
Interplay among Oxidative Stress, Autophagy, and the Endocannabinoid System in Neurodegenerative Diseases: Role of the Nrf2- p62/SQSTM1 Pathway and Nutraceutical Activation

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Triggering of the p62/SQSTM1-Keap1-NRF2 Pathway
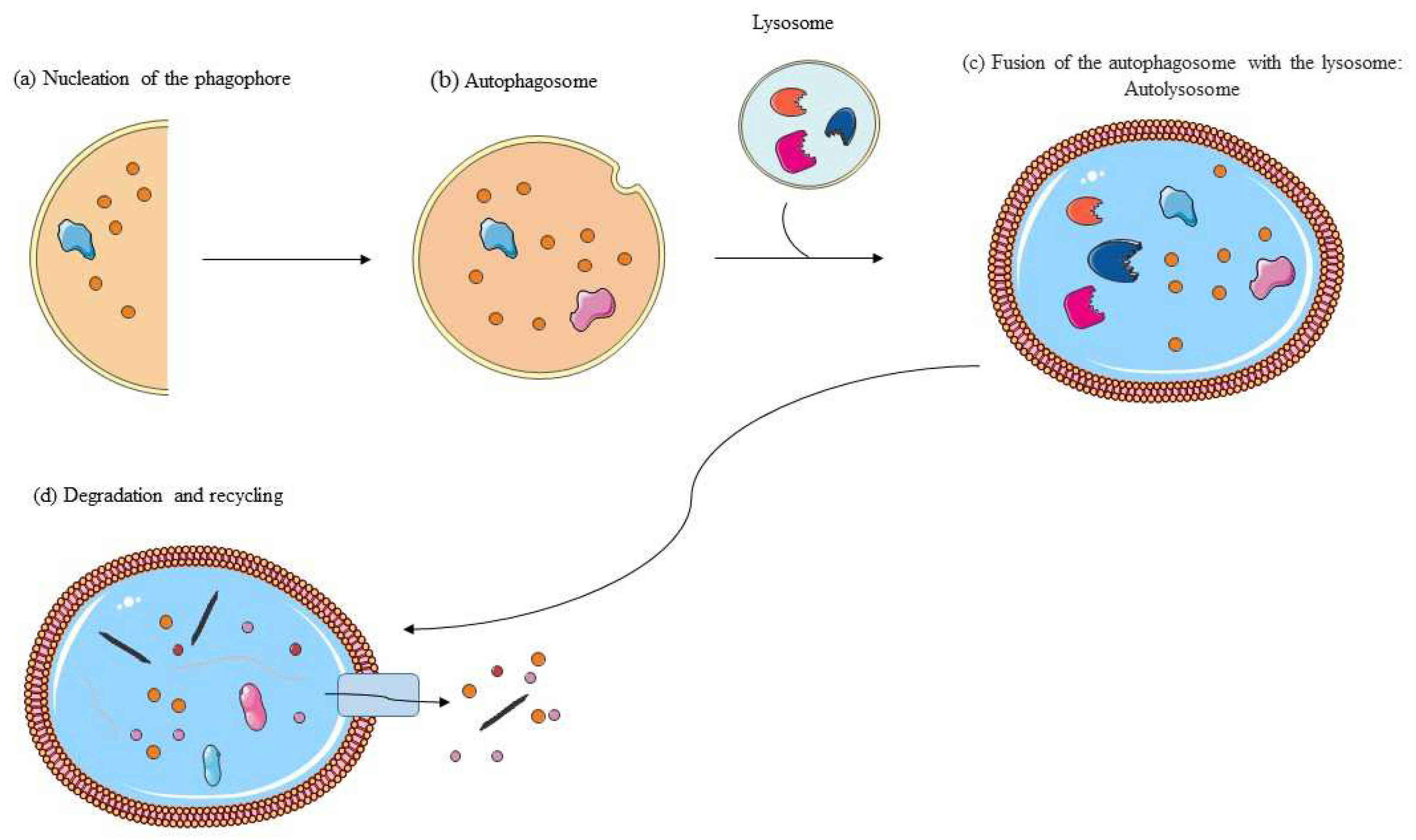
3. Autophagy: Molecular Mechanisms
4. p62/SQSTM1-Nrf2 Pathway: A Target in Neurodegenerative Disease Therapeutic Approaches
5. Endocannabinoid Activity and the Nrf2 Pathway
6. Stimulation of the Nrf2 Regulatory Pathway as a Mechanism for Maintaining Homeostasis
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hensley, K.; Harris-White, M.E. Redox Regulation of Autophagy in Healthy Brain and Neurodegeneration. Neurobiol. Dis. 2015, 84, 50–59. [Google Scholar] [CrossRef]
- Hensley, K. Neuroinflammation in Alzheimer’s Disease: Mechanisms, Pathologic Consequences, and Potential for Therapeutic Manipulation. JAD 2010, 21, 1–14. [Google Scholar] [CrossRef]
- Giordano, S.; Darley-Usmar, V.; Zhang, J. Autophagy as an Essential Cellular Antioxidant Pathway in Neurodegenerative Disease. Redox Biol. 2014, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Bongioanni, P.; Del Carratore, R.; Corbianco, S.; Diana, A.; Cavallini, G.; Masciandaro, S.M.; Dini, M.; Buizza, R. Climate Change and Neurodegenerative Diseases. Environ. Res. 2021, 201, 111511. [Google Scholar] [CrossRef]
- Bórquez, D.A.; Urrutia, P.J.; Wilson, C.; Van Zundert, B.; Núñez, M.T.; González-Billault, C. Dissecting the Role of Redox Signaling in Neuronal Development. J. Neurochem. 2016, 137, 506–517. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Neurodegeneration in Parkinson’s Disease: Interactions of Oxidative Stress, Tryptophan Catabolites and Depression with Mitochondria and Sirtuins. Mol. Neurobiol. 2014, 49, 771–783. [Google Scholar] [CrossRef]
- Aslan, M.; Ozben, T. Reactive Oxygen and Nitrogen Species in Alzheimers Disease. Curr. Alzheimer Res. 2004, 1, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Tanji, K.; Wakabayashi, K.; Matsuura, S.; Itoh, K. Role of the K Eap1/ N Rf2 Pathway in Neurodegenerative Diseases. Pathol. Int. 2015, 65, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Liddell, J. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Hubbs, A.F.; Benkovic, S.A.; Miller, D.B.; O’Callaghan, J.P.; Battelli, L.; Schwegler-Berry, D.; Ma, Q. Vacuolar Leukoencephalopathy with Widespread Astrogliosis in Mice Lacking Transcription Factor Nrf2. Am. J. Pathol. 2007, 170, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Cai, Z.; Zhou, Y.; Xiao, M.; Yan, L.-J.; He, W. Activation of mTOR: A Culprit of Alzheimer’s Disease? NDT 2015, 2015, 1015–1030. [Google Scholar] [CrossRef]
- Babu, J.R.; Geetha, T.; Wooten, M.W. Sequestosome 1/P62 Shuttles Polyubiquitinated Tau for Proteasomal Degradation. J. Neurochem. 2005, 94, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Kuusisto, E.; Salminen, A.; Alafuzoff, I. Early Accumulation of P62 in Neurofibrillary Tangles in Alzheimer’s Disease: Possible Role in Tangle Formation. Neuropathol. Appl. Neurobiol. 2002, 28, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Cui, M.-C.; Hu, W.-Z.; Zeng, Q.; Wang, Y.-L.; Zhang, W.; Huang, Y. Genetic and Molecular Evaluation of SQSTM1/P62 on the Neuropathologies of Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 829232. