
Sickle Cell Disease: Current Drug Treatments and Functional Foods with Therapeutic Potential

 ,
,  ,
,  ,
,  and
and 
Abstract
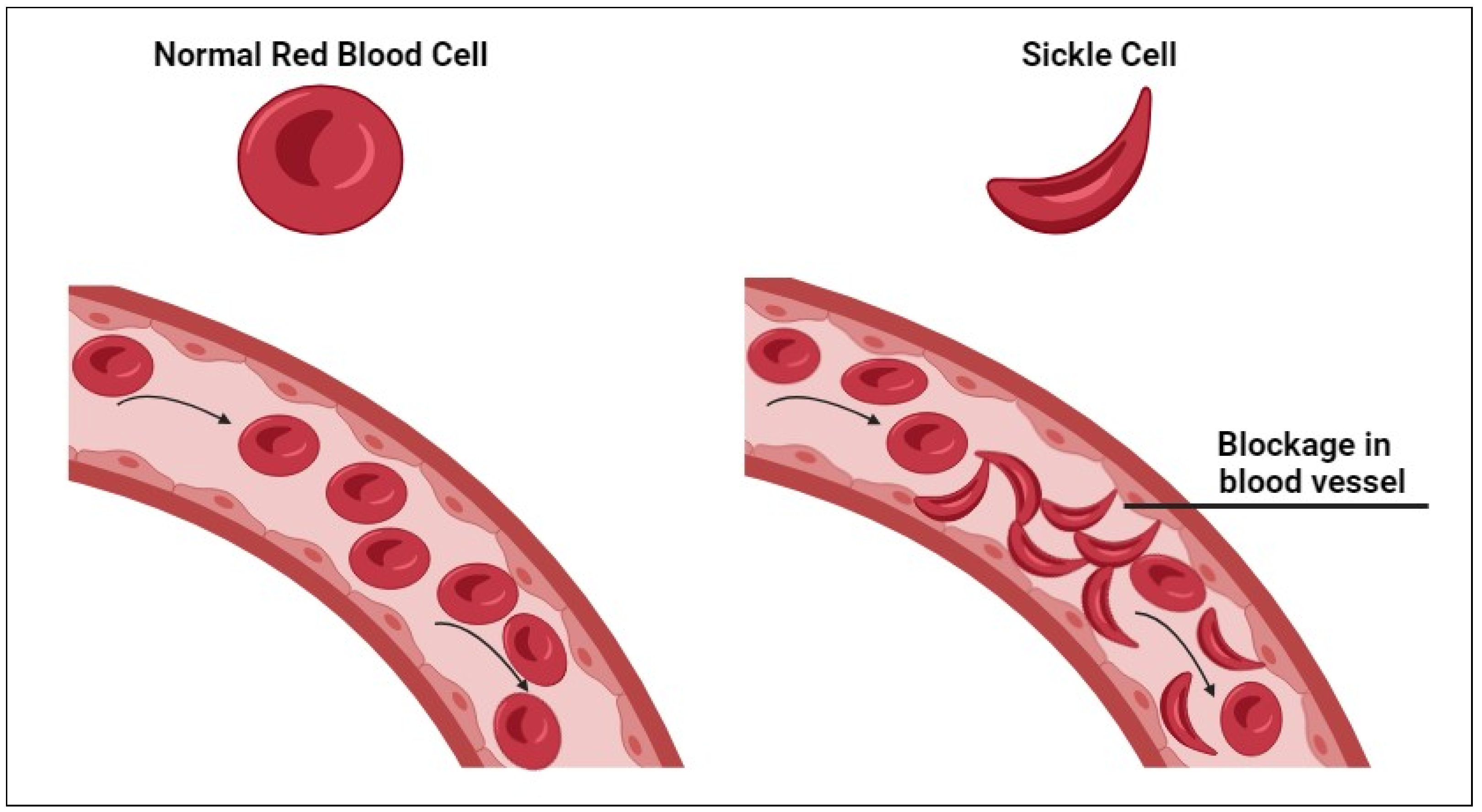
1. Introduction
2. Methods
3. Review of Randomized, Double-Blind Clinical Trials for Sickle Cell Disease Treatments
4. Functional Food Exploited in SCD Management
5. Impact of Bioactive Compounds in Sickle Cell Disease Management
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tebbi, C.K. Sickle Cell Disease, a Review. Hemato 2022, 3, 341–366. [Google Scholar] [CrossRef]
- Serjeant, G.R. The emerging understanding of sickle cell disease. Br. J. Haematol. 2001, 112, 3–18. [Google Scholar] [CrossRef] [PubMed]
- WHO. Sickle Cell Disease. Available online: https://www.afro.who.int/health-topics/sickle-cell-disease (accessed on 9 June 2024).
