
Genomic Evolution Strategy in SARS-CoV-2 Lineage B: Coevolution of Cis Elements

 , ,
, ,  , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of Cis Elements in the Genome of Human Pathogenic Coronaviruses
2.2. Secondary Structures of Cis and ORF Elements of Coronavirus Genomes
2.3. Mutational Distance and Evolutionary Relationships of Cis Elements in Coronaviruses
3. Results
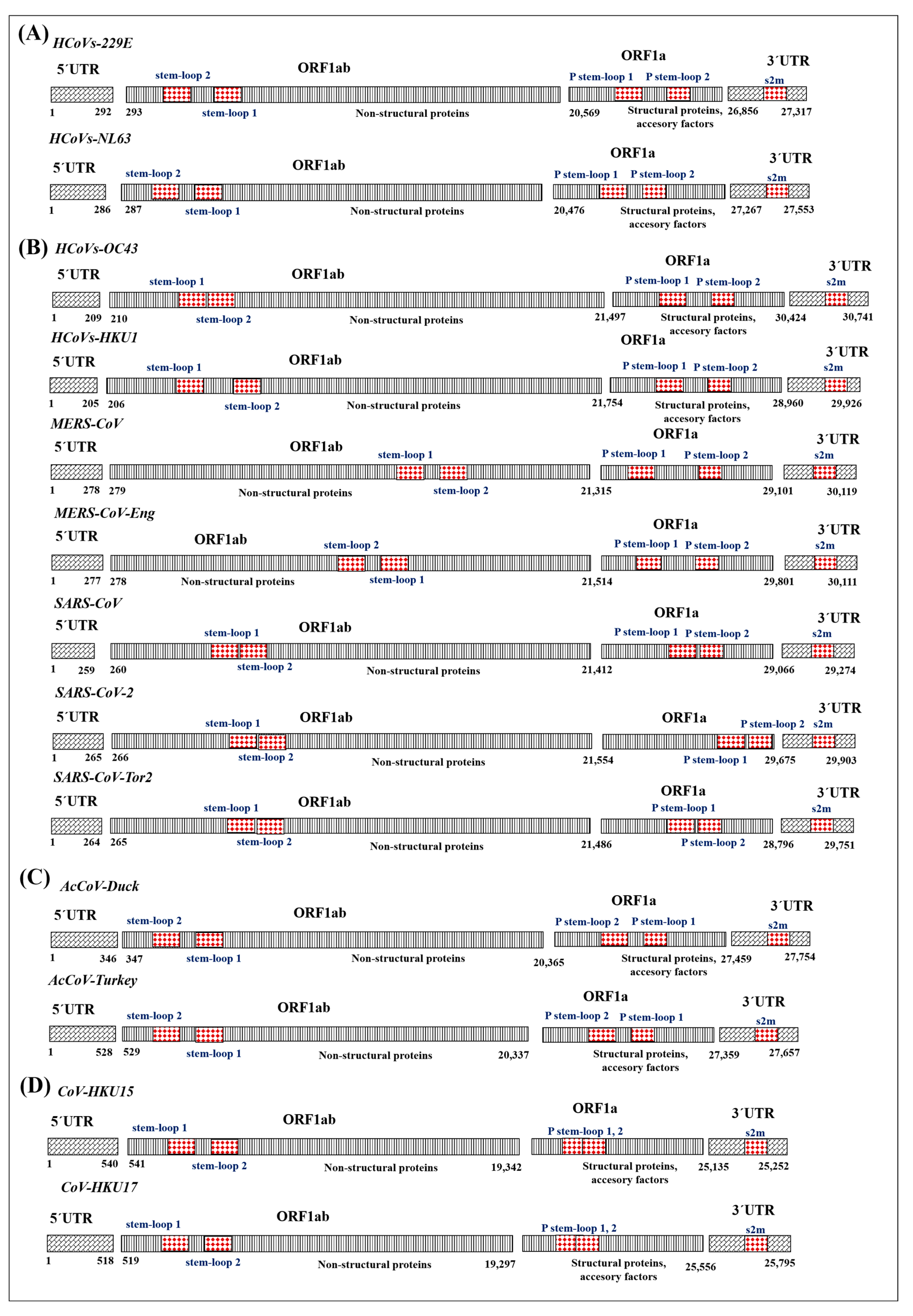
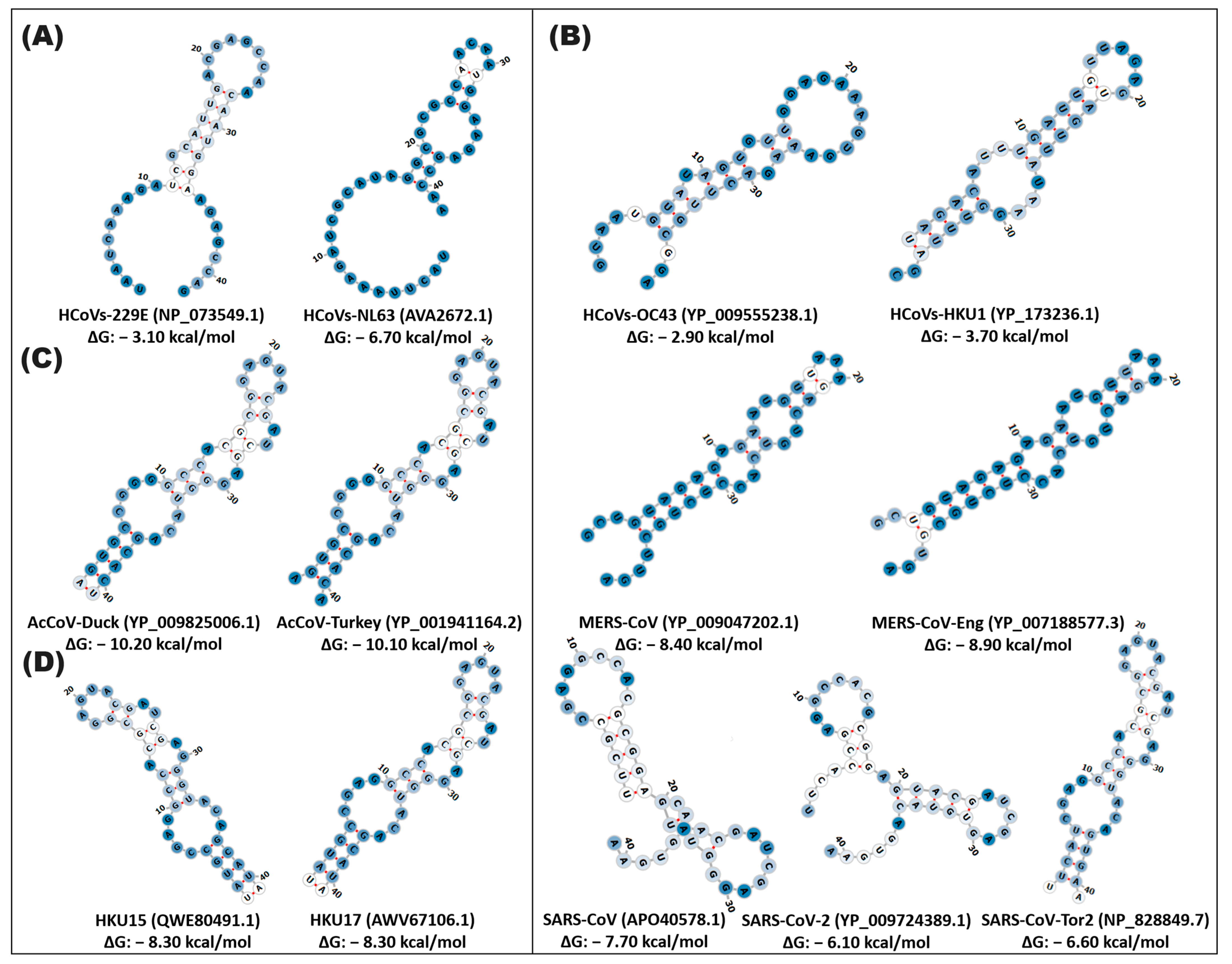
3.1. Identification and Organization of Cis Elements in Human Pathogenic Coronaviruses
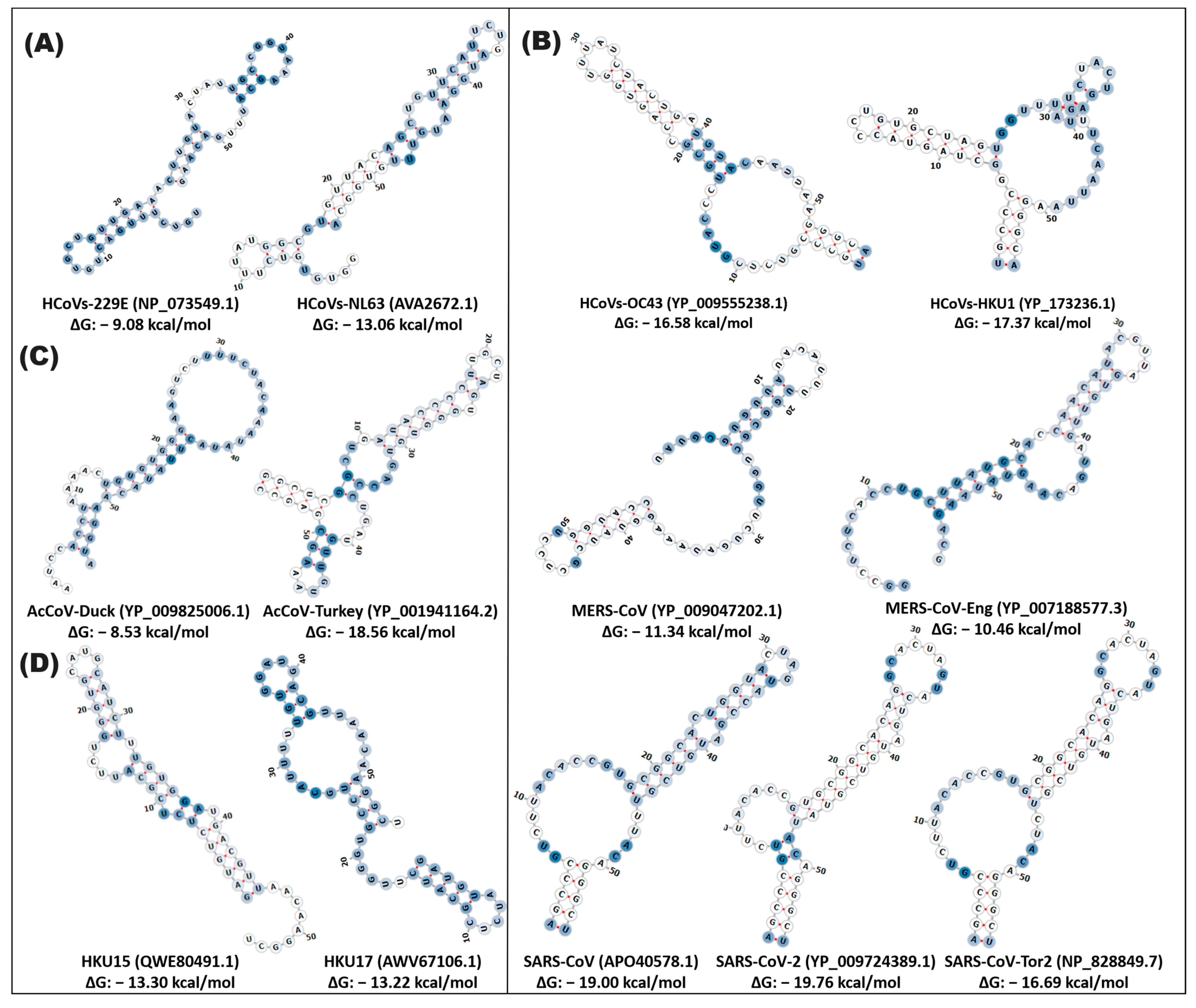
3.2. Diversity and Structural Functionality of Cis Elements in Coronaviruses
3.3. Cis Elements in the SARS-CoV Lineage
3.4. Evolutionary Relationships of Cis Elements of Coronavirus Pathogens of Humans
4. Discussion
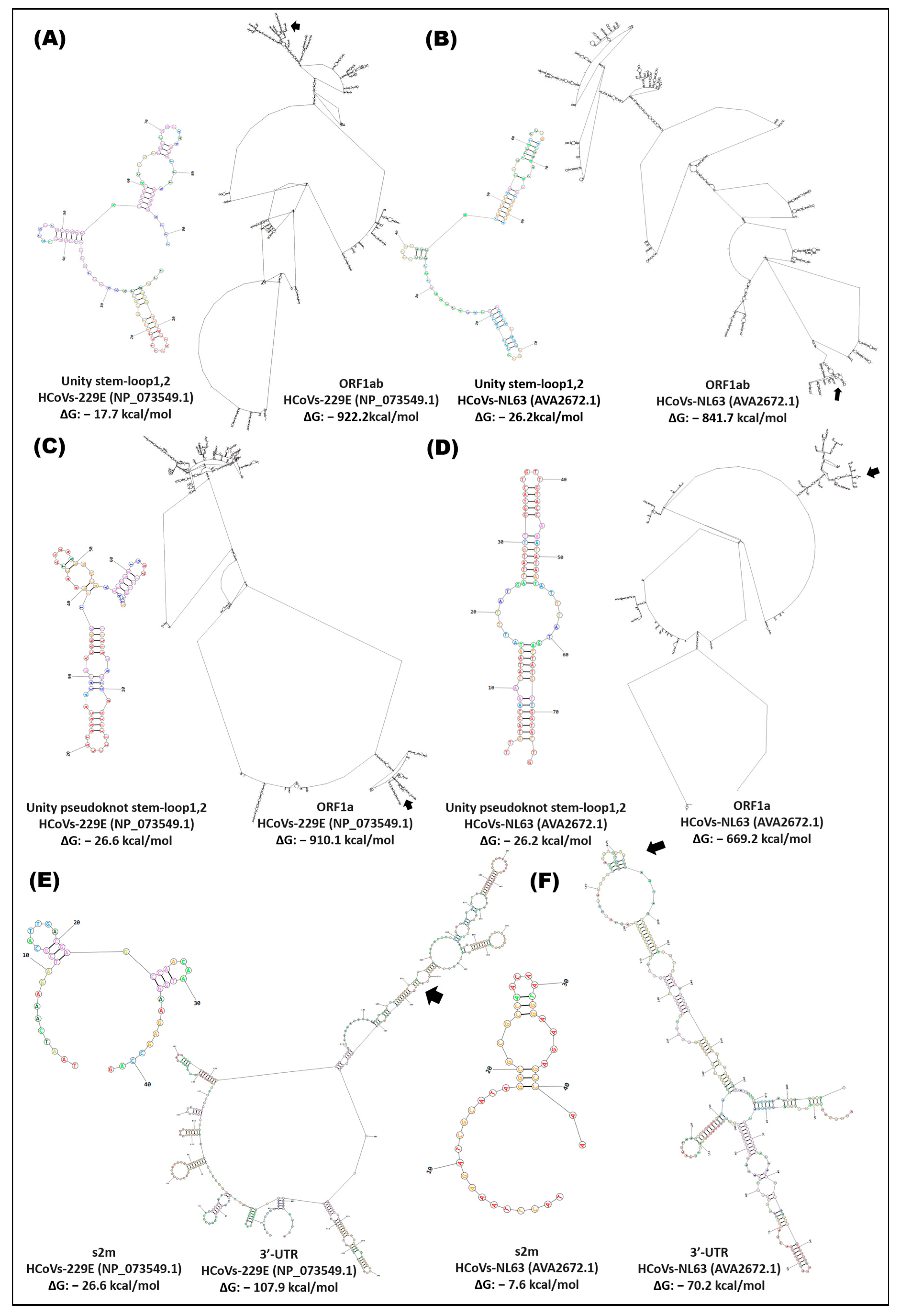
4.1. Importance of Cis Elements in Human Pathogenic Coronaviruses
4.2. Coevolution of Cis Elements in the Genome of Human Pathogenic Coronaviruses
4.3. Functional Adaptation of Cis Elements in Coronaviruses
4.4. Future Research
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Imperatore, J.A.; Cunningham, C.L.; Pellegrene, K.A.; Brinson, R.G.; Marino, J.P.; Evanseck, J.D.; Mihailescu, M.R. Highly conserved s2m element of SARS-CoV-2 dimerizes via a kissing complex and interacts with host miRNA-1307-3p. Nucleic Acids Res. 2022, 50, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Sharma, M.; Al-Ghamdi, A.A.; Al-Yami, S.M.; Al-Salami, A.M.; Refai, M.Y.; Baeshen, M.N. A comprehensive analysis of cis-acting RNA elements in the SARS-CoV-2 genome by a bioinformatics approach. Front. Genet. 2020, 11, 572702. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Olsthoorn, R.C. Group-specific structural features of the 5′-proximal sequences of coronavirus genomic RNAs. Virology 2010, 401, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Dalton, K.; Casais, R.; Shaw, K.; Stirrups, K.; Evans, S.; Britton, P.; Cavanagh, D. Cis-acting sequences required for coronavirus infectious bronchitis virus defective-RNA replication and packaging. J. Virol. 