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Huang, H.-C.; Yang, C.S.; Pickett, C.B. Increased Protein Stability as a Mechanism That Enhances Nrf2-Mediated Transcriptional Activation of the Antioxidant Response Element. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct Evidence That Sulfhydryl Groups of Keap1 Are the Sensors Regulating Induction of Phase 2 Enzymes That Protect against Carcinogens and Oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the Cysteine-Based Mammalian Intracellular Sensor for Electrophiles and Oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef]
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic Evidence That Small Maf Proteins Are Essential for the Activation of Antioxidant Response Element-Dependent Genes. Mol. Cell. Biol. 2005, 25, 8044–8051. [Google Scholar] [CrossRef]
- Ishii, T.; Warabi, E.; Siow, R.C.M.; Mann, G.E. Sequestosome1/P62: A Regulator of Redox-Sensitive Voltage-Activated Potassium Channels, Arterial Remodeling, Inflammation, and Neurite Outgrowth. Free Radic. Biol. Med. 2013, 65, 102–116. [Google Scholar] [CrossRef]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 Degradation by Autophagy for the Maintenance of Redox Homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the Context-Dependent Role for Autophagy in Cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H.; Alafuzoff, I. Emerging Role of P62/Sequestosome-1 in the Pathogenesis of Alzheimer’s Disease. Prog. Neurobiol. 2012, 96, 87–95. [Google Scholar] [CrossRef]
- Joshi, G.; Gan, K.A.; Johnson, D.A.; Johnson, J.A. Increased Alzheimer’s Disease–like Pathology in the APP/ PS1ΔE9 Mouse Model Lacking Nrf2 through Modulation of Autophagy. Neurobiol. Aging 2015, 36, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and Molecular Mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and Diversity in Autophagy Mechanisms: Lessons from Yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, X.; Song, Y.-Q.; Tu, J. Autophagy in Alzheimer’s Disease Pathogenesis: Therapeutic Potential and Future Perspectives. Ageing Res. Rev. 2021, 72, 101464. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. Role of the Apg12 Conjugation System in Mammalian Autophagy. Int. J. Biochem. Cell Biol. 2003, 35, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 Conjugation System in Mammalian Autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. The Function of Autophagy in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 26797–26812. [Google Scholar] [CrossRef] [PubMed]
- Dello Russo, C.; Lisi, L.; Feinstein, D.L.; Navarra, P. mTOR Kinase, a Key Player in the Regulation of Glial Functions: Relevance for the Therapy of Multiple Sclerosis. Glia 2013, 61, 301–311. [Google Scholar] [CrossRef]
- Yamamoto, A.; Yue, Z. Autophagy and Its Normal and Pathogenic States in the Brain. Annu. Rev. Neurosci. 2014, 37, 55–78. [Google Scholar] [CrossRef]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of Selective Autophagy: The P62/SQSTM1 Paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef]