- Sanyaolu, A.; Agiri, E.; Bertram, C.; Brookes, L.; Choudhury, J.; Datt, D.; Ibrahim, A.; Maciejko, A.; Mansfield, A.; Nkrumah, J. Current modalities of sickle cell disease management. Blood Sci. 2020, 2, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Alabi, O.J.; Adegboyega, F.N.; Olawoyin, D.S.; Babatunde, O.A. Functional foods: Promising therapeutics for Nigerian Children with sickle cell diseases. Heliyon 2022, 8, e09630. [Google Scholar] [CrossRef] [PubMed]
- Adigwe, O.P.; Onoja, S.O.; Onavbavba, G. A Critical Review of Sickle Cell Disease Burden and Challenges in Sub-Saharan Africa. J. Blood Med. 2023, 14, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Trabuchet, G.; Elion, J.; Baudot, G.; Pagnier, J.; Bouhass, R.; Nigon, V.M.; Labie, D.; Krishnamoorthy, R. Origin and spread of β-globin gene mutations in India, Africa, and Mediterranea: Analysis of the 5′flanking and intragenic sequences of β s and β c genes. Hum. Biol. 1991, 63, 241–252. [Google Scholar] [PubMed]
- Zago, M.A.; Silva, W.A., Jr.; Dalle, B.; Gualandro, S.; Hutz, M.H.; Lapoumeroulie, C.; Tavella, M.H.; Araujo, A.G.; Krieger, J.E.; Elion, J.; et al. Atypical βs haplotypes are generated by diverse genetic mechanisms. Am. J. Hematol. 2000, 63, 79–84. [Google Scholar] [CrossRef]
- Jang, T.; Poplawska, M.; Cimpeanu, E.; Mo, G.; Dutta, D.; Lim, S.H. Vaso-occlusive crisis in sickle cell disease: A vicious cycle of secondary events. J. Transl. Med. 2021, 19, 397. [Google Scholar] [CrossRef] [PubMed]
- da Guarda, C.C.; Yahouédéhou, S.; Santiago, R.P.; Neres, J.; Fernandes, C.F.L.; Aleluia, M.M.; Figueiredo, C.V.B.; Fiuza, L.M.; Carvalho, S.P.; Oliveira, R.M.; et al. Sickle cell disease: A distinction of two most frequent genotypes (HbSS and HbSC). PLoS ONE 2020, 15, e0228399. [Google Scholar] [CrossRef] [PubMed]
- Tonin, F.S.; Ginete, C.; Ferreira, J.; Delgadinho, M.; Santos, B.; Fernandez-Llimos, F.; Brito, M. Efficacy and safety of pharmacological interventions for managing sickle cell disease complications in children and adolescents: Systematic review with network meta-analysis. Pediatr. Blood Cancer 2023, 70, e30294. [Google Scholar] [CrossRef]
- Starlard-Davenport, A.; Gu, Q.; Pace, B.S. Targeting Genetic Modifiers of HBG Gene Expression in Sickle Cell Disease: The miRNA Option. Mol. Diagn. Ther. 2022, 26, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Change, S.S.C. Impact of Sickle Cell on the Body. 2024. Available online: https://www.sparksicklecellchange.com/what-is-sickle-cell/symptoms-complications (accessed on 9 June 2024).
- Arishi, W.A.; Alhadrami, H.A.; Zourob, M. Techniques for the Detection of Sickle Cell Disease: A Review. Micromachines 2021, 12, 519. [Google Scholar] [CrossRef] [PubMed]
- Salinas Cisneros, G.; Thein, S.L. Recent Advances in the Treatment of Sickle Cell Disease. Front. Physiol. 2020, 11, 435. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hadi, L.; Ventura Carmenate, Y.; Castillo-Aleman, Y.M.; Sheikh, S.; Zakaria, A.; Phillips, J. Treatment of sickle cell disease—Options and perspective. Am. J. Blood Res. 2023, 13, 61–70. [Google Scholar] [PubMed]
- Eaton, W.A.; Bunn, H.F. Treating sickle cell disease by targeting HbS polymerization. Blood 2017, 129, 2719–2726. [Google Scholar] [CrossRef] [PubMed]
- Oniyangi, O.; Cohall, D.H. Phytomedicines (medicines derived from plants) for sickle cell disease. Cochrane Database Syst. Rev. 2020, 9, Cd004448. [Google Scholar] [CrossRef] [PubMed]
- Yembeau, N.L.; Biapa Nya, P.C.; Pieme, C.A.; Tchouane, K.D.; Kengne Fotsing, C.B.; Nya Nkwikeu, P.J.; Feudjio, A.F.; Telefo, P.B. Ethnopharmacological Study of the Medicinal Plants Used in the Treatment of Sickle Cell Anemia in the West Region of Cameroon. Evid. Based Complement. Altern. Med. 2022, 2022, 5098428. [Google Scholar] [CrossRef] [PubMed]
- Famojuro, T.; Moody, J. Survey of medicinal plants used in the management of sickle cell disease by traditional medical practitioners of gbonyin local government area of Ekiti state, Nigeria. Niger. J. Nat. Prod. Med. 2015, 19, 78–84. [Google Scholar] [CrossRef]
- Kaptchuk, T.J. The double-blind, randomized, placebo-controlled trial: Gold standard or golden calf? J. Clin. Epidemiol. 2001, 54, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Giriraja, K.V.; Bhatnagar, S.K.; Tomlinson, L.; Sancilio, F. An open-label, multicenter, phase 2 study of a food enriched with docosahexaenoic acid in adults with sickle cell disease. Prostaglandins Leukot. Essent. Fat. Acids 2023, 193, 102574. [Google Scholar] [CrossRef] [PubMed]
- Abdelhalim, S.M.; Murphy, J.E.; Meabed, M.H.; Elberry, A.A.; Gamaleldin, M.M.; Shaalan, M.S.; Hussein, R.R.S. Comparative effectiveness of adding Omega-3 or Vitamin D to standard therapy in preventing and treating episodes of painful crisis in pediatric sickle cell patients. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 5043–5052. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.N.; Schall, J.I.; McAnlis, C.R.; Smith-Whitley, K.; Norris, C.F.; Stallings, V.A. Effect of High-dose Vitamin A Supplementation in Children With Sickle Cell Disease: A Randomized, Double-blind, Dose-finding Pilot Study. J. Pediatr. Hematol. Oncol. 2020, 42, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Muga, R.; Ajwang, A.; Ouma, J.; Ojigo, J.; Otieno, J.; Okoth, P.; Wafula, C.; Ajwang, S.; Ogolla, D.; Hollist, A. Efficacy of the Nutritional Supplement, EvenFlo, in the Management of Sickle Cell Disease: A Randomized Controlled Trial. Nurs. Health Sci. Res. J. 2020, 3, 35–45. [Google Scholar] [CrossRef]
