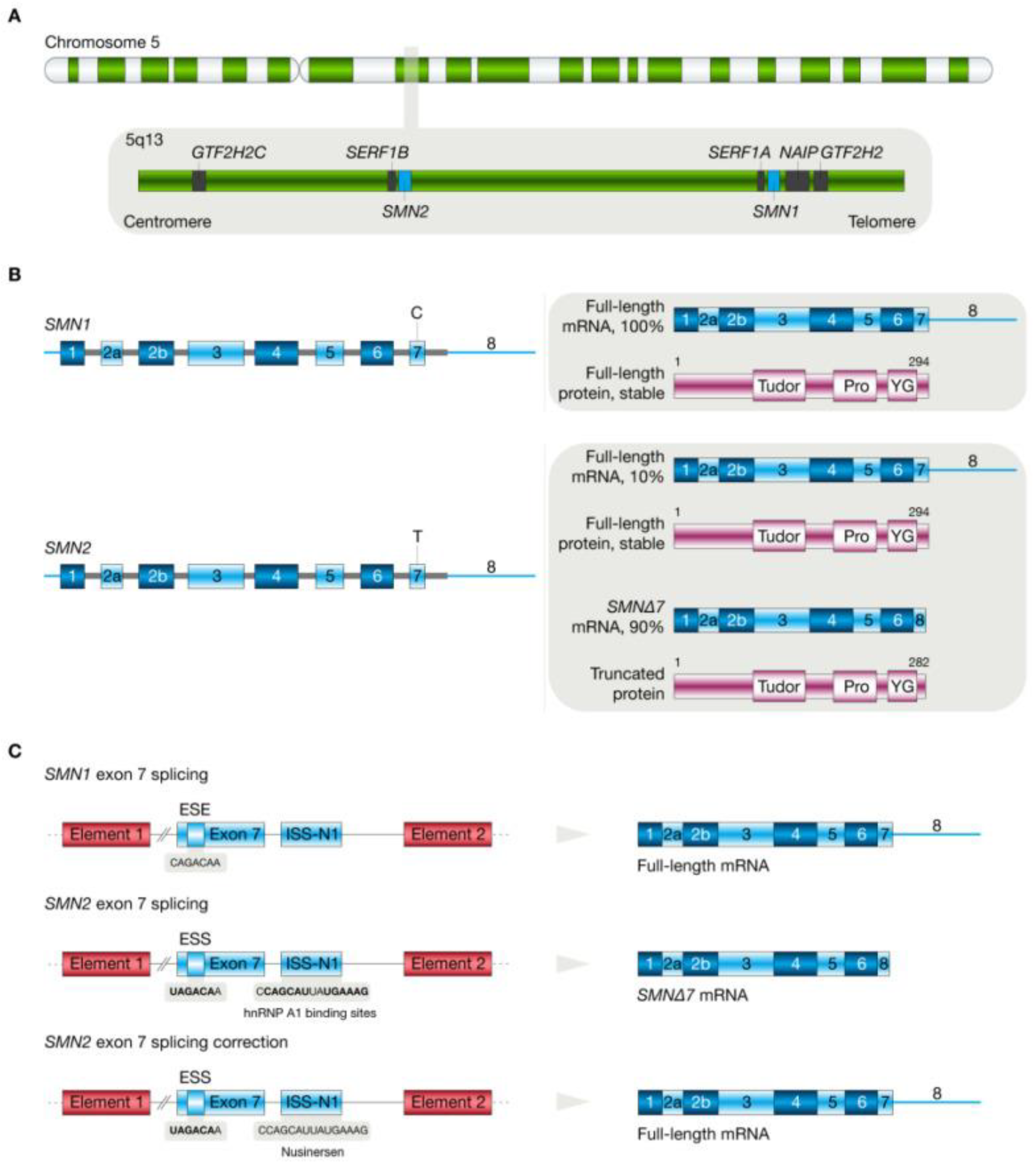
5. Spinal Muscular Atrophy
5.1. Molecular Pathogenesis of Spinal Muscular Atrophy
5.2. Clinical Symptoms and Course of Spinal Muscular Atrophy
5.3. Potential Treatments for Spinal Muscular Atrophy
5.4. Novel Treatment Options for Spinal Muscular Atrophy
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Heemels, M.-T. Neurodegenerative Diseases. Nature 2016, 539, 179. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Rabal, P.; González-Fuentes, J.; Castro-Vázquez, L.; Lozano, M.V.; Rodríguez-Robledo, V.; Santander-Ortega, M.J.; Selva-Clemente, J.; Villaseca-González, N.; Del Mar Arroyo-Jiménez, M. Neurodegenerative diseases: A multidisciplinary approach. CPD 2021, 27, 3305–3336. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, H.; Yin, Y. Microglia polarization from M1 to M2 in neurodegenerative diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef] [PubMed]
- Agnello, L.; Ciaccio, M. Neurodegenerative diseases: From molecular basis to therapy. Int. J. Mol. Sci. 2022, 23, 12854. [Google Scholar] [CrossRef] [PubMed]
- Schmoldt, A.; Benthe, H.F.; Haberland, G. Digitoxin metabolism by rat liver microsomes. Biochem. Pharmacol. 1975, 24, 1639–1641. [Google Scholar] [CrossRef] [PubMed]
- Hang, Z.; Zhou, L.; Xing, C.; Wen, Y.; Du, H. The blood-brain barrier, a key bridge to treat neurodegenerative diseases. Ageing Res. Rev. 2023, 91, 102070. [Google Scholar] [CrossRef] [PubMed]
- Nowak, I.; Madej, M.; Secemska, J.; Sarna, R.; Strzalka-Mrozik, B. Virus-based biological systems as next-generation carriers for the therapy of central nervous system diseases. Pharmaceutics 2023, 15, 1931. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.; Elman, L. Amyotrophic lateral sclerosis and other motor neuron diseases. CONTINUUM Lifelong Learn. Neurol. 2020, 26, 1323–1347. [Google Scholar] [CrossRef] [PubMed]
- Guennoc, A.; Pallix-Guyot, M.; Le Page, E.; Le Port, D.; Daryabin, M.; Hergesheimer, R.; Beltran, S.; Tourbah, A.; Edan, G.; Corcia, P. Co-occurrence of MS and ALS: A clue in favor of common pathophysiological findings? Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Deeb, O.; Nabulsi, M. Exploring multiple sclerosis (MS) and amyotrophic lateral sclerosis (ALS) as neurodegenerative diseases and their treatments: A review study. CTMC 2020, 20, 2391–2403. [Google Scholar] [CrossRef] [PubMed]
- Witzel, S.; Mayer, K.; Oeckl, P. Biomarkers for amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2022, 35, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Amezcua, L. Progressive multiple sclerosis. CONTINUUM Lifelong Learn. Neurol. 2022, 28, 1083–1103. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T. Amyotrophic lateral sclerosis (ALS)—Diagnosis, course of disease and treatment options. Dtsch. Med. Wochenschr. 2021, 146, 1613–1618. [Google Scholar] [PubMed]