- Abdelhamid, R.F.; Nagano, S. Crosstalk between Oxidative Stress and Aging in Neurodegeneration Disorders. Cells 2023, 12, 753. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. P62 Links Autophagy and Nrf2 Signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 Signaling in Oxidative and Reductive Stress. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of P62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol Cell 2013, 51, 618–631. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins Activate Nrf2 by Promoting P62-Dependent Autophagic Degradation of Keap1 and Prevent Oxidative Liver Damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef]
- Gureev, A.P.; Sadovnikova, I.S.; Starkov, N.N.; Starkov, A.A.; Popov, V.N. P62-Nrf2-P62 Mitophagy Regulatory Loop as a Target for Preventive Therapy of Neurodegenerative Diseases. Brain Sci. 2020, 10, 847. [Google Scholar] [CrossRef]
- Ma, S.; Attarwala, I.Y.; Xie, X.-Q. SQSTM1/P62: A Potential Target for Neurodegenerative Disease. ACS Chem. Neurosci. 2019, 10, 2094–2114. [Google Scholar] [CrossRef]
- Ramesh Babu, J.; Lamar Seibenhener, M.; Peng, J.; Strom, A.; Kemppainen, R.; Cox, N.; Zhu, H.; Wooten, M.C.; Diaz-Meco, M.T.; Moscat, J.; et al. Genetic Inactivation of P62 Leads to Accumulation of Hyperphosphorylated Tau and Neurodegeneration. J. Neurochem. 2008, 106, 107–120. [Google Scholar] [CrossRef]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of Amyloid Precursor Protein C-Terminal Fragments Triggers Mitochondrial Structure, Function, and Mitophagy Defects in Alzheimer’s Disease Models and Human Brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H.; Ylä-Herttuala, S.; Tanila, H.; Levonen, A.-L.; Koistinaho, M.; et al. Intrahippocampal Injection of a Lentiviral Vector Expressing Nrf2 Improves Spatial Learning in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505–16510. [Google Scholar] [CrossRef]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and Alzheimer’s Disease. Cell. Mol. Neurobiol. 2017, 37, 377–388. [Google Scholar] [CrossRef]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial Defects and Oxidative Stress in Alzheimer Disease and Parkinson Disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of Mitochondrial ROS in the Brain: From Physiology to Neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Businaro, R.; Maggi, E.; Armeli, F.; Murray, A.; Laskin, D.L. Nutraceuticals as Potential Therapeutics for Vesicant-induced Pulmonary Fibrosis. Ann. N. Y. Acad. Sci. 2020, 1480, 5–13. [Google Scholar] [CrossRef]
- Wang, H.; Cao, G.; Prior, R.L. Oxygen Radical Absorbing Capacity of Anthocyanins. J. Agric. Food Chem. 1997, 45, 304–309. [Google Scholar] [CrossRef]
- Huang, W.; Zhu, Y.; Li, C.; Sui, Z.; Min, W. Effect of Blueberry Anthocyanins Malvidin and Glycosides on the Antioxidant Properties in Endothelial Cells. Oxidative Med. Cell. Longev. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- De Caris, M.G.; Grieco, M.; Maggi, E.; Francioso, A.; Armeli, F.; Mosca, L.; Pinto, A.; D’Erme, M.; Mancini, P.; Businaro, R. Blueberry Counteracts BV-2 Microglia Morphological and Functional Switch after LPS Challenge. Nutrients 2020, 12, 1830. [Google Scholar] [CrossRef]
- Carey, A.N.; Fisher, D.R.; Rimando, A.M.; Gomes, S.M.; Bielinski, D.F.; Shukitt-Hale, B. Stilbenes and Anthocyanins Reduce Stress Signaling in BV-2 Mouse Microglia. J. Agric. Food Chem. 2013, 61, 5979–5986. [Google Scholar] [CrossRef]
