
Skin Development and Disease: A Molecular Perspective

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
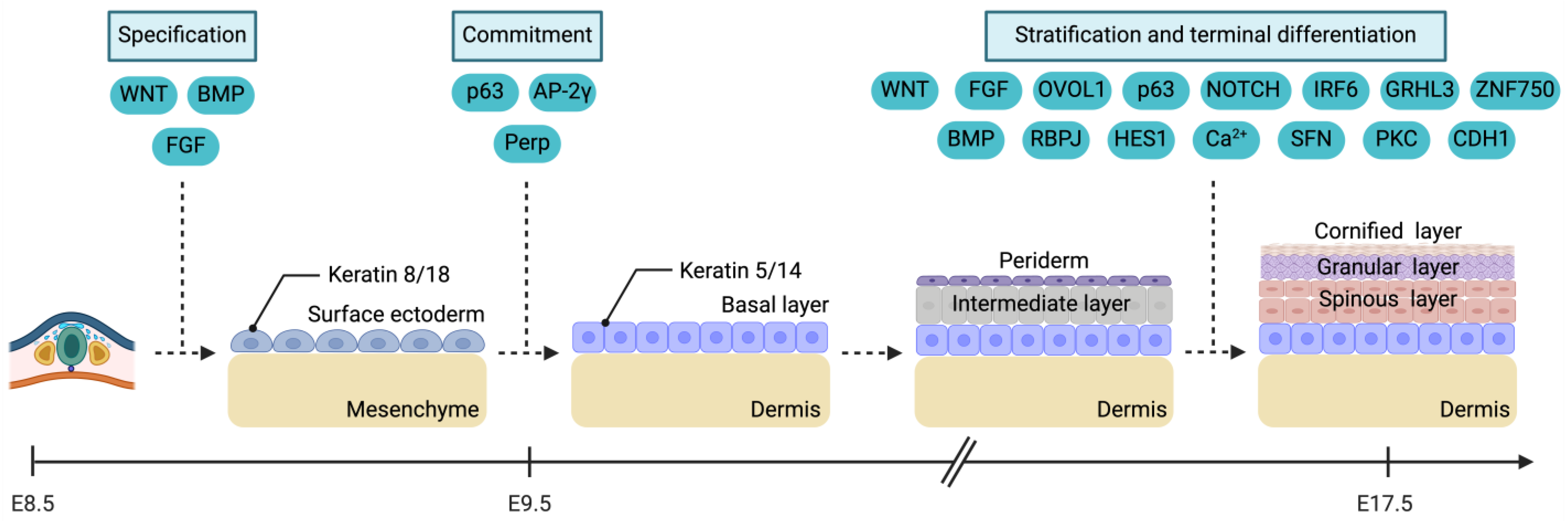
2. Embryonic Development of the Skin
2.1. Specification
2.2. Commitment
2.3. Stratification and Terminal Differentiation
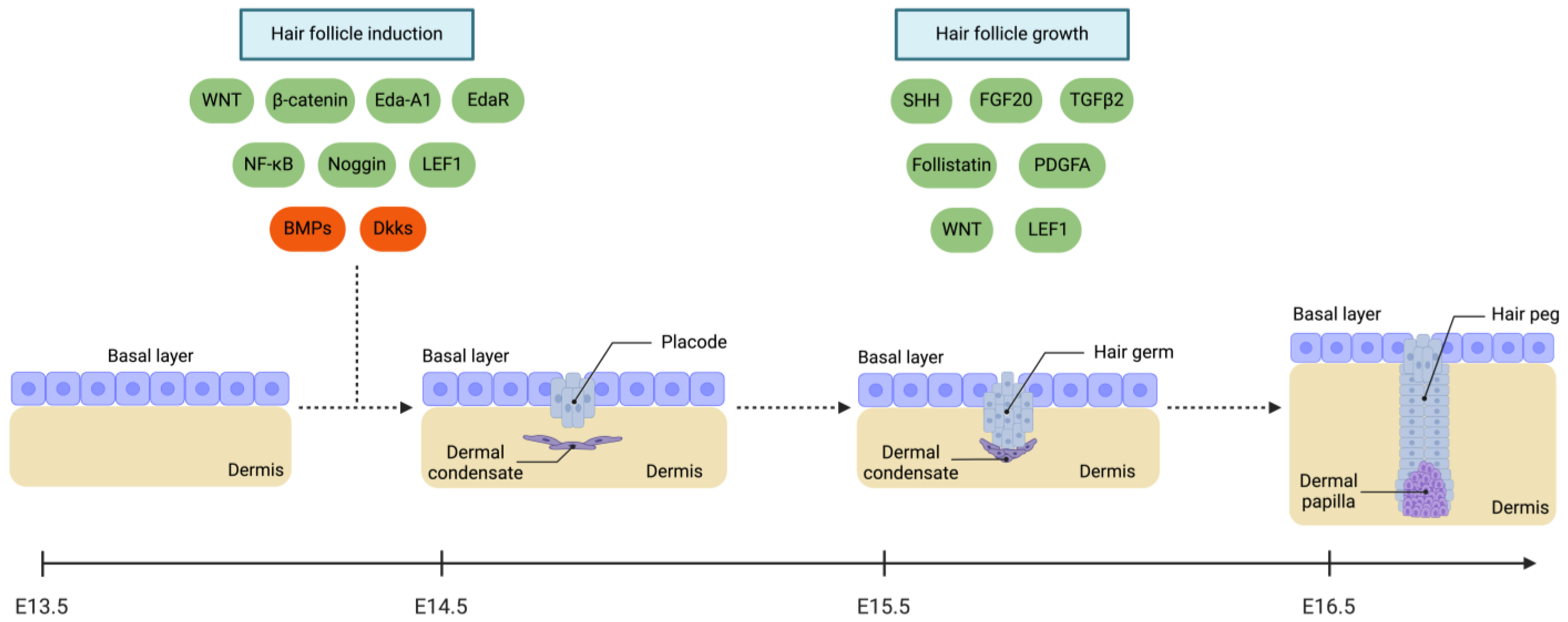
2.4. Appendageal Growth
2.4.1. Hair Follicle (HF)
2.4.2. Sweat Glands
2.5. Migration of Cells within the Epidermis
3. Molecular Basis of Congenital Skin Diseases and Cancer
3.1. Congenital Skin Disorders
3.1.1. Nevoid Basal Cell Carcinoma Syndrome (NBCCS)
3.1.2. Epidermolysis Bullosa (EB)
3.1.3. Hypohidrotic Ectodermal Dysplasia (HED)
3.2. Skin Cancer
3.2.1. Basal Cell Carcinoma (BCC)
3.2.2. Squamous Cell Carcinoma (SCC)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AP-2 | Activating protein 2 |
| BCC | Basal cell carcinoma |
| BMP | Bone morphogenetic protein |
| Ca2+ | Calcium ion |
| CAS9 | CRISPR-associated protein 9 |
| CCND1 | Cyclin D1 |
| CD151 | Cluster of differentiation 151 |
| CDH1 | Cadherin 1 |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| cDNA | Complementary deoxyribonucleic acid |
| CLDN23 | Claudin 23 |
| CLDN6 | Claudin 6 |
| COL17A1 | Collagen type XVII alpha 1 chain |
| COL7A1 | Collagen type VII alpha 1 chain |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CSF1R | Colony-stimulating factor 1 receptor |
| DCs | Dermal condensates |
| DEB | Dystrophic epidermolysis bullosa |
| DKK | Dickkopf WNT signalling pathway inhibitor |
| DPH3 | Diphthamide biosynthesis 3 |
| DSG | Desmoglein |
| DST | Dystonin |
| E | Embryonic day |
| EB | Epidermolysis bullosa |
| EBS | Epidermolysis bullosa simplex |
| EDA-A1 | Ectodysplasin-A1 |
| EDAR | Ectodysplasin A receptor |
| EDARADD | EDAR-associated death domain |
| EDI200 | Ectodysplasin-A1 replacement therapy |
| EDNRB | Endothelin receptor type B |
| EGFR | Epidermal growth factor receptor |
| EN1 | Engrailed homeobox 1 |
| EXPH5 | Exiphilin 5 |
| FAT1 | FAT atypical cadherin 1 |
| FBXW7 | F-box and WD repeat domain containing 7 |
| FERMT1 | FERM domain containing kindlin 1 |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| FOXA1 | Forkhead box protein A1 |
| FOXD3 | Forkhead box D3 |
| FOXI1 | Forkhead box I1 |
| GLI | Glioma-associated oncogene |
| GPCR | G protein-coupled receptor-like protein |
| GRHL3 | Grainyhead-like transcription factor 3 |