- Grégoire-Pelchat, P.; Pastore, Y.; Robitaille, N.; LeMay, S.; Khamessan, A.; Kleiber, N.; Nyalendo, C.; Gagné, N.; Alos, N.; Mailhot, G. Comparison of two vitamin D supplementation strategies in children with sickle cell disease: A randomized controlled trial. Br. J. Haematol. 2021, 192, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Onalo, R.; Cooper, P.; Cilliers, A.; Vorster, B.C.; Uche, N.-A.; Oluseyi, O.O.; Onalo, V.D.; Zubairu, Y.; Ayodele-Kehinde, A.U.; Damilare, O.M.; et al. Randomized control trial of oral arginine therapy for children with sickle cell anemia hospitalized for pain in Nigeria. Am. J. Hematol. 2021, 96, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Onalo, R.; Cilliers, A.; Cooper, P.; Morris, C.R. Arginine therapy and cardiopulmonary hemodynamics in hospitalized children with sickle cell anemia: A prospective, double-blinded, randomized placebo-controlled clinical trial. Am. J. Respir. Crit. Care Med. 2022, 206, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Hutchaleelaha, A.; Patel, M.; Washington, C.; Siu, V.; Allen, E.; Oksenberg, D.; Gretler, D.D.; Mant, T.; Lehrer-Graiwer, J. Pharmacokinetics and pharmacodynamics of voxelotor (GBT440) in healthy adults and patients with sickle cell disease. Br. J. Clin. Pharmacol. 2019, 85, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C. A phase 3 randomized trial of voxelotor in sickle cell disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, S.U.; Jibir, B.W.; Bello-Manga, H.; Gambo, S.; Inuwa, H.; Tijjani, A.G.; Idris, N.; Galadanci, A.; Hikima, M.S.; Galadanci, N.; et al. Hydroxyurea for primary stroke prevention in children with sickle cell anaemia in Nigeria (SPRING): A double-blind, multicentre, randomised, phase 3 trial. Lancet Haematol. 2022, 9, e26–e37. [Google Scholar] [CrossRef]
- Panda, P.C.; Mishra, N.R.; Patra, C.S.; Nayak, B.K.; Panda, S.K. Intravenous Acetaminophen vs Intravenous Diclofenac Sodium in Management of Skeletal Vaso-occlusive Crisis Among Children with Homozygous Sickle Cell Disease: A Randomized Controlled Trial. Indian Pediatr. 2021, 58, 229–232. [Google Scholar] [CrossRef]
- Rees, D.C.; Kilinc, Y.; Unal, S.; Dampier, C.; Pace, B.S.; Kaya, B.; Trompeter, S.; Odame, I.; Mahlangu, J.; Unal, S. A randomized, placebo-controlled, double-blind trial of canakinumab in children and young adults with sickle cell anemia. Blood J. Am. Soc. Hematol. 2022, 139, 2642–2652. [Google Scholar] [CrossRef] [PubMed]
- Heeney, M.M.; Abboud, M.R.; Githanga, J.; Inusa, B.P.D.; Kanter, J.; Michelson, A.D.; Nduba, V.; Musiime, V.; Apte, M.; Inati, A.; et al. Ticagrelor vs placebo for the reduction of vaso-occlusive crises in pediatric sickle cell disease: The HESTIA3 study. Blood 2022, 140, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Alshahrani, M.S.; AlSulaibikh, A.H.; ElTahan, M.R.; AlFaraj, S.Z.; Asonto, L.P.; AlMulhim, A.A.; AlAbbad, M.F.; Almaghraby, N.; AlJumaan, M.A.; AlJunaid, T.O.; et al. Ketamine administration for acute painful sickle cell crisis: A randomized controlled trial. Acad. Emerg. Med. 2022, 29, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Lizarralde-Iragorri, M.A.; Parachalil Gopalan, B.; Merriweather, B.; Brooks, J.; Hill, M.; Lovins, D.; Pierre-Charles, R.; Cullinane, A.; Dulau-Florea, A.; Lee, D.-Y.; et al. Isoquercetin for thromboinflammation in sickle cell disease: A randomized double-blind placebo-controlled trial. Blood Adv. 2024, 8, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, A.; Lawrence, R.; Shetty, R.; Chaithanya, A.; Jhaveri, S.; Pichardo, B.V.; Mujakari, A. Role of gene therapy in sickle cell disease. Dis. Mon. 2024, 101689. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; McGann, P.T. Changing the Clinical Paradigm of Hydroxyurea Treatment for Sickle Cell Anemia Through Precision Medicine. Clin. Pharmacol. Ther. 2021, 109, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Yahouédéhou, S.C.M.A.; Adorno, E.V.; da Guarda, C.C.; Ndidi, U.S.; Carvalho, S.P.; Santiago, R.P.; Aleluia, M.M.; de Oliveira, R.M.; Gonçalves, M.d.S. Hydroxyurea in the management of sickle cell disease: Pharmacogenomics and enzymatic metabolism. Pharmacogenomics J. 2018, 18, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Atmapoojya, P.; Colah, R.; Lodha, P. Sickle Cell Disease and Pregnancy. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019040. [Google Scholar] [CrossRef] [PubMed]
- McGann, P.T.; Ware, R.E. Hydroxyurea therapy for sickle cell anemia. Expert Opin. Drug Saf. 2015, 14, 1749–1758. [Google Scholar] [CrossRef]
- Bell, V.; Varzakas, T.; Psaltopoulou, T.; Fernandes, T. Sickle Cell Disease Update: New Treatments and Challenging Nutritional Interventions. Nutrients 2024, 16, 258. [Google Scholar] [CrossRef]
- Erkelens, M.N.; Mebius, R.E. Retinoic Acid and Immune Homeostasis: A Balancing Act. Trends Immunol. 2017, 38, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Schall, J.I.; Zemel, B.S.; Kawchak, D.A.; Ohene-Frempong, K.; Stallings, V.A. Vitamin A status, hospitalizations, and other outcomes in young children with sickle cell disease. J. Pediatr. 2004, 145, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Chandrakar, D.; Patel, S.; Wasnik, P.N.; Mohapatra, E.; Nanda, R.; Shah, S.; Gupta, D.L. Effect of Unmetabolized Folic Acid on Immunoinflammatory Markers in Sickle Cell Disease Patients Taking Folic Acid Supplementation. Indian J. Clin. Biochem. 2024, 2024, 1–8. [Google Scholar] [CrossRef]