- Wu, T.; Gao, Y.; Guo, X.; Zhang, M.; Gong, L. Blackberry and Blueberry Anthocyanin Supplementation Counteract High-Fat-Diet-Induced Obesity by Alleviating Oxidative Stress and Inflammation and Accelerating Energy Expenditure. Oxidative Med. Cell. Longev. 2018, 2018, 4051232. [Google Scholar] [CrossRef]
- Xie, C.; Kang, J.; Ferguson, M.E.; Nagarajan, S.; Badger, T.M.; Wu, X. Blueberries Reduce Pro-inflammatory Cytokine TNF-α and IL-6 Production in Mouse Macrophages by Inhibiting NF-κB Activation and the MAPK Pathway. Mol. Nutr. Food Res. 2011, 55, 1587–1591. [Google Scholar] [CrossRef]
- Sobolev, A.P.; Ciampa, A.; Ingallina, C.; Mannina, L.; Capitani, D.; Ernesti, I.; Pinto, A. Blueberry-Based Meals for Obese Patients with Metabolic Syndrome: A Multidisciplinary Metabolomic Pilot Study. Metabolites 2019, 9, 138. [Google Scholar] [CrossRef]
- Nair, A.R.; Mariappan, N.; Stull, A.J.; Francis, J. Blueberry Supplementation Attenuates Oxidative Stress within Monocytes and Modulates Immune Cell Levels in Adults with Metabolic Syndrome: A Randomized, Double-Blind, Placebo-Controlled Trial. Food Funct. 2017, 8, 4118–4128. [Google Scholar] [CrossRef]
- Zafra-Stone, S.; Yasmin, T.; Bagchi, M.; Chatterjee, A.; Vinson, J.A.; Bagchi, D. Berry Anthocyanins as Novel Antioxidants in Human Health and Disease Prevention. Mol. Nutr. Food Res. 2007, 51, 675–683. [Google Scholar] [CrossRef]
- Lippai, M.; Lőw, P. The Role of the Selective Adaptor P62 and Ubiquitin-Like Proteins in Autophagy. BioMed Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. P62/SQSTM1 Is Required for Parkin-Induced Mitochondrial Clustering but Not Mitophagy; VDAC1 Is Dispensable for Both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-Mediated Mitophagy Is Dependent on VDAC1 and P62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- George, M.; Tharakan, M.; Culberson, J.; Reddy, A.P.; Reddy, P.H. Role of Nrf2 in Aging, Alzheimer’s and Other Neurodegenerative Diseases. Ageing Res. Rev. 2022, 82, 101756. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.-C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and Amyloid Beta-Induced Defective Autophagy, Mitophagy, Mitochondrial Structural and Functional Changes and Synaptic Damage in Hippocampal Neurons from Alzheimer’s Disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, V.S.; Berman, S.B. Mitochondrial Dynamics in Parkinson’s Disease. Exp. Neurol. 2009, 218, 247–256. [Google Scholar] [CrossRef]
- Anis, E.; Zafeer, M.F.; Firdaus, F.; Islam, S.N.; Khan, A.A.; Hossain, M.M. Perillyl Alcohol Mitigates Behavioural Changes and Limits Cell Death and Mitochondrial Changes in Unilateral 6-OHDA Lesion Model of Parkinson’s Disease Through Alleviation of Oxidative Stress. Neurotox. Res. 2020, 38, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The Selective Autophagy Substrate P62 Activates the Stress Responsive Transcription Factor Nrf2 through Inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A Noncanonical Mechanism of Nrf2 Activation by Autophagy Deficiency: Direct Interaction between Keap1 and P62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef]