- Yang, X.; Ji, Y.; Wang, W.; Zhang, L.; Chen, Z.; Yu, M.; Shen, Y.; Ding, F.; Gu, X.; Sun, H. Amyotrophic Lateral Sclerosis: Molecular Mechanisms, Biomarkers, and Therapeutic Strategies. Antioxidants 2021, 10, 1012. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Freischmidt, A. Update on genetics of amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2022, 35, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Porras, G.; Arribas-Blázquez, M.; Maestro, I.; Borrego-Hernández, D.; Boya, P.; Cerdán, S.; García-Redondo, A.; Martínez, A.; Martin-Requero, Á. Molecular Alterations in Sporadic and SOD1-ALS Immortalized Lymphocytes: Towards a Personalized Therapy. Int. J. Mol. Sci. 2021, 22, 3007. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Ticozzi, N.; Vance, C.; Leclerc, A.L.; Keagle, P.; Glass, J.D.; McKenna-Yasek, D.; Sapp, P.C.; Silani, V.; Bosco, D.A.; Shaw, C.E.; et al. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Van Daele, S.H.; Moisse, M.; van Vugt, J.J.F.A.; Zwamborn, R.A.J.; van der Spek, R.; van Rheenen, W.; Van Eijk, K.; Kenna, K.; Corcia, P.; Vourc’h, P.; et al. Genetic variability in sporadic amyotrophic lateral sclerosis. Brain 2023, 146, 3760–3769. [Google Scholar] [CrossRef] [PubMed]
- Siddique, N.; Siddique, T. Amyotrophic lateral sclerosis overview. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Dilliott, A.A.; Andary, C.M.; Stoltz, M.; Petropavlovskiy, A.A.; Farhan, S.M.K.; Duennwald, M.L. DnaJC7 in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2022, 23, 4076. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Cree, B.A.C. Treatment of multiple sclerosis: A review. Am. J. Med. 2020, 133, 1380–1390.e2. [Google Scholar] [CrossRef] [PubMed]
- Pokrishevsky, E.; Grad, L.I.; Cashman, N.R. TDP-43 or FUS-induced misfolded human wild-type SOD1 can propagate intercellularly in a prion-like fashion. Sci. Rep. 2016, 6, 22155. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Dighriri, I.M.; Aldalbahi, A.A.; Albeladi, F.; Tahiri, A.A.; Kinani, E.M.; Almohsen, R.A.; Alamoudi, N.H.; Alanazi, A.A.; Alkhamashi, S.J.; Althomali, N.A.; et al. An Overview of the History, Pathophysiology, and Pharmacological Interventions of Multiple Sclerosis. Cureus 2023, 15, e33242. [Google Scholar] [CrossRef] [PubMed]
- Vasileiou, E.S.; Fitzgerald, K.C. Multiple Sclerosis Pathogenesis and Updates in Targeted Therapeutic Approaches. Curr. Allergy Asthma Rep. 2023, 23, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Haki, M.; Al-Biati, H.A.; Al-Tameemi, Z.S.; Ali, I.S.; Al-Hussaniy, H.A. Review of multiple sclerosis: Epidemiology, etiology, pathophysiology, and treatment. Medicine 2024, 103, e37297. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.; Goldman, M.D. Epidemiology and Pathophysiology of Multiple Sclerosis. Continuum 2022, 28, 988–1005. [Google Scholar] [CrossRef] [PubMed]
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.; Riley, C. Treatment of multiple sclerosis. Continuum 2022, 28, 1025–1051. [Google Scholar] [CrossRef] [PubMed]
- Pozzilli, C.; Pugliatti, M.; Vermersch, P.; Grigoriadis, N.; Alkhawajah, M.; Airas, L.; Oreja-Guevara, C. Diagnosis and treatment of progressive multiple sclerosis: A position paper. Eur. J. Neurol. 2023, 30, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amytrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Fucito, M.; Pieragostino, D. Molecular research in multiple sclerosis. Int. J. Mol. Sci. 2022, 23, 2792. [Google Scholar] [CrossRef] [PubMed]