- Soe, H.H.K.; Abas, A.B.; Than, N.N.; Ni, H.; Singh, J.; Said, A.; Osunkwo, I. Vitamin D supplementation for sickle cell disease. Cochrane Database Syst. Rev. 2020, 5, Cd010858. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, N.; Morris, C.R. The role of the arginine metabolome in pain: Implications for sickle cell disease. J. Pain Res. 2016, 167–175. [Google Scholar]
- Morris, S.M. Chapter 11—Regulation of Arginine Availability and Its Impact on NO Synthesis. In Nitric Oxide; Ignarro, L.J., Ed.; Academic Press: San Diego, CA, USA, 2000; pp. 187–197. [Google Scholar]
- Morris, C.R.; Hamilton-Reeves, J.; Martindale, R.G.; Sarav, M.; Ochoa Gautier, J.B. Acquired Amino Acid Deficiencies: A Focus on Arginine and Glutamine. Nutr. Clin. Pract. 2017, 32, 30S–47S. [Google Scholar] [CrossRef] [PubMed]
- Tayyaba Rehan, S.; Hussain, H.U.; Malik, F.; Usama, R.M.; Tahir, M.J.; Asghar, M.S. Voxelotor versus other therapeutic options for sickle cell disease: Are we still lagging behind in treating the disease? Health Sci. Rep. 2022, 5, e713. [Google Scholar] [CrossRef] [PubMed]
- Barriteau, C.M.; Badawy, S.M. Practical Guidance for the Use of Voxelotor in the Management of Sickle Cell Disease. J. Blood Med. 2022, 13, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Shastri, N. Intravenous acetaminophen use in pediatrics. Pediatr. Emerg. Care 2015, 31, 444–448. [Google Scholar] [CrossRef]
- Dampier, C.D.; Setty, B.; Logan, J.; Ioli, J.G.; Dean, R. Intravenous morphine pharmacokinetics in pediatric patients with sickle cell disease. J. Pediatr. 1995, 126, 461–467. [Google Scholar] [CrossRef]
- Riley, T.R.; Riley, T.T. Profile of crizanlizumab and its potential in the prevention of pain crises in sickle cell disease: Evidence to date. J. Blood Med. 2019, 10, 307–311. [Google Scholar] [CrossRef]
- Kutlar, A.; Kanter, J.; Liles, D.K.; Alvarez, O.A.; Cançado, R.D.; Friedrisch, J.R.; Knight-Madden, J.M.; Bruederle, A.; Shi, M.; Zhu, Z.; et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: A SUSTAIN study analysis. Am. J. Hematol. 2019, 94, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.H.; Buddharaju, R.; Idowu, M. Real-world experience of patients with sickle cell disease treated with crizanlizumab. J. Investig. Med. 2024, 72, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Wang, X.; Fu, Q.; Cao, B. Progress in the clinical effects and adverse reactions of ticagrelor. Thromb. J. 2024, 22, 8. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Ren, S.; Zhang, L.; Liu, W.; Zhao, Y.; Chen, C.; Mao, X.; Chen, Z.; Gu, X. A Review of the Role of the Antiplatelet Drug Ticagrelor in the Management of Acute Coronary Syndrome, Acute Thrombotic Disease, and Other Diseases. Med. Sci. Monit. 2022, 28, e935664. [Google Scholar] [CrossRef] [PubMed]
- Heeney, M.M.; Abboud, M.R.; Amilon, C.; Andersson, M.; Githanga, J.; Inusa, B.; Kanter, J.; Leonsson-Zachrisson, M.; Michelson, A.D.; Berggren, A.R.; et al. Ticagrelor versus placebo for the reduction of vaso-occlusive crises in pediatric sickle cell disease: Rationale and design of a randomized, double-blind, parallel-group, multicenter phase 3 study (HESTIA3). Contemp. Clin. Trials 2019, 85, 105835. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Filho, J.; Yahouédéhou, S.C.M.A.; Pitanga, T.N.; Santana, S.S.; Adorno, E.V.; Barbosa, C.G.; Ferreira, J.R.D.; Pina, E.T.G.; Neres, J.S.d.S.; Leite, I.P.R.; et al. An evaluation of ticagrelor for the treatment of sickle cell anemia. Expert Rev. Hematol. 2020, 13, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ye, F.; Zhang, T.; Lv, S.; Zhou, L.; Du, D.; Lin, H.; Guo, F.; Luo, C.; Zhu, S. Structural basis of ketamine action on human NMDA receptors. Nature 2021, 596, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Davoudian, P.A.; Wilkinson, S.T. Chapter Four—Clinical overview of NMDA-R Antagonists and Clinical Practice. In Advances in Pharmacology; Duman, R.S., Krystal, J.H., Eds.; Academic Press: San Diego, CA, USA, 2020; Volume 89, pp. 103–129. [Google Scholar]
- Syed, M.M.; Doshi, P.J.; Dhavale, D.D.; Doshi, J.B.; Kate, S.L.; Kulkarni, G.; Sharma, N.; Uppuladinne, M.; Sonavane, U.; Joshi, R.; et al. Potential of isoquercitrin as antisickling agent: A multi-spectroscopic, thermophoresis and molecular modeling approach. J. Biomol. Struct. Dyn. 2020, 38, 2717–2736. [Google Scholar] [CrossRef]
- Lizarralde, M.A.; Merriweather, B.; Conrey, A.; Saxena, A.; Shet, A.S. Effects of Flavonoid Quercetin on Thrombo-Inflammatory Processes in Patients with Sickle Cell Disease. Blood 2021, 138, 2020. [Google Scholar] [CrossRef]
- Telen, M.J.; Batchvarova, M.; Shan, S.; Bovee-Geurts, P.H.; Zennadi, R.; Leitgeb, A.; Brock, R.; Lindgren, M. Sevuparin binds to multiple adhesive ligands and reduces sickle red blood cell-induced vaso-occlusion. Br. J. Haematol. 2016, 175, 935–948. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Lindgren, M.; Liu, K.; Gao, X.; Jendeberg, L.; Hines, P. Sevuparin blocks sickle blood cell adhesion and sickle-leucocyte rolling on immobilized L-selectin in a dose dependent manner. Br. J. Haematol. 2019, 184, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Biemond, B.J.; Tombak, A.; Kilinc, Y.; Al-Khabori, M.; Abboud, M.R.; Nafea, M.; Inati, A.; Wali, Y.A.M.S.; Kristensen, J.; Donnelly, E.; et al. Efficacy and Safety of Sevuparin, a Novel Non-Anti-Coagulant Heparinoid, in Patients with Acute Painful Vaso-Occlusive Crisis; A Global, Multicenter Double-Blind, Randomized, Placebo-Controlled Phase 2 Trial (TVOC01). Blood 2019, 134, 614. [Google Scholar] [CrossRef]