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and Functional Interaction of Sequestosome 1 with Keap1 Regulates the Keap1-Nrf2 Cell Defense Pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjøttem, E.; Bowitz Larsen, K.; Atesoh Awuh, J.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. P62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-Driven Gene Transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Wang, Z.; Fu, Z.; Ma, H.; Jiang, M.; Xu, A.; Zhang, W. P62/SQSTM1 Protects against Cisplatin-Induced Oxidative Stress in Kidneys by Mediating the Cross Talk between Autophagy and the Keap1-Nrf2 Signalling Pathway. Free Radic. Res. 2019, 53, 800–814. [Google Scholar] [CrossRef]
- Armeli, F.; Bonucci, A.; Maggi, E.; Pinto, A.; Businaro, R. Mediterranean Diet and Neurodegenerative Diseases: The Neglected Role of Nutrition in the Modulation of the Endocannabinoid System. Biomolecules 2021, 11, 790. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Ferrer, I. CB2 Cannabinoid Receptor As Potential Target against Alzheimer’s Disease. Front. Neurosci. 2016, 10, 243. [Google Scholar] [CrossRef]
- Gómez-Gálvez, Y.; Palomo-Garo, C.; Fernández-Ruiz, J.; García, C. Potential of the Cannabinoid CB2 Receptor as a Pharmacological Target against Inflammation in Parkinson’s Disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 64, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Bilkei-Gorzo, A. The Endocannabinoid System in Normal and Pathological Brain Ageing. Phil. Trans. R. Soc. B 2012, 367, 3326–3341. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Ferrer, I. Cannabinoids for Treatment of Alzheimerâ€TMs Disease: Moving toward the Clinic. Front. Pharmacol. 2014, 5, 37. [Google Scholar] [CrossRef]
- Monsalvo-Maraver, L.A.; Ovalle-Noguez, E.A.; Nava-Osorio, J.; Maya-López, M.; Rangel-López, E.; Túnez, I.; Tinkov, A.A.; Tizabi, Y.; Aschner, M.; Santamaría, A.; et al. Interactions Between the Ubiquitin–Proteasome System, Nrf2, and the Cannabinoidome as Protective Strategies to Combat Neurodegeneration: Review on Experimental Evidence. Neurotox. Res. 2024, 42, 18. [Google Scholar] [CrossRef]
- Elmazoglu, Z.; Rangel-López, E.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Túnez, I.; Aschner, M.; Santamaría, A.; Karasu, Ç. Cannabinoid-Profiled Agents Improve Cell Survival via Reduction of Oxidative Stress and Inflammation, and Nrf2 Activation in a Toxic Model Combining hyperglycemia+Aβ1-42 Peptide in Rat Hippocampal Neurons. Neurochem. Int. 2020, 140, 104817. [Google Scholar] [CrossRef] [PubMed]
- Tadijan, A.; Vlašić, I.; Vlainić, J.; Đikić, D.; Oršolić, N.; Jazvinšćak Jembrek, M. Intracellular Molecular Targets and Signaling Pathways Involved in Antioxidative and Neuroprotective Effects of Cannabinoids in Neurodegenerative Conditions. Antioxidants 2022, 11, 2049. [Google Scholar] [CrossRef]
- Wang, M.; Liu, M.; Ma, Z. Cannabinoid Type 2 Receptor Activation Inhibits MPP+-Induced M1 Differentiation of Microglia through Activating PI3K/Akt/Nrf2 Signal Pathway. Mol. Biol. Rep. 2023, 50, 4423–4433. [Google Scholar] [CrossRef] [PubMed]
- Komorowska-Müller, J.A.; Schmöle, A.-C. CB2 Receptor in Microglia: The Guardian of Self-Control. Int. J. Mol. Sci. 2020, 22, 19. [Google Scholar] [CrossRef]
- Young, A.P.; Denovan-Wright, E.M. The Dynamic Role of Microglia and the Endocannabinoid System in Neuroinflammation. Front. Pharmacol. 2022, 12, 806417. [Google Scholar] [CrossRef] [PubMed]