- Hartog, L.; Dekeyzer, S.; Bossche, S.V. Alexander disease. J. Belg. Soc. Radiol. 2021, 105, 61. [Google Scholar] [CrossRef] [PubMed]
- Bachetti, T.; Zanni, E.D.; Adamo, A.; Rosamilia, F.; Sechi, M.M.; Solla, P.; Bozzo, M.; Ceccherini, I.; Sechi, G. Beneficial effect of phenytoin and carbamazepine on GFAP gene expression and mutant GFAP folding in a cellular model of Alexander’s disease. Front. Pharmacol. 2021, 12, 723218. [Google Scholar] [CrossRef] [PubMed]
- Mura, E.; Nicita, F.; Masnada, S.; Battini, R.; Ticci, C.; Montomoli, M.; Berardinelli, A.; Pantaleoni, C.; Ardissone, A.; Foiadelli, T.; et al. Alexander disease evolution over time: Data from an Italian cohort of pediatric-onset patients. Mol. Genet. Metab. 2021, 134, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, A.C.; McCall, D.M.; Lange, H.; Renaud, D.; Brown, T.; Zaccariello, M.J. Neuropsychological functioning in Alexander disease: A case series. Child Neurol. Open 2021, 8, 2329048X2110486. [Google Scholar] [CrossRef] [PubMed]
- Devos, J.; Devriendt, K.; Richter, J.; Jansen, K.; Baldewijns, M.; Thal, D.R.; Aertsen, M. Fetal-onset Alexander disease with radiological-neuropathological correlation. Pediatr. Radiol. 2023, 53, 2149–2153. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.; Cascella, M. Alexander disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Srivastava, S.; Waldman, A.; Naidu, S. Alexander Disease. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Helman, G.; Takanohashi, A.; Hagemann, T.L.; Perng, M.D.; Walkiewicz, M.; Woidill, S.; Sase, S.; Cross, Z.; Du, Y.; Zhao, L.; et al. Type II Alexander disease caused by splicing errors and aberrant overexpression of an uncharacterized GFAP isoform. Hum. Mutat. 2020, 41, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, V.C.; Tian, R.; Long, H.; Der Perng, M.; Brenner, M.; Quinlan, R.; Goldman, E.J. Alexander-disease mutation of GFAP causes filament disorganization and decreased solubility of GFAP. J. Cell Sci. 2005, 118 Pt 9, 2057–2065. [Google Scholar] [CrossRef] [PubMed]
- Der Perng, M.; Su, M.; Fang Wen, S.; Li, R.; Gibbon, T.; Prescott, A.R.; Brenner, M.; Quinlan, R.A. The Alexander disease-causing glial fibrillary acidic protein mutant, R416W, accumulates into Rosenthal fibers by a pathway that involves filament aggregation and the association of alpha B-crystallin and HSP27. Am. J. Hum. Genet. 2006, 79, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Messing, A. Refining the concept of GFAP toxicity in Alexander disease. J. Neurodev. Disord. 2019, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Viedma-Poyatos, Á.; González-Jiménez, P.; Pajares, M.A.; Pérez-Sala, D. Alexander disease GFAP R239C mutant shows increased susceptibility to lipoxidation and elicits mitochondrial dysfunction and oxidative stress. Redox Biol. 2022, 55, 102415. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Messing, A. Regulation of GFAP Expression. ASN Neuro 2021, 13, 1759091420981206. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.L. Alexander disease: Models, mechanisms, and medicine. Curr. Opin. Neurobiol. 