- Subramanian, P.; Anandharamakrishnan, C. Chapter One—Introduction to Functional Foods and Nutraceuticals. In Industrial Application of Functional Foods, Ingredients and Nutraceuticals; Anandharamakrishnan, C., Subramanian, P., Eds.; Academic Press: San Diego, CA, USA, 2023; pp. 3–43. [Google Scholar]
- Granato, D.; Barba, F.J.; Bursać Kovačević, D.; Lorenzo, J.M.; Cruz, A.G.; Putnik, P. Functional Foods: Product Development, Technological Trends, Efficacy Testing, and Safety. Annu. Rev. Food Sci. Technol. 2020, 11, 93–118. [Google Scholar] [CrossRef] [PubMed]
- Luvián-Morales, J.; Varela-Castillo, F.O.; Flores-Cisneros, L.; Cetina-Pérez, L.; Castro-Eguiluz, D. Functional foods modulating inflammation and metabolism in chronic diseases: A systematic review. Crit. Rev. Food Sci. Nutr. 2022, 62, 4371–4392. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Muhammad, S.A. Antioxidant-Rich Nutraceutical as a Therapeutic Strategy for Sickle Cell Disease. J. Am. Nutr. Assoc. 2023, 42, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Teguem Tchoulegheu, A.; Nya Nkwikeu, P.J.; Lena Yembeau, N.; Choupo, A.C.; Nkenmeni Djamnou, C.; Feudjio, A.F.; Chetcha Chemegni, B.; Biapa Nya, P.C.; Pieme, C.A. Antisickling and Antihemolytic Mechanism of Spirulina platensis (Oscillatoriaceae): A Nutraceutical Commonly Used in Cameroon. Evid. Based Complement. Altern. Med. 2023, 2023, 1260169. [Google Scholar] [CrossRef]
- Ejiofor, E.U.; Ako, A.C.; Kube, M.T.; Agwamba, E.C.; Alala, C.; Maduabuchi, K.; Ejiofor, M. Phytochemistry, Mineral Estimation, Nutritional, and the In Vitro Anti-Sickling Potentials of Oil Extracted from the Seeds of Mucuna Flagellipes. J. Mex. Chem. Soc. 2024, 68, 220–233. [Google Scholar] [CrossRef]
- de Paula, R.G.; Ribeiro, H.M.; de Melo Borges, L.; Barreto, O.A.C.; Montel, A.L.B.; Scapin, E.; Silva, K.L.F.; Seibert, C.S. The Use of Natural Products in the Treatment of Sickle Cell Disease. Rev. Bras. Farmacogn. 2024, 2024, 1–13. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Bartosz, G. Peroxiredoxin 2: An Important Element of the Antioxidant Defense of the Erythrocyte. Antioxidants 2023, 12, 1012. [Google Scholar] [CrossRef]
- Kotue, T. Functional foods and nutraceuticals in the primary prevention of sickle cell disease crises. Indian J. Nutr. 2018, 5, 186. [Google Scholar]
- Famojuro, T.I.; Adeyemi, A.A.; Ajayi, T.O.; Fasola, F.A.; Fukushi, Y.; Omotade, O.O.; Moody, J.O. Anti-sickling activities of two isolated compounds from the root of Combretum racemosum P. beauv. (Combretaceae). J. Ethnopharmacol. 2021, 273, 113992. [Google Scholar] [CrossRef] [PubMed]
- Ahajumobi, N.E.; Asika, J.C. Afro Medicinal Plants a Promising Remedy for Sickle Cell Anemia. Int. Blood Res. Rev. 2024, 15, 26–37. [Google Scholar] [CrossRef]
- Wang, Q.; Zennadi, R. The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants 2021, 10, 1608. [Google Scholar] [CrossRef] [PubMed]
- Ilboudo, Y. The genetics of red blood cell density, a biomarker of clinical severity in sickle cell disease. Master’s Thesis, Université de Montréal, Montreal, QC, Canda, 2017. [Google Scholar]
- Liu, M.; Huang, Y.; Zhang, H.; Aitken, D.; Nevitt, M.C.; Rockel, J.S.; Pelletier, J.-P.; Lewis, C.E.; Torner, J.; Rampersaud, Y.R. Restricting branched-chain amino acids within a high-fat diet prevents obesity. Metabolites 2022, 12, 334. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, Y.; Zhou, Y.; Xu, Q.; Cheng, S.; Yan, J.; Xiao, Y.; Han, L.; Wang, Y.; Cai, W. Targeted metabolomics unravels altered phenylalanine levels in piglets receiving total parenteral nutrition. Faseb J. 2023, 37, e23014. [Google Scholar] [CrossRef] [PubMed]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Daak, A.A.; Lopez-Toledano, M.A.; Heeney, M.M. Biochemical and therapeutic effects of Omega-3 fatty acids in sickle cell disease. Complement. Ther. Med. 2020, 52, 102482. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Sposi, N.M.; Mattia, L.; Gambardella, L.; Straface, E.; Pietraforte, D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants 2021, 10, 296. [Google Scholar] [CrossRef] [PubMed]
- Karafin, M.S.; Field, J.J.; Ilich, A.; Li, L.; Qaquish, B.F.; Shevkoplyas, S.S.; Yoshida, T. Hypoxic storage of donor red cells preserves deformability after exposure to plasma from adults with sickle cell disease. Transfusion 2023, 63, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Hajizamani, S.; Atarodi, K.; Deyhim, M.R.; Kermani, F.R.; Hosseini, K.M. Antioxidative effects of α-tocopherol on stored human red blood cell units. Asian J. Transfus. Sci. 2023. [Google Scholar] [CrossRef]
- Wong-Roushar, J. Biochemical and Biophysical Characterization of Multi-Targeted Inhibitors of One-Carbon Metabolism for Cancer Treatment. Ph.D. Thesis, Indiana University, Bloomington, IN, USA, 2021. [Google Scholar]
- Bhatt, S.; Argueta, D.A.; Gupta, K.; Kundu, S. Red Blood Cells as Therapeutic Target to Treat Sickle Cell Disease. Antioxid. Redox. Signal 2024. [Google Scholar] [CrossRef] [PubMed]
- Ballas, S.K. The Evolving Pharmacotherapeutic Landscape for the Treatment of Sickle Cell Disease. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020010. [Google Scholar] [CrossRef] [PubMed]
- Doganci, M.; Doganay, G.E. Magnesium levels and mortality relationship in patients with Acinetobacter baumannii detected in the Intensive Care Unit. Eur. Rev. Med. Pharmacol. Sci. 2024, 28, 1295–1305. [Google Scholar] [CrossRef]