- Kozela, E.; Pietr, M.; Juknat, A.; Rimmerman, N.; Levy, R.; Vogel, Z. Cannabinoids Δ9-Tetrahydrocannabinol and Cannabidiol Differentially Inhibit the Lipopolysaccharide-Activated NF-κB and Interferon-β/STAT Proinflammatory Pathways in BV-2 Microglial Cells. J. Biol. Chem. 2010, 285, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting Molecular Cross-Talk between Nrf2 and NF-κB Response Pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Solas, M.; Francis, P.T.; Franco, R.; Ramirez, M.J. CB2 Receptor and Amyloid Pathology in Frontal Cortex of Alzheimer’s Disease Patients. Neurobiol. Aging 2013, 34, 805–808. [Google Scholar] [CrossRef]
- Ehrhart, J.; Obregon, D.; Mori, T.; Hou, H.; Sun, N.; Bai, Y.; Klein, T.; Fernandez, F.; Tan, J.; Shytle, R.D. Stimulation of Cannabinoid Receptor 2 (CB2) Suppresses Microglial Activation. J. Neuroinflamm. 2005, 2, 29. [Google Scholar] [CrossRef]
- Ramírez, B.G.; Blázquez, C.; Del Pulgar, T.G.; Guzmán, M.; De Ceballos, M.L. Prevention of Alzheimer’s Disease Pathology by Cannabinoids: Neuroprotection Mediated by Blockade of Microglial Activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef]
- Martín-Moreno, A.M.; Reigada, D.; Ramírez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; De Ceballos, M.L. Cannabidiol and Other Cannabinoids Reduce Microglial Activation In Vitro and In Vivo: Relevance to Alzheimer’s Disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef]
- Esposito, G.; Iuvone, T.; Savani, C.; Scuderi, C.; De Filippis, D.; Papa, M.; Di Marzo, V.; Steardo, L. Opposing Control of Cannabinoid Receptor Stimulation on Amyloid-β-Induced Reactive Gliosis: In Vitro and in Vivo Evidence. J. Pharmacol. Exp. Ther. 2007, 322, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Galán-Ganga, M.; del Río, R.; Jiménez-Moreno, N.; Díaz-Guerra, M.; Lastres-Becker, I. Cannabinoid CB2 Receptor Modulation by the Transcription Factor NRF2 Is Specific in Microglial Cells. Cell. Mol. Neurobiol. 2020, 40, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Assis, L.C.; Straliotto, M.R.; Engel, D.; Hort, M.A.; Dutra, R.C.; de Bem, A.F. β-Caryophyllene Protects the C6 Glioma Cells against Glutamate-Induced Excitotoxicity through the Nrf2 Pathway. Neuroscience 2014, 279, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Garduño, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-Deacetylation of the Transcription Factor Nrf2 (Nuclear Factor Erythroid 2-Related Factor 2) Regulates Its Transcriptional Activity and Nucleocytoplasmic Localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef]
- Grzeczka, A.; Kordowitzki, P. Resveratrol and SIRT1: Antiaging Cornerstones for Oocytes? Nutrients 2022, 14, 5101. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α (PGC-1α): Transcriptional Coactivator and Metabolic Regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef]
- Olmos, Y.; Sánchez-Gómez, F.J.; Wild, B.; García-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SirT1 Regulation of Antioxidant Genes Is Dependent on the Formation of a FoxO3a/PGC-1α Complex. Antioxid. Redox Signal. 2013, 19, 1507–1521. [Google Scholar] [CrossRef]
- Rajaram, S.; Jones, J.; Lee, G.J. Plant-Based Dietary Patterns, Plant Foods, and Age-Related Cognitive Decline. Adv. Nutr. 2019, 10, S422–S436. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxidative Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D.; Reed, T.T. Cellular Stress Response (Hormesis) in Response to Bioactive Nutraceuticals with Relevance to Alzheimer Disease. Antioxid. Redox Signal. 2023, 38, ars.2022.0214. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Wang, Z.; Luo, Y.; Zhang, Y.; He, W.; Mei, Y.; Xue, J.; Li, M.; Pan, H.; Li, W.; et al. FA-97, a New Synthetic Caffeic Acid Phenethyl Ester Derivative, Protects against Oxidative Stress-Mediated Neuronal Cell Apoptosis and Scopolamine-Induced Cognitive Impairment by Activating Nrf2/HO-1 Signaling. Oxidative Med. Cell. Longev. 2019, 2019, 8239642. [Google Scholar] [CrossRef]