2022, 72, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Messing, A.; Brenner, M. GFAP at 50. ASN Neuro 2020, 12, 1759091420949680. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Hernández-Gerez, E.; Pekny, M.; Pérez-Sala, D. Alexander disease: The road ahead. Neural Regen. Res. 2023, 18, 2156. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tian, E.; Chen, X.; Chao, J.; Klein, J.; Qu, Q.; Sun, G.; Huang, Y.; Warden, C.D.; Ye, P.; et al. GFAP Mutations in Astrocytes Impair Oligodendrocyte Progenitor Proliferation and Myelination in an hiPSC Model of Alexander Disease. Cell Stem Cell 2018, 23, 239–251.e6. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.M.; Litscher, S.J.; Seitz, L.L.; Messing, A.; Hagemann, T.L.; Collier, L.S. Pexidartinib treatment in Alexander disease model mice reduces macrophage numbers and increases glial fibrillary acidic protein levels, yet has minimal impact on other disease phenotypes. J. Neuroinflamm. 2021, 18, 67. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.L.; Powers, B.; Lin, N.-H.; Mohamed, A.F.; Dague, K.L.; Hannah, S.C.; Bachmann, G.; Mazur, C.; Rigo, F.; Olsen, A.L.; et al. Antisense therapy in a rat model of Alexander disease reverses GFAP pathology, white matter deficits, and motor impairment. Sci. Transl. Med. 2021, 13, eabg4711. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.L.; Powers, B.; Mazur, C.; Kim, A.; Wheeler, S.; Hung, G.; Swayze, E.; Messing, A. Antisense suppression of glial fibrillary acidic protein as a treatment for Alexander disease. Ann. Neurol. 2018, 83, 27–39. [Google Scholar] [CrossRef] [PubMed]
- McCall, M.A.; Gregg, R.G.; Behringer, R.R.; Brenner, M.; Delaney, C.L.; Galbreath, E.J.; Zhang, C.L.; Pearce, R.A.; Chiu, S.Y.; Messing, A. Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc. Natl. Acad. Sci. USA 1996, 93, 6361–6366. [Google Scholar] [CrossRef] [PubMed]
- Stowe, N.A.; Singh, A.P.; Barnett, B.R.; Yi, S.Y.; Frautschi, P.C.; Messing, A.; Hagemann, T.L.; Yu, J.J. Quantitative diffusion imaging and genotype-by-sex interactions in a rat model of Alexander disease. Magn. Reson. Med. 2024, 91, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Schobel, S.; Gantman, E.C.; Mansbach, A.; Borowsky, B.; Konstantinova, P.; Mestre, T.A.; Panagoulias, J.; Ross, C.A.; Zauderer, M.; et al. A biological classification of Huntington’s disease: The integrated staging system. Lancet Neurol. 2022, 21, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Handley, R.R.; Lehnert, K.; Snell, R.G. From pathogenesis to therapeutics: A review of 150 years of Huntington’s disease research. Int. J. Mol. Sci. 2023, 24, 13021. [Google Scholar] [CrossRef] [PubMed]
- Stoker, T.B.; Mason, S.L.; Greenland, J.C.; Holden, S.T.; Santini, H.; Barker, R.A. Huntington’s disease: Diagnosis and management. Pract. Neurol. 2022, 22, 32–41. [Google Scholar] [CrossRef]
- Pellegrini, M.; Bergonzoni, G.; Perrone, F.; Squitieri, F.; Biagioli, M. Current diagnostic methods and non-coding RNAs as possible biomarkers in Huntington’s disease. Genes 2022, 13, 2017. [Google Scholar] [CrossRef]