- Aliev, G.; Li, Y.; Chubarev, V.N.; Lebedeva, S.A.; Parshina, L.N.; Trofimov, B.A.; Sologova, S.S.; Makhmutova, A.; Avila-Rodriguez, M.F.; Klochkov, S.G.; et al. Application of Acyzol in the Context of Zinc Deficiency and Perspectives. Int. J. Mol. Sci. 2019, 20, 2104. [Google Scholar] [CrossRef] [PubMed]
- Kotue, T.; Pieme, A.; Fokou, E. Ethnobotanicals usages in the management of sickle cell disease (SDC) in some localities of Cameroon. Pharmacophore 2016, 7, 192–200. [Google Scholar]
- Öztaş, Y.; Boşgelmez, İ.İ. Oxidative Stress in Sickle Cell Disease and Emerging Roles for Antioxidants in Treatment Strategies. In Pathology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 65–75. [Google Scholar]
- Bleizgys, A. Zinc, Magnesium and Vitamin K Supplementation in Vitamin D Deficiency: Pathophysiological Background and Implications for Clinical Practice. Nutrients 2024, 16, 834. [Google Scholar] [CrossRef] [PubMed]
- Awor, S.; Bongomin, F.; Kaggwa, M.M.; Pebalo, F.P.; Musoke, D. Prevalence of Use of Herbal Medicines for the Treatment of Sickle Cell Disease in Africa: A Systematic Review and Meta-analysis. J. Herb. Med. 2023, 42, 100735. [Google Scholar] [CrossRef]
- Cotoraci, C.; Ciceu, A.; Sasu, A.; Hermenean, A. Natural Antioxidants in Anemia Treatment. Int. J. Mol. Sci. 2021, 22, 1883. [Google Scholar] [CrossRef]
- Ameh, S.J.; Tarfa, F.D.; Ebeshi, B.U. Traditional herbal management of sickle cell anemia: Lessons from Nigeria. Anemia 2012, 2012, 607436. [Google Scholar] [CrossRef]
- Takasu, J.; Uykimpang, R.; Sunga, M.A.; Amagase, H.; Niihara, Y. Aged Garlic Extract Is a Potential Therapy for Sickle-Cell Anemia13. J. Nutr. 2006, 136, 803S–805S. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, S.T.; Ohnishi, T. In Vitro Effects of Aged Garlic Extract and Other Nutritional Supplements on Sickle Erythrocytes. J. Nutr. 2001, 131, 1085S–1092S. [Google Scholar] [CrossRef] [PubMed]
- Fokou, E.; Arumugam, N. HPLC profiling, in vitro antisickling and antioxidant activities of phenolic compound extracts from black bean seeds (Phaseolus vulgarus L.) used in the management of sickle cell disease in the West Region of Cameroon. Nutr. Res. 2019, 3, 30. [Google Scholar]
- Afolabi, I.S.; Osikoya, I.O.; Fajimi, O.D.; Usoro, P.I.; Ogunleye, D.O.; Bisi-Adeniyi, T.; Adeyemi, A.O.; Adekeye, B.T. Solenostemon monostachyus, Ipomoea involucrata and Carica papaya seed oil versus Glutathione, or Vernonia amygdalina: Methanolic extracts of novel plants for the management of sickle cell anemia disease. BMC Complement. Altern. Med. 2012, 12, 262. [Google Scholar] [CrossRef] [PubMed]
- Oduola, T.; Adeniyi, F.; Ogunyemi, E.; Idowu, T.; Bello, I. Evaluation of the effects of intake of extract of unripe pawpaw (Carica papaya) on liver function in sickle cell patients. World J. Med. Sci. 2007, 2, 28–32. [Google Scholar]
- Imaga, N.; Gbenle, G.; Okochi, V.; Akanbi, S.; Edeoghon, S.; Oigbochie, V.; Kehinde, M.; Bamiro, S. Antisickling property of Carica papaya leaf extract. Afr. J. Biochem. Res. 2009, 3, 102–106. [Google Scholar]
- Christianah, C.-O.M.; Ajayi, D.O.; Odunowo, O.O. Ethno medicinal survey and evaluation of two recipes used in managing sickle cell disease in Ile-Ife community of Osun-State, Nigeria. Afr. J. Tradit. Complement. Altern. Med. 2020, 17, 37–54. [Google Scholar]
- Anorue, E.C.; Joshua, P.E. Evaluation of anti-sickling effects of two varieties of Cajanus cajan (L.) Huth on sickle cell beta thalassemia. J. Ethnopharmacol. 2024, 331, 118280. [Google Scholar] [CrossRef] [PubMed]
- Elemo, G.N.; Erukainure, O.L.; Okafor, J.N.C.; Banerjee, P.; Preissner, R.; Nwachukwu Nicholas-Okpara, V.A.; Atolani, O.; Omowunmi, O.; Ezeanyanaso, C.S.; Awosika, A.; et al. Underutilized legumes, Cajanus cajan and Glycine max may bring about antisickling effect in sickle cell disease by modulation of redox homeostasis in sickled erythrocytes and alteration of its functional chemistry. J. Food Biochem. 2022, 46, e14322. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-E.; Vo, T.-L.T.; Chen, C.-L.; Yang, N.-C.; Chen, C.-I.; Song, T.-Y. Nutritional Composition, Bioactive Compounds and Functional Evaluation of Various Parts of Cajanus cajan (L.) Millsp. Agriculture 2020, 10, 558. [Google Scholar] [CrossRef]
- Ohiagu, F.; Chikezie, P.; Chikezie, C. Sickle hemoglobin polymerization inhibition and antisickling medicinal plants. J. Phytopharm. 2021, 10, 126–133. [Google Scholar] [CrossRef]
- Pauline, N.; Cabral, B.N.P.; Anatole, P.C.; Jocelyne, A.M.V.; Bruno, M.; Jeanne, N.Y. The in vitro antisickling and antioxidant effects of aqueous extracts Zanthoxyllum heitzii on sickle cell disorder. BMC Complement. Altern. Med. 2013, 13, 162. [Google Scholar] [CrossRef]
- Okagu, I.U.; Ndefo, J.C.; Aham, E.C.; Udenigwe, C.C. Zanthoxylum Species: A Review of Traditional Uses, Phytochemistry and Pharmacology in Relation to Cancer, Infectious Diseases and Sickle Cell Anemia. Front. Pharmacol. 2021, 12, 713090. [Google Scholar] [CrossRef] [PubMed]
- Iyekowa, O.; Okieimen, F.; Ehisuoria, C.O. In-vitro Antisickling Activity of Pergularia daemia, Canna indica and Petiveria alliacea Plants used in the Treatment of Sickle Cell Anaemia in Edo State, Nigeria. Tanzan. J. Sci. 2023, 49, 433–445. [Google Scholar] [CrossRef]
- Danladi, B.Y.; Geetha, P. Ways to improve Life Expectancy in Sickle Cell Anaemia Patients using Herbs. Asian J. Res. Pharm. Sci. 2017, 7, 205. [Google Scholar] [CrossRef]