2001, 75, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liao, C.L.; Lai, M.M. Coronavirus leader RNA regulates and initiates subgenomic mRNA transcription both in trans and in cis. J. Virol. 1994, 68, 4738–4746. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; van den Born, E.; van den Worm, S.H.; Pleij, C.W.; Snijder, E.J.; Olsthoorn, R.C. New structure model for the packaging signal in the genome of group IIa coronaviruses. J. Virol. 2007, 81, 6771–6774. [Google Scholar] [CrossRef] [PubMed]
- Morales, L.; Mateos-Gomez, P.A.; Capiscol, C.; Del Palacio, L.; Enjuanes, L.; Sola, I. Transmissible gastroenteritis coronavirus genome packaging signal is located at the 5′ end of the genome and promotes viral RNA incorporation into virions in a replication-independent process. J. Virol. 2013, 87, 11579–11590. [Google Scholar] [CrossRef] [PubMed]
- Namy, O.; Moran, S.J.; Stuart, D.I.; Gilbert, R.J.; Brierley, I. A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature 2006, 441, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Madhugiri, R.; Fricke, M.; Marz, M.; Ziebuhr, J. RNA structure analysis of alphacoronavirus terminal genome regions. Virus Res. 2014, 194, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, L.; Millership, J.J.; Kang, H.; Leibowitz, J.L.; Giedroc, D.P. A U-turn motif-containing stem–loop in the coronavirus 5′ untranslated region plays a functional role in replication. RNA 2007, 13, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wimmer, E.; Paul, A.V. Cis-acting RNA elements in human and animal plus-strand RNA viruses. BBA-Gene Regul. Mech. 2009, 1789, 495–517. [Google Scholar] [CrossRef] [PubMed]