| HED | Hypohidrotic ectodermal dysplasia |
| HES1 | Hes family bHLH transcription factor 1 |
| HF | Hair follicle |
| HH | Hedgehog |
| HPV | Human papillomavirus |
| ID2 | Inhibitor of DNA binding 2 |
| IKBKG | Inhibitor of nuclear factor kappa B kinase regulatory subunit gamma |
| IL-34 | Interleukin 34 |
| iPSCs | Induced pluripotent stem cells |
| IRF6 | Interferon regulatory factor 6 |
| ITGA3 | Integrin subunit alpha 3 |
| ITGA6 | Integrin subunit alpha 6 |
| ITGB4 | Integrin subunit beta 4 |
| JEB | Junctional epidermolysis bullosa |
| KIT | KIT proto-oncogene |
| KLF4 | Krüppel-like factor 4 |
| KLHL24 | Kelch-like member 24 |
| KRT | Keratin |
| KS | Kindler syndrome |
| LAMA3 | Laminin subunit alpha 3 |
| LAMB3 | Laminin subunit beta 3 |
| LAMC3 | Laminin subunit gamma 3 |
| LEF1 | Lymphoid enhancer binding factor 1 |
| MAPK | Mitogen-activated protein kinases |
| MITF | Melanocyte-inducing transcription factor |
| MYC | MYC proto-oncogene, bHLH transcription factor |
| MYCN | MYCN proto-oncogene |
| MYH14 | Myosin heavy chain 14 |
| NBCCS | Nevoid basal cell carcinoma syndrome |
| NF-κB | Nuclear factor kappa B |
| NFE2L2 | Nuclear factor-erythroid 2-like bZIP transcription factor 2 |
| NICD | Notch intracellular domain |
| NOTCH1 | Neurogenic locus Notch homolog protein 1 |
| OVOL1 | Ovo like transcriptional repressor 1 |
| P | Postnatal day |
| PAX3 | Paired box 3 |
| PDGF | Platelet-derived growth factor |
| PDGFRA | Platelet-derived growth factor receptor alpha |
| PERP | P53 apoptosis effector related to PMP22 |
| PI3K | Phosphatidylinositol-3-kinase |
| PKC | Protein kinase C |
| PLEC | Plectin |
| PTCH1 | Patched 1 |
| PTCH2 | Patched 2 |
| Rb | Retinoblastoma |
| RBPJ | Recombination signal binding protein for immunoglobulin kappa J region |
| RUNX | RUNX family transcription factor |
| SCC | Squamous cell carcinoma |
| scRNA-seq | Small conditional ribonucleic acid sequencing |
| SDC1 | Syndecan 1 |
| SFN | Stratifin |
| SHH | Sonic Hedgehog |
| SMO | Smoothened, frizzled class receptor |
| SOX | SRY-box transcription factor |
| SUFU | Suppressor of fused homolog |
| TALEN | Transcription activator-like effector nuclease |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| TERT | Telomerase reverse transcriptase |
| TGF | Transforming growth factor |
| TNF | Tumour necrosis factor |
| TP53 | Tumour protein p53 |
| TP63 | Tumour protein p63 |