- Wambebe, C.; Ogunyale, P.; Gamaniel, K.; Nasipuri, R.; Okogun, J.; Samuel, B.; Olusola, A.; Orisadipe, A. Piper Guineense, Pterocarpus Osun, Eugenia Caryophyllata, and Sorghum Bicolor Extracts for Treating Sickle Cell Disease. U.S. Patent 5,800,819A, 1 September 1998. [Google Scholar]
- Akinsulie, A.; Temiye, E.; Akanmu, A.; Lesi, F.; Whyte, C. Clinical evaluation of extract of Cajanus cajan (Ciklavit®) in sickle cell anaemia. J. Trop. Pediatr. 2005, 51, 200–205. [Google Scholar] [CrossRef]
- Tavares, L.; Smaoui, S.; Pinilla, C.M.B.; Ben Hlima, H.; Lopes Barros, H. Ginger: A systematic review of clinical trials and recent advances in encapsulation of its bioactive compounds. Food Funct. 2022, 13, 1078–1091. [Google Scholar] [CrossRef]
- Tavares, L.; Santos, L.; Zapata Noreña, C.P. Bioactive compounds of garlic: A comprehensive review of encapsulation technologies, characterization of the encapsulated garlic compounds and their industrial applicability. Trends Food Sci. Technol. 2021, 114, 232–244. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Product | Objective | Formulation and Dose | Trial Length | Patients | Phases of Clinical Trials | Main Results | Reference |
|---|---|---|---|---|---|---|---|
| Omega-3 fatty acid docosahexaenoic acid as a triglyceride ester | Create a food product enriched with omega-3 fatty acids to examine its influence on fatty acid composition of blood cell membranes. | EPA: 570 mg/pouch DHA: 1900 mg/pouch. | 28 days | 10 | Phase II | Consumption of the food product was well-tolerated by participants and resulted in a statistically significant decrease in C-reactive protein levels. | [22] |
| Omega-3 fish oil | Investigate the relative efficacy of HU and folic acid (standard therapy) compared to the addition of omega-3 or vitamin D supplementation in managing the frequency and severity of painful crises in individuals with sickle cell disease. | Capsules of either 1000 mg omega-3 fish oil (400 mg EPA and 300 mg DHA) or 1.5 mL vitamin D (2800 IU/7 mL) | 12 months | 165 | Phase III | The omega-3 group exhibited a significant differential change in serum lipid profile compared to the control group, characterized by an increase in high-density lipoprotein (HDL) and low-density lipoprotein (LDL). | [23] |
| Vitamin A | Investigate the potential benefits of high-dose vitamin A supplementation in mitigating clinical complications associated with SCD in children. | 3000 or 6000 IU/d | 8 weeks | 42 | Pilot Study | Neither 3000 nor 6000 IU/d of vitamin A were sufficient to increase serum vitamin A concentrations in children with SCD. | [24] |
| EvenFlo and folic acid | Evaluate the potential benefits of a combined nutraceutical supplement (EvenFlo + folic acid) compared to standard folic acid supplementation alone in managing the clinical manifestations of SCD. | Folic Acid 500 microgram (mcg) OD and EvenFlo 500 milligram (mg) BID. | 4 weeks | 61 | Phase I | EvenFlo in addition to folic acid is an effective agent in the management of SCD. | [25] |
| Vitamin D | Evaluate the potential benefits of a combined nutraceutical supplement (EvenFlo + folic acid) compared to standard folic acid supplementation alone in managing the clinical manifestations of sickle cell disease (SCD). | Daily 1000 IU vitamin D3 | 10 months | 369 | Phase III | In children with SCD, combining a high-dose vitamin D bolus with daily 1000 IU vitamin D3 supplementation demonstrated greater efficacy in elevating 25-hydroxyvitamin D levels compared to daily supplementation alone. | [26] |
| Arginine | Evaluate the efficacy and safety of oral L-arginine (Arg) therapy in reducing the severity of vaso-occlusive painful crises in children with sickle cell anemia within the Nigerian population. | 100 mg/kg/dose | 24 moths | 68 | Phase II | Children receiving oral L-arginine therapy experienced a significant reduction in both the duration of vaso-occlusive crisis resolution and the length of their hospital stay. | [27] |
| Arginine | Investigate the impact of L-arginine supplementation on Doppler-derived indices of cardiopulmonary hemodynamics in children with sickle cell anemia experiencing vaso-occlusive pain crisis. | 300 mg/kg/d in three divided doses (100 mg/kg/dose) | 22 months | 66 | Phase II | L-arginine supplementation demonstrably improves cardiopulmonary hemodynamics in children with SCD experiencing vaso-occlusive pain crisis and acute chest syndrome. | [28] |
| Voxelotor | Investigate the differential pharmacokinetic and pharmacodynamic profiles of voxelotor in healthy adults compared to patients with SCD. | Doses of 100, 400, 1000, 2000, or 2800 mg. | 72 days | 48 | Phase I/II | Voxelotor acts by increasing hemoglobin’s affinity for oxygen, promoting the stabilization of the non-sickling, high-oxygen affinity (oxy) conformation of hemoglobin S, thereby reducing its propensity to polymerize and form sickle-shaped red blood cells. | [29] |
| Voxelotor | Conduct a comparative evaluation of voxelotor’s efficacy and safety against placebo in adolescents and adults with SCD, assessing its impact on hemoglobin levels, hemolysis markers, and the frequency and severity of vaso-occlusive crises. | 1500 mg and 900 mg | 72 weeks | 274 | Phase III | Voxelotor treatment resulted in a statistically significant increase in hemoglobin levels and a concurrent reduction in markers associated with hemolysis in patients with SCD. | [30] |