- Han, X.-D.; Zhang, Y.-Y.; Wang, K.-L.; Huang, Y.-P.; Yang, Z.-B.; Liu, Z. The Involvement of Nrf2 in the Protective Effects of (-)-Epigallocatechin-3-Gallate (EGCG) on NaAsO2-Induced Hepatotoxicity. Oncotarget 2017, 8, 65302–65312. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 Signaling Pathway: Pivotal Roles in Inflammation. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Leonardo, C.C.; Doré, S. Dietary Flavonoids Are Neuroprotective through Nrf2-Coordinated Induction of Endogenous Cytoprotective Proteins. Nutr. Neurosci. 2011, 14, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Unno, K.; Pervin, M.; Nakagawa, A.; Iguchi, K.; Hara, A.; Takagaki, A.; Nanjo, F.; Minami, A.; Nakamura, Y. Blood–Brain Barrier Permeability of Green Tea Catechin Metabolites and Their Neuritogenic Activity in Human Neuroblastoma SH-SY5Y Cells. Mol. Nutr. Food Res. 2017, 61, 1700294. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; Mithieux, G.; Vega, F.V. Resveratrol Protects Primary Rat Hepatocytes against Oxidative Stress Damage. Eur. J. Pharmacol. 2008, 591, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Bian, Z.; Zhang, M. Targeting the Nrf2 Signaling Pathway Using Phytochemical Ingredients: A Novel Therapeutic Road Map to Combat Neurodegenerative Diseases. Phytomedicine 2023, 109, 154582. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Chang, J.-C.; Lin, W.-Y.; Li, C.-C.; Hsieh, M.; Chen, H.-W.; Wang, T.-S.; Wu, W.-T.; Liu, C.-S.; Liu, K.-L. Caffeic Acid and Resveratrol Ameliorate Cellular Damage in Cell and Drosophila Models of Spinocerebellar Ataxia Type 3 through Upregulation of Nrf2 Pathway. Free Radic. Biol. Med. 2018, 115, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Yuan, T.; Diao, Z.; Zhang, C.; Liu, Z.; Liu, X. Protective Effects of Sesamol on Systemic Oxidative Stress-Induced Cognitive Impairments via Regulation of Nrf2/Keap1 Pathway. Food Funct. 2018, 9, 5912–5924. [Google Scholar] [CrossRef]
- Baluchnejadmojarad, T.; Rabiee, N.; Zabihnejad, S.; Roghani, M. Ellagic Acid Exerts Protective Effect in Intrastriatal 6-Hydroxydopamine Rat Model of Parkinson’s Disease: Possible Involvement of ERβ/Nrf2/HO-1 Signaling. Brain Res. 2017, 1662, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; He, L.; Wang, W.; Xie, Z.; Zhang, X.; Wang, P.; Wang, L.; Yan, C.; Liu, Z.; Zhao, J.; et al. Activation of Atg7-Dependent Autophagy by a Novel Inhibitor of the Keap1–Nrf2 Protein–Protein Interaction from Penthorum chinense Pursh. Attenuates 6-Hydroxydopamine-Induced Ferroptosis in Zebrafish and Dopaminergic Neurons. Food Funct. 2022, 13, 7885–7900. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S.; Khalifeh, S.; Shajiee, H.; Ashabi, G. Dietary Uptake of Salvia Macilenta Extract Improves Nrf2 Antioxidant Signaling Pathway and Diminishes Inflammation and Apoptosis in Amyloid Beta-Induced Rats. Mol. Biol. Rep. 2021, 48, 7667–7676. [Google Scholar] [CrossRef] [PubMed]