- Hong, E.P.; MacDonald, M.E.; Wheeler, V.C.; Jones, L.; Holmans, P.; Orth, M.; Monckton, D.G.; Long, J.D.; Kwak, S.; Gusella, J.F.; et al. Huntington’s disease pathogenesis: Two sequential components. JHD 2021, 10, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Jurcau, C.M. Mitochondria in Huntington’s disease: Implications in pathogenesis and mitochondrial-targeted therapeutic strategies. Neural Regen. Res. 2023, 18, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fu, Z.; Wu, S.; Wang, X.; Zhang, S.; Chu, C.; Hong, Y.; Wu, W.; Chen, S.; Jiang, Y.; et al. Mitochondrial HSF1 triggers mitochondrial dysfunction and neurodegeneration in Huntington’s disease. EMBO Mol. Med. 2022, 14, e15851. [Google Scholar] [CrossRef] [PubMed]
- Šonský, I.; Vodička, P.; Vodičková Kepková, K.; Hansíková, H. Mitophagy in Huntington’s disease. Neurochem. Int. 2021, 149, 105147. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A. Molecular pathophysiological mechanisms in Huntington’s disease. Biomedicines 2022, 10, 1432. [Google Scholar] [CrossRef] [PubMed]
- Caron, N.S.; Southwell, A.L.; Brouwers, C.C.; Cengio, L.D.; Xie, Y.; Black, H.F.; Anderson, L.M.; Ko, S.; Zhu, X.; van Deventer, S.J.; et al. Potent and sustained huntingtin lowering via AAV5 encoding miRNA preserves striatal volume and cognitive function in a humanized mouse model of Huntington disease. Nucleic Acids Res. 2019, gkz976. [Google Scholar] [CrossRef] [PubMed]
- Alkanli, S.S.; Alkanli, N.; Ay, A.; Albeniz, I. CRISPR/Cas9 mediated therapeutic approach in Huntington’s disease. Mol. Neurobiol. 2023, 60, 1486–1498. [Google Scholar] [CrossRef] [PubMed]
- Fields, E.; Vaughan, E.; Tripu, D.; Lim, I.; Shrout, K.; Conway, J.; Salib, N.; Lee, Y.; Dhamsania, A.; Jacobsen, M.; et al. Gene targeting techniques for Huntington’s disease. Ageing Res. Rev. 2021, 70, 101385. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Jurcau, M.C. Therapeutic strategies in Huntington’s disease: From genetic defect to gene therapy. Biomedicines 2022, 10, 1895. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Cong, S. MicroRNAs in Huntington’s disease: Diagnostic biomarkers or therapeutic agents? Front. Cell. Neurosci. 2021, 15, 705348. [Google Scholar] [CrossRef] [PubMed]
- Spronck, E.; Vallès, A.; Lampen, M.; Montenegro-Miranda, P.; Keskin, S.; Heijink, L.; Evers, M.; Petry, H.; Deventer, S.; Konstantinova, P.; et al. Intrastriatal administration of AAV5-MiHTT in non-human primates and rats is well tolerated and results in miHTT transgene expression in key areas of Huntington disease pathology. Brain Sci. 2021, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Bräuer, S.; Falkenburger, B. Gentherapie der Huntington-krankheit [Gene therapy for Huntington disease]. Fortschr. Neurol. Psychiatr. 2023, 91, 141–146. [Google Scholar] [PubMed]