| Hydroxyurea | Investigate the efficacy of hydroxyurea as a primary stroke prevention strategy in children with SCD within the Nigerian population. | Low-dose (10 mg/kg per day) or moderate-dose (20 mg/kg per day) | 20 months | 220 | Phase III | A low to moderate dose of hydroxyurea reduces the frequency of vaso-occlusive episodes in the hospital and acute pain incidents at home. | [31] |
| Acetaminophen and Diclofenac Sodium | Evaluate the effectiveness of intravenous acetaminophen versus intravenous diclofenac sodium in treating skeletal VOCs in children with SCD. | Intravenous acetaminophen at 10 mg/kg/dose 8 hourly and intravenous diclofenac sodium at 1 mg/kg/dose 8 hourly | 2 months | 104 | Phase I | Intravenous acetaminophen offers a more effective treatment option than intravenous diclofenac for managing skeletal VOCs in children. | [32] |
| Canakinumab | Examine the hypothesis that persistent inflammatory processes associated with SCA contribute to the spectrum of clinical manifestations experienced by patients in an ambulatory setting. | 300 mg (or 4 mg/kg for participants < 40 kg) canakinumab (provided as 150 mg/1 mL of liquid in vials) or matching placebo | 6 months | 47 | Phase II | This study proposes that canakinumab’s targeted inhibition of IL-1β-driven inflammation in adolescents and young adults with SCA might offer therapeutic advantages with minimal safety risks. | [33] |
| Ticagrelor | Evaluate the efficacy and safety of ticagrelor, a reversible P2Y12 inhibitor, compared to placebo in preventing VOCs in pediatric patients with SCD. | Three different doses of ticagrelor were given based on body weight: 12 to 24 kg: 15 mg (1 tablet) twice daily; 24 to 48 kg: 30 mg (2 tablets) twice daily; 48 kg: 45 mg (3 tablets) twice daily. | 8 moths | 193 | Phase III | Ticagrelor treatment did not demonstrate a significant reduction in the rate of VOCs compared to placebo in pediatric patients with SCD. | [34] |
| Ketamine | Investigate the potential benefits and safety profile of single-dose ketamine infusion for managing acute VOCs in adult patients with SCD. | Ketamine (0.3 mg/kg) in 100 mL of normal saline. | 12 months | 278 | Phase III | Single-dose ketamine administration in adult SCD patients with acute VOCs led to a significant decrease in pain scores within a 2-h timeframe. | [35] |
| Isoquercetin | Investigate the potential of isoquercetin in mitigating thromboinflammation associated with SCD. | 28 to 35 doses of 1000 mg | 32 months | 46 | Phase II | Potential to reduce a range of thromboinflammatory biomarkers within the context of SCD management. | [36] |
| Herbal Products | Bioactive Compounds | Main Functions | References |
|---|---|---|---|
| Garlic | Organosulfur compounds (such as allicin, alliin, methylsulfanylalmethane, and diallyl disulfide). Flavonoids (such as quercetin, catechin, and epicatechin). | Targeting hemolysis-mediated endothelial dysfunction. Antioxidant potentials. Infective conditions especially respiratory infections in SCA. Significant enhancement in erythrocyte deformability by stabilizing the membranes of non-sickled red blood cells. | [5,98,99,100] |
| Black beans (Phaseolus vulgaris L.) | Anthocyanins, flavonols, flavones, and tannins. | Antioxidant properties. Inhibitory and reversibility activities on sickling. Stability effect on the membranes of erythrocytes. Targeting HbS polymerization. | [5,98,101] |
| Carica papaya seed oil | Phenolic compounds, fatty acids, tocopherols, and carotenoids. | Antioxidant activity. Effective in enhancing human sickle cell blood polymerization inhibition in females. Increase catalase activity in female human sickle cell blood. | [98,102,103,104] |
| Garcinia kola Heckel | Saponins, phenolic compounds, and flavonoids. | Targeting vaso-occlusion: modulate inflammatory responses. Effective membrane stabilization effects. Antisickling and antioxidant activities. | [5,71,98,105] |
| Cajanus cajan L. (seed) | Flavonoids and phenolic compounds. | Increase in β-globin synthesis and oxyhemoglobin concentration. Decrease in rate of polymerization and percentage hemolysis of the red blood cells. Antisickling effect. | [106,107,108,109] |
| Zanthoxyllum heitzii (Rutaceae) | Phenolic compounds (syringic acid, vanillic acid, protocatechuic acid, and p-hydroxybenzoic acid) and alkaloid compounds. | Antisickling property. Antioxidant properties. Antiradical activity. Sickle cell disease polymerization inhibition and sickle erythrocyte membrane stabilization. | [109,110,111] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, E.; Smaoui, S.; Brito, M.; Oliveira, J.M.; Arez, A.P.; Tavares, L. Sickle Cell Disease: Current Drug Treatments and Functional Foods with Therapeutic Potential. Curr. Issues Mol. Biol. 2024, 46, 5845-5865. https://doi.org/10.3390/cimb46060349
Gonçalves E, Smaoui S, Brito M, Oliveira JM, Arez AP, Tavares L. Sickle Cell Disease: Current Drug Treatments and Functional Foods with Therapeutic Potential. Current Issues in Molecular Biology. 2024; 46(6):5845-5865. https://doi.org/10.3390/cimb46060349
Chicago/Turabian StyleGonçalves, Elisângela, Slim Smaoui, Miguel Brito, J. M. Oliveira, Ana Paula Arez, and Loleny Tavares. 2024. "Sickle Cell Disease: Current Drug Treatments and Functional Foods with Therapeutic Potential" Current Issues in Molecular Biology 46, no. 6: 5845-5865. https://doi.org/10.3390/cimb46060349
APA StyleGonçalves, E., Smaoui, S., Brito, M., Oliveira, J. M., Arez, A. P., & Tavares, L. (2024). Sickle Cell Disease: Current Drug Treatments and Functional Foods with Therapeutic Potential. Current Issues in Molecular Biology, 46(6), 5845-5865. https://doi.org/10.3390/cimb46060349