- Hajiluian, G.; Karegar, S.J.; Shidfar, F.; Aryaeian, N.; Salehi, M.; Lotfi, T.; Farhangnia, P.; Heshmati, J.; Delbandi, A.-A. The Effects of Ellagic Acid Supplementation on Neurotrophic, Inflammation, and Oxidative Stress Factors, and Indoleamine 2, 3-Dioxygenase Gene Expression in Multiple Sclerosis Patients with Mild to Moderate Depressive Symptoms: A Randomized, Triple-Blind, Placebo-Controlled Trial. Phytomedicine 2023, 121, 155094. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Lam, C.W.K.; Cheung, S.K.-K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-Month Randomized, Placebo-Controlled, Double-Blind, Pilot Clinical Trial of Curcumin in Patients With Alzheimer Disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Lee, Y.Z.; Kow, A.S.F.; Lee, Q.L.; Cheng Lim, L.W.; Yusof, R.; Tham, C.L.; Ho, Y.-C.; Ming Tatt, L. Neuroprotective Effects of Gypenosides: A Review on Preclinical Studies in Neuropsychiatric Disorders. Eur. J. Pharmacol. 2024, 978, 176766. [Google Scholar] [CrossRef]
- Chiu, H.-F.; Venkatakrishnan, K.; Wang, C.-K. The Role of Nutraceuticals as a Complementary Therapy against Various Neurodegenerative Diseases: A Mini-Review. J. Tradit. Complement. Med. 2020, 10, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Tomata, Y.; Kakizaki, M.; Nakaya, N.; Tsuboya, T.; Sone, T.; Kuriyama, S.; Hozawa, A.; Tsuji, I. Green Tea Consumption and the Risk of Incident Functional Disability in Elderly Japanese: The Ohsaki Cohort 2006 Study. Am. J. Clin. Nutr. 2012, 95, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Witte, A.V.; Kerti, L.; Margulies, D.S.; Floel, A. Effects of Resveratrol on Memory Performance, Hippocampal Functional Connectivity, and Glucose Metabolism in Healthy Older Adults. J. Neurosci. 2014, 34, 7862–7870. [Google Scholar] [CrossRef]
- Wang, K.; Chen, X. Protective Effect of Flavonoids on Oxidative Stress Injury in Alzheimer’s Disease. Nat. Prod. Res. 2024, 23, 1–28. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armeli, F.; Mengoni, B.; Laskin, D.L.; Businaro, R. Interplay among Oxidative Stress, Autophagy, and the Endocannabinoid System in Neurodegenerative Diseases: Role of the Nrf2- p62/SQSTM1 Pathway and Nutraceutical Activation. Curr. Issues Mol. Biol. 2024, 46, 6868-6884. https://doi.org/10.3390/cimb46070410
Armeli F, Mengoni B, Laskin DL, Businaro R. Interplay among Oxidative Stress, Autophagy, and the Endocannabinoid System in Neurodegenerative Diseases: Role of the Nrf2- p62/SQSTM1 Pathway and Nutraceutical Activation. Current Issues in Molecular Biology. 2024; 46(7):6868-6884. https://doi.org/10.3390/cimb46070410
Chicago/Turabian StyleArmeli, Federica, Beatrice Mengoni, Debra L. Laskin, and Rita Businaro. 2024. "Interplay among Oxidative Stress, Autophagy, and the Endocannabinoid System in Neurodegenerative Diseases: Role of the Nrf2- p62/SQSTM1 Pathway and Nutraceutical Activation" Current Issues in Molecular Biology 46, no. 7: 6868-6884. https://doi.org/10.3390/cimb46070410
APA StyleArmeli, F., Mengoni, B., Laskin, D. L., & Businaro, R. (2024). Interplay among Oxidative Stress, Autophagy, and the Endocannabinoid System in Neurodegenerative Diseases: Role of the Nrf2- p62/SQSTM1 Pathway and Nutraceutical Activation. Current Issues in Molecular Biology, 46(7), 6868-6884. https://doi.org/10.3390/cimb46070410