- DiFiglia, M.; Sena-Esteves, M.; Chase, K.; Sapp, E.; Pfister, E.; Sass, M.; Yoder, J.; Reeves, P.; Pandey, R.K.; Rajeev, K.G.; et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Estevez-Fraga, C.; Van Roon-Mom, W.M.C.; Flower, M.D.; Scahill, R.I.; Wild, E.J.; Muñoz-Sanjuan, I.; Sampaio, C.; Rosser, A.E.; Leavitt, B.R. Potential disease-modifying therapies for Huntington’s disease: Lessons learned and future opportunities. Lancet Neurol. 2022, 21, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Shin, J.H.; Lee, J.; Kim, D.; Hwang, H.-Y.; Nam, B.-G.; Lee, J.; Kim, H.H.; Cho, S.-R. DNA Double-strand break-free CRISPR interference delays Huntington’s disease progression in mice. Commun. Biol. 2023, 6, 466. [Google Scholar] [CrossRef] [PubMed]
- Oura, S.; Noda, T.; Morimura, N.; Hitoshi, S.; Nishimasu, H.; Nagai, Y.; Nureki, O.; Ikawa, M. Precise CAG repeat contraction in a Huntington’s disease mouse model is enabled by gene editing with SpCas9-NG. Commun. Biol. 2021, 4, 771. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Jiang, L.; Yu, Y.; Cui, Y.; Chen, Y.; Zhou, D.; Gao, F.; Mao, S. Optimized MLPA workflow for spinal muscular atrophy diagnosis: Identification of a novel variant, NC_000005.10:g.(70919941_70927324)del in isolated exon 1 of SMN1 gene through long-range PCR. BMC Neurol. 2024, 24, 93. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Eulate, G.; Theuriet, J.; Record, C.J.; Querin, G.; Masingue, M.; Leonard-Louis, S.; Behin, A.; Le Forestier, N.; Pegat, A.; Michaud, M.; et al. Phenotype presentation and molecular diagnostic yield in non-5q spinal muscular atrophy. Neurol. Genet. 2023, 9, e200087. [Google Scholar] [CrossRef] [PubMed]
- Aasdev, A.R.S.S.; Iyer, V.R.; Moharir, S.C. Spinal muscular atrophy: Molecular mechanism of pathogenesis, diagnosis, therapeutics, and clinical trials in the Indian context. J. Biosci. 2024, 49, 36. [Google Scholar] [CrossRef]
- Chen, T.H. New and developing therapies in spinal muscular atrophy: From genotype to phenotype to treatment and where do we stand? Int. J. Mol. Sci. 2020, 21, 3297. [Google Scholar] [CrossRef] [PubMed]
- Angilletta, I.; Ferrante, R.; Giansante, R.; Lombardi, L.; Babore, A.; Dell’Elice, A.; Alessandrelli, E.; Notarangelo, S.; Ranaudo, M.; Palmarini, C.; et al. Spinal muscular atrophy: An evolving scenario through new perspectives in diagnosis and advances in therapies. Int. J. Mol. Sci. 2023, 24, 14873. [Google Scholar] [CrossRef] [PubMed]
- López-Cortés, A.; Echeverría-Garcés, G.; Ramos-Medina, M.J. Molecular pathogenesis and new therapeutic dimensions for spinal muscular atrophy. Biology 2022, 11, 894. [Google Scholar] [CrossRef] [PubMed]
- Nishio, H.; Niba, E.T.E.; Saito, T.; Okamoto, K.; Takeshima, Y.; Awano, H. Spinal muscular atrophy: The past, present, and future of diagnosis and treatment. Int. J. Mol. Sci. 2023, 24, 11939. [Google Scholar] [CrossRef] [PubMed]
- Babić, M.; Banović, M.; Berečić, I.; Banić, T.; Babić Leko, M.; Ulamec, M.; Junaković, A.; Kopić, J.; Sertić, J.; Barišić, N.; et al. Molecular biomarkers for the diagnosis, prognosis, and pharmacodynamics of spinal muscular atrophy. J. Clin. Med. 2023, 12, 5060. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.B.; Ahmed, H.; Zaman, A.; Ali, A.M.N.; Shah, H.H.; Rauf, S.A.; Dave, T. Spinal muscular atrophy type 1: A fatal case in a 1-year-old girl with delayed diagnosis. Clin. Case Rep. 2024, 12, e8513. [Google Scholar] [CrossRef] [PubMed]