- Madhugiri, R.; Karl, N.; Petersen, D.; Lamkiewicz, K.; Fricke, M.; Wend, U.; Ziebuhr, J. Structural and functional conservation of cis-acting RNA elements in coronavirus 5′-terminal genome regions. Virology 2018, 517, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, P.; Wudeck, E.V.; Giedroc, D.P.; Leibowitz, J.L. SHAPE analysis of the RNA secondary structure of the Mouse Hepatitis Virus 5’untranslated region and N-terminal nsp1 coding sequences. Virology 2015, 475, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Luytjes, W.; Gerritsma, H.; Spaan, W.J. Replication of synthetic defective interfering RNAs derived from coronavirus mouse hepatitis virus-A59. Virology 1996, 216, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Hsue, B.; Hartshorne, T.; Masters, P.S. Characterization of an essential RNA secondary structure in the 3′ untranslated region of the murine coronavirus genome. J. Virol. 2000, 74, 6911–6921. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Johnson, R.F.; Leibowitz, J.L. Secondary structural elements within the 3′ untranslated region of mouse hepatitis virus strain JHM genomic RNA. J. Virol. 2001, 75, 12105–12113. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.-J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Farris, J.S. Methods for computing Wagner trees. Syst. Zool. 1970, 19, 83–92. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.D.K.; Brierley, I. The Coronavirus Nonstructural Proteins. In The Coronaviridae. The Viruses; Siddell, S.G., Ed.; Springer: Boston, MA, USA, 1995. [Google Scholar]
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene 2002, 299, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Madhugiri, R.; Fricke, M.; Marz, M.; Ziebuhr, J. Coronavirus cis-acting RNA elements. Adv. Virus Res. 2016, 96, 127–163. [Google Scholar] [PubMed]