| YAP | Yes1-associated transcriptional regulator |
| ZNF750 | Zinc finger protein 750 |
References
- Wysocki, A.B. Skin Anatomy, Physiology, and Pathophysiology. Nurs. Clin. N. Am. 1999, 34, 777–797. [Google Scholar] [CrossRef]
- Romanovsky, A.A. Skin Temperature: Its Role in Thermoregulation. Acta Physiol. 2014, 210, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Merana, G.R.; Harris-Tryon, T.; Scharschmidt, T.C. Skin Immunity: Dissecting the Complex Biology of Our Body’s Outer Barrier. Mucosal Immunol. 2022, 15, 551–561. [Google Scholar] [CrossRef]
- Lumpkin, E.A.; Caterina, M.J. Mechanisms of Sensory Transduction in the Skin. Nature 2007, 445, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Gilaberte, Y.; Prieto-Torres, L.; Pastushenko, I.; Juarranz, Á. Chapter 1—Anatomy and Function of the Skin. In Nanoscience in Dermatology; Hamblin, M.R., Avci, P., Prow, T.W., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 1–14. ISBN 978-0-12-802926-8. [Google Scholar]
- Baroni, A.; Buommino, E.; De Gregorio, V.; Ruocco, E.; Ruocco, V.; Wolf, R. Structure and Function of the Epidermis Related to Barrier Properties. Clin. Dermatol. 2012, 30, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Singh, R. Basal Cells in the Epidermis and Epidermal Differentiation. Stem Cell Rev. Rep. 2022, 18, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Castellano-Pellicena, I.; Morrison, C.G.; Bell, M.; O’Connor, C.; Tobin, D.J. Melanin Distribution in Human Skin: Influence of Cytoskeletal, Polarity, and Centrosome-Related Machinery of Stratum Basale Keratinocytes. Int. J. Mol. Sci. 2021, 22, 3143. [Google Scholar] [CrossRef]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tymińska, A. Skin Melanocytes: Biology and Development. Adv. Dermatol. Allergol. Dermatol. Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef]
- Abraham, J.; Mathew, S. Merkel Cells: A Collective Review of Current Concepts. Int. J. Appl. Basic Med. Res. 2019, 9, 9–13. [Google Scholar] [CrossRef]
- Merad, M.; Ginhoux, F.; Collin, M. Origin, Homeostasis and Function of Langerhans Cells and Other Langerin-Expressing Dendritic Cells. Nat. Rev. Immunol. 2008, 8, 935–947. [Google Scholar] [CrossRef]
- Simpson, C.L.; Patel, D.M.; Green, K.J. Deconstructing the Skin: Cytoarchitectural Determinants of Epidermal Morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Hoober, J.K.; Eggink, L.L. The Discovery and Function of Filaggrin. Int. J. Mol. Sci. 2022, 23, 1455. [Google Scholar] [CrossRef]