- Keinath, M.C.; Prior, D.E.; Prior, T.W. Spinal muscular atrophy: Mutations, testing, and clinical relevance. Appl. Clin. Genet. 2021, 14, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Fang, C.Y.; Chang, C.C.; Chiang, C.K.; Chen, Y.W. A rapid molecular diagnostic method for spinal muscular atrophy. J. Neurogenet. 2021, 35, 29–32. [Google Scholar] [CrossRef]
- Kleinle, S.; Scholz, V.; Benet-Pagés, A.; Wohlfrom, T.; Gehling, S.; Scharf, F.; Rost, S.; Prott, E.C.; Grinzinger, S.; Hotter, A.; et al. Closing the gap—Detection of 5q-spinal muscular atrophy by short-read next-generation sequencing and unexpected results in a diagnostic patient cohort. J. Neuromuscul. Dis. 2023, 10, 835–846. [Google Scholar] [CrossRef]
- Theil, D.; Valdez, R.; Darribat, K.; Doelemeyer, A.; Sivasankaran, R.; Hartmann, A. Orally administered branaplam does not impact neurogenesis in juvenile mice, rats, and dogs. Biol. Open 2021, 10, bio058551. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W.; Singh, R.N. Synergistic effect of an antisense oligonucleotide and small molecule on splicing correction of the spinal muscular atrophy gene. Neurosci. Insights 2024, 19, 26331055241233596. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, Y.; Wong, M.S.; Martí-Gómez, C.; Ayaz, A.; Kooshkbaghi, M.; Hanson, S.M.; McCandlish, D.M.; Krainer, A.R.; Kinney, J.B. Specificity, synergy, and mechanisms of splice-modifying drugs. Nat. Commun. 2024, 15, 1880. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Opazo, A.; Garrison, S.; Waite, M. Molecular biomarkers for spinal muscular atrophy: A systematic review. Neurol. Clin. Pract. 2021, 11, e524–e536. [Google Scholar] [CrossRef] [PubMed]
- Rech, J.; Getinger-Panek, A.; Gałka, S.; Bednarek, I. Origin and composition of exosomes as crucial factors in designing drug delivery systems. Appl. Sci. 2022, 12, 12259. [Google Scholar] [CrossRef]
- Kurowska, N.; Strzalka-Mrozik, B.; Madej, M.; Pająk, K.; Kruszniewska-Rajs, C.; Kaspera, W.; Gola, J.M. Differences in the expression patterns of TGFβ isoforms and associated genes in astrocytic brain tumors. Cancers 2022, 14, 1876. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: First Regulatory Approvals for CRISPR-Cas9 Therapeutic Gene Editing for Sickle Cell Disease and Transfusion-Dependent β-Thalassemia. Med. Sci. Monit. 2024, 30, e944204. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Type of Therapy | Therapeutic | Model Organism | Delivery Strategy | Therapeutic Effect | Ref. |
|---|---|---|---|---|---|
| miRNA | AAV5-miHTT | Mice R6/2 HD | Bilateral AAV5-miHTT injection into the thalamus, intradurally | Improved motor coordination, increased survival time, alleviation of neuronal dysfunction, reduced changes in striatum and cortex | [74] |
| miRNA | AAV5-miHTT | Mice Q175 HD | Injection of AAV5-miHTT into the striatum | Dose-dependent reduction in mHTT aggregates in striatum and cortex, improved dyskinesia, prolonged survival time media | [74] |
| miRNA | AMT-130 | Patients HD | Injection of AAV5-miHTT into the striatum | Improved motor coordination and long-term reduction in mHTT, high specificity and no off-target effects observed | [75,76] |