- Buchan, J.R.; Stansfield, I. Halting a cellular production line: Responses to ribosomal pausing during translation. Biol. Cell. 2007, 99, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Tamayo-Ordóñez, M.C.; Rosas-García, N.M.; Ayil-Gutiérrez, B.A.; Bello-López, J.M.; Tamayo-Ordóñez, F.A.; Anguebes-Franseschi, F.; Damas-Damas, S.; Tamayo-Ordóñez, Y.J. Non-Structural Proteins (Nsp): A Marker for Detection of Human Coronavirus Families. Pathogens 2023, 12, 1185. [Google Scholar] [CrossRef] [PubMed]
- Tengs, T.; Kristoffersen, A.B.; Bachvaroff, T.R.; Jonassen, C.M. A mobile genetic element with unknown function found in distantly related viruses. Virol. J. 2013, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Tengs, T.; Jonassen, C. Distribution and evolutionary history of the mobile genetic element s2m in coronaviruses. Diseases 2016, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Keep, S.; Dowgier, G.; Lulla, V.; Britton, P.; Oade, M.; Freimanis, G.; Bickerton, E. Deletion of the s2m RNA structure in the avian coronavirus infectious bronchitis virus and human astrovirus results in sequence insertions. J. Virol. 2023, 97, e00038-23. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, C.M. Detection and sequence characterization of the 3-end of coronavirus genomes harboring the highly conserved RNA motif s2m. Methods Mol. Biol. 2008, 454, 27–34. [Google Scholar] [PubMed]
- Williams, G.D.; Chang, R.Y.; Brian, D.A. A phylogenetically conserved hairpin-type 3′ untranslated region pseudoknot functions in coronavirus RNA replication. J. Virol. 1999, 73, 8349–8355. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.; Pei, Y. Full-Length Genomic Sequence of Bovine Coronavirus (31kb). In The Nidoviruses; Lavi, E., Weiss, S.R., Hingley, S.T., Eds.; Springer: Boston, MA, USA, 2001; Volume 494. [Google Scholar]