- Kezic, S.; O’Regan, G.M.; Yau, N.; Sandilands, A.; Chen, H.; Campbell, L.E.; Kroboth, K.; Watson, R.; Rowland, M.; McLean, W.H.I.; et al. Levels of Filaggrin Degradation Products Are Influenced by Both Filaggrin Genotype and Atopic Dermatitis Severity. Allergy 2011, 66, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T. Epidermal Barrier Development via Corneoptosis: A Unique Form of Cell Death in Stratum Granulosum Cells. J. Dev. Biol. 2023, 11, 43. [Google Scholar] [CrossRef]
- Madison, K.C.; Swartzendruber, D.C.; Wertz, P.W.; Downing, D.T. Lamellar Granule Extrusion and Stratum Corneum Intercellular Lamellae in Murine Keratinocyte Cultures. J. Investig. Dermatol. 1988, 90, 110–116. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Soulika, A.M. The Dynamics of the Skin’s Immune System. Int. J. Mol. Sci. 2019, 20, 1811. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.R. The Stratum Corneum: Structure and Function in Health and Disease. Dermatol. Ther. 2004, 17 (Suppl. S1), 6–15. [Google Scholar] [CrossRef]
- Brown, T.M.; Krishnamurthy, K. Histology, Dermis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Benias, P.C.; Wells, R.G.; Sackey-Aboagye, B.; Klavan, H.; Reidy, J.; Buonocore, D.; Miranda, M.; Kornacki, S.; Wayne, M.; Carr-Locke, D.L.; et al. Structure and Distribution of an Unrecognized Interstitium in Human Tissues. Sci. Rep. 2018, 8, 4947. [Google Scholar] [CrossRef]
- Yousef, H.; Miao, J.H.; Alhajj, M.; Badri, T. Histology, Skin Appendages. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Mittal, B. Subcutaneous Adipose Tissue & Visceral Adipose Tissue. Indian J. Med. Res. 2019, 149, 571–573. [Google Scholar] [CrossRef]
- Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Miliaras, D.; Kesidou, E.; Boziki, M.; Petratos, S.; Grigoriadis, N.; Theotokis, P. Developmental Cues and Molecular Drivers in Myelinogenesis: Revisiting Early Life to Re-Evaluate the Integrity of CNS Myelin. Curr. Issues Mol. Biol. 2022, 44, 3208–3237. [Google Scholar] [CrossRef]
- Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Tremblay, M.-È.; Petratos, S.; Zoupi, L.; Boziki, M.; Kesidou, E.; Simeonidou, C.; Theotokis, P. Origin and Emergence of Microglia in the CNS—An Interesting (Hi)Story of an Eccentric Cell. Curr. Issues Mol