| siRNA | cc-siRNA-Htt | Mice | Injection into the right striatum | Weakening of motor abnormalities and reduction in mHtt expression | [77,78] |
| ASO | MkHuASO | Rhesus monkeys | Administration of Aso into the cerebrospinal fluid | Sustained reduction in Huntington mRNA in most areas of the brain and spinal cord | [78,79] |
| CRISPR-Cas9 | dCas9-sgRNA | Mice R6/2 HD | Injection into the striatum | Improves motor functions and delays their progression | [80] |
| CRISPR-Cas9 | SpCas9-NG | Mice R6/2 HD | Microinjection into the mouse zygote | Contraction of CAG repeats, improvement of HD phenotype | [81] |
| Research Title | Drug/Molecule | Status | Application Route | NCT Number | Phase | Participants |
|---|---|---|---|---|---|---|
| A Study to Evaluate Higher Dose (HD) Nusinersen (BIIB058) in Participants with Spinal Muscular Atrophy Previously Treated with Risdiplam (ASCEND) | Higher Dose Nusinersen (BIIB058) | Recruiting | Intrathecally | NCT05067790 | III | 45 |
| Study of the Functional Effects of Nusinersen in 5q-Spinal Muscular Amyotrophy Adults (SMA Type 2 or 3 Forms) (NUSI-AD-5qSM) | Nusinersen | Recruiting | Intrathecally | NCT04576494 | N/A | 24 |
| Clinical Trial to Assess the Safety and Efficacy of EXG001-307 in Patients with Spinal Muscular Atrophy Type 1 | EXG001-307 | Recruiting | Intravenously injection | NCT05614531 | I/II | 12 |
| A Clinical Study Evaluating the Safety and Efficacy of SKG0201 Injection in Patients with Spinal Muscular Atrophy Type 1 | SKG0201 | Recruiting | Injection | NCT06191354 | N/A | 12 |
| Evaluation of Safety and Efficacy of Gene Therapy Drug in the Treatment of Spinal Muscular Atrophy (SMA) Type 1 Patients | GC101 | Recruiting | Intrathecally | NCT05824169 | I/II | 18 |
| Phase IIIb, Open-label, Multicenter Study to Evaluate Safety, Tolerability and Efficacy of OAV101 Administered Intrathecally to Participants with SMA Who Discontinued Treatment with Nusinersen or Risdiplam (STRENGTH) | OAV101 | Active, non-recruiting | Intrathecally | NCT05386680 | IIIb | 27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowak, I.; Paździor, M.; Sarna, R.; Madej, M. Molecular Mechanisms in the Design of Novel Targeted Therapies for Neurodegenerative Diseases. Curr. Issues Mol. Biol. 2024, 46, 5436-5453. https://doi.org/10.3390/cimb46060325
Nowak I, Paździor M, Sarna R, Madej M. Molecular Mechanisms in the Design of Novel Targeted Therapies for Neurodegenerative Diseases. Current Issues in Molecular Biology. 2024; 46(6):5436-5453. https://doi.org/10.3390/cimb46060325
Chicago/Turabian StyleNowak, Ilona, Marlena Paździor, Robert Sarna, and Marcel Madej. 2024. "Molecular Mechanisms in the Design of Novel Targeted Therapies for Neurodegenerative Diseases" Current Issues in Molecular Biology 46, no. 6: 5436-5453. https://doi.org/10.3390/cimb46060325
APA StyleNowak, I., Paździor, M., Sarna, R., & Madej, M. (2024). Molecular Mechanisms in the Design of Novel Targeted Therapies for Neurodegenerative Diseases. Current Issues in Molecular Biology, 46(6), 5436-5453. https://doi.org/10.3390/cimb46060325