- Züst, R.; Miller, T.B.; Goebel, S.J.; Thiel, V.; Masters, P.S. Genetic interactions between an essential 3′ cis-acting RNA pseudoknot, replicase gene products, and the extreme 3′ end of the mouse coronavirus genome. J. Virol. 2008, 82, 1214–1228. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Rawal, R.K.; Singh, P.K. RdRp (RNA-dependent RNA polymerase): A key target providing anti-virals for the management of various viral diseases. J. Mol. Struct. 2022, 1250, 131756. [Google Scholar] [CrossRef] [PubMed]
- Goebel, S.J.; Hsue, B.; Dombrowski, T.F.; Masters, P.S. Characterization of the RNA components of a putative molecular switch in the 3′ untranslated region of the murine coronavirus genome. J. Virol. 2004, 78, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.; Jia, N.; Zhang, Y.; Shum, M.H.; Jiang, J.; Zhu, H.; Tong, Y.; Shi, Y.; Ni, X.; Liao, Y.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Flores-Alanis, A.; Sandner-Miranda, L.; Delgado, G.; Cravioto, A.; Morales-Espinosa, R. The receptor binding domain of SARS-CoV-2 spike protein is the result of an ancestral recombination between the bat-CoV RaTG13 and the pangolin-CoV MP789. BMC Res. 2020, 13, 398. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Matsui, N.; Noguchi, K.; Kuwata, R.; Shimoda, H.; Soma, T.; Mochizuki, M.; Maeda, K. Emergence of pathogenic coronaviruses in cats by homologous recombination between feline and canine coronaviruses. PLoS ONE 2014, 9, e106534. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, N.; Honce, R.; Schultz-Cherry, S. Astrovirus evolution and emergence. Infect. Genet. Evol. 2019, 69, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Kofstad, T.; Jonassen, C.M. Screening of feral and Wood pigeons for viruses harbouring a conserved mobile viral element: Characterization of novel astroviruses and picornaviruses. PLoS ONE 2011, 6, e25964. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.Y.; Contreras, G.P. Emerging viral mutants in Australia suggest RNA recombination event in the SARS-CoV-2 genome. Med. J. Aus. 2020, 213, 44. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.Y.; Contreras, G.P. Viral transmission and evolution dynamics of SARS-CoV-2 in shipboard quarantine. Bull. World Health Organ. 2021, 99, 486. [Google Scholar] [CrossRef] [PubMed]
- Eden, J.S.; Rockett, R.; Carter, I.; Rahman, H.; De Ligt, J.; Hadfield, J.; Storey, M.; Ren, X.; Tulloch, R.; Basile, K.; et al. An emergent clade of SARS-CoV-2 linked to returned travellers from Iran. Virus Evol, 2020; 6, veaa027. [Google Scholar]
- de Wilde, A.H.; Snijder, E.J.; Kikkert, M.; van Hemert, M.J. Host factors in coronavirus replication. Curr. Top. Microbiol. Immunol. 2018, 419, 1–42. [Google Scholar] [PubMed]
- Alam, T.; Lipovich, L. miRcovid-19: Potential targets of human miRNAs in SARS-CoV-2 for RNA-based drug discovery. Non-coding RNA 2021, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Balmeh, N.; Mahmoudi, S.; Mohammadi, N.; Karabedianhajiabadi, A. Predicted therapeutic targets for COVID-19 disease by inhibiting SARS-CoV-2 and its related receptors. Inform. Med. Unlocked 2020, 20, 100407. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Gardner, C.L.; Sun, C.; Haddow, A.D.; Wang, E.; Chapnik, E.; Mildner, A.; Weaver, S.C.; Ryman, K.D.; Klimstra, W.B. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 2014, 506, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Klimstra, W.B. MicroRNA regulation of RNA virus replication and pathogenesis. Trends Mol. Med. 2017, 23, 80–93. [Google Scholar] [CrossRef] [PubMed]
- de la Rica, R.; Borges, M.; Gonzalez-Freire, M. COVID-19: In the eye of the cytokine storm. Front. Immunol. 2020, 11, 558898. [Google Scholar] [CrossRef] [PubMed]
- Lulla, V.; Wandel, M.P.; Bandyra, K.J.; Ulferts, R.; Wu, M.; Dendooven, T.; Luisi, B.F. Targeting the conserved stem loop 2 motif in the SARS-CoV-2 genome. J. Virol. 2021, 95, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Alhatlani, B.Y. In silico identification of conserved cis-acting RNA elements in the SARS-CoV-2 genome. Future Virol. 2020, 15, 409–417. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Coronavirus | Denomination | Host | Accession of Genome | 1ab Polyprotein (ORF1ab) | Length of Genome (nt) |
|---|---|---|---|---|---|
| Alphacoronavirus | |||||
| HCoVs-229E | Human coronavirus 229E | Human, vertebrate strain: 229E | NC_002645 | NP_073549.1 | 27,317 |
| HCoVs-NL63 | Human coronavirus NL63 | Human strain: Amsterdam I | NC_005831 | AVA2672.1 | 27,553 |
| Betacoronavirus | |||||
| HCoVs-OC43 | Human coronavirus OC43 | Human, vertebrates strain: ATCC VR-759; serotype: OC43 | NC_006213 | YP_009555238.1 | 30,741 |
| HCoVs-HKU1 | Human coronavirus HKU1 | Human isolate: HKU1 | NC_006577 | YP_173236.1 | 29,926 |
| MERS-CoV | Middle East respiratory syndrome-related coronavirus | Human, vertebrate strain: HCoV-EMC; isolate: HCoV-EMC/2012 | NC_19843 | YP_009047202.1 | 30,119 |
| MERS-CoV-Eng | Middle East respiratory syndrome-related coronavirus, England | Human, vertebrate strain: England 1; isolate: H123990006 | NC_038294 | YP_007188577.3 | 30,111 |
| SARS-CoV | Severe acute respiratory syndrome-related coronavirus | Human, vertebrate Strain: BtKY72 | KY352407 a | APO40578.1 | 29,274 |
| SARS-CoV-2 | Severe acute respiratory syndrome-related coronavirus 2 | Human, vertebrate isolate: Wuhan-Hu-1 | NC_045512 | YP_009724389.1 | 29,903 |
| SARS-CoV-Tor2 | Severe acute respiratory syndrome-related coronavirus Tor2 | Human isolate: patient #2 with severe acute respiratory syndrome (SARS) | NC_04718 | NP_828849.7 | 29,751 |
| Gammacoronavirus b | |||||
| AcCoV-Duck | Duck avian coronavirus | Human, vertebrate isolate: DK/GD/27/2014 | NC_048214 | YP_009825006.1 | 27,754 |
| AcCoV-Turkey | Turkey avian coronavirus | Human, vertebrate isolate: MG10 | NC_010800 | YP_001941164.2 | 27,657 |
| Deltacoronavirus c | |||||
| Porcine coronavirus | Porcine coronavirus HKU15 | Porcine isolate: PDCoV/Haiti/Human/0081-4/2014 | MW685622 | QWE80491.1 | 25,444 |
| Sparrow deltacoronavirus | Sparrow deltacoronavirus HKU17 | Passeridae isolate PDCoV/Haiti/Human/0081-4/2014 | MG812375 | AWV67106.1 | 25,795 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamayo-Ordóñez, Y.d.J.; Rosas-García, N.M.; Tamayo-Ordoñez, F.A.; Ayil-Gutiérrez, B.A.; Bello-López, J.M.; Sosa-Santillán, G.d.J.; Acosta-Cruz, E.; Anguebes-Franseschi, F.; Damas-Damas, S.; Domínguez-May, A.V.; et al. Genomic Evolution Strategy in SARS-CoV-2 Lineage B: Coevolution of Cis Elements. Curr. Issues Mol. Biol. 2024, 46, 5744-5776. https://doi.org/10.3390/cimb46060344
Tamayo-Ordóñez YdJ, Rosas-García NM, Tamayo-Ordoñez FA, Ayil-Gutiérrez BA, Bello-López JM, Sosa-Santillán GdJ, Acosta-Cruz E, Anguebes-Franseschi F, Damas-Damas S, Domínguez-May AV, et al. Genomic Evolution Strategy in SARS-CoV-2 Lineage B: Coevolution of Cis Elements. Current Issues in Molecular Biology. 2024; 46(6):5744-5776. https://doi.org/10.3390/cimb46060344
Chicago/Turabian StyleTamayo-Ordóñez, Yahaira de J., Ninfa M. Rosas-García, Francisco A. Tamayo-Ordoñez, Benjamín A. Ayil-Gutiérrez, Juan M. Bello-López, Gerardo de J. Sosa-Santillán, Erika Acosta-Cruz, Francisco Anguebes-Franseschi, Siprian Damas-Damas, Angel V. Domínguez-May, and et al. 2024. "Genomic Evolution Strategy in SARS-CoV-2 Lineage B: Coevolution of Cis Elements" Current Issues in Molecular Biology 46, no. 6: 5744-5776. https://doi.org/10.3390/cimb46060344
APA StyleTamayo-Ordóñez, Y. d. J., Rosas-García, N. M., Tamayo-Ordoñez, F. A., Ayil-Gutiérrez, B. A., Bello-López, J. M., Sosa-Santillán, G. d. J., Acosta-Cruz, E., Anguebes-Franseschi, F., Damas-Damas, S., Domínguez-May, A. V., Córdova-Quiroz, A. V., & Tamayo-Ordóñez, M. C. (2024). Genomic Evolution Strategy in SARS-CoV-2 Lineage B: Coevolution of Cis Elements. Current Issues in Molecular Biology, 46(6), 5744-5776. https://doi.org/10.3390/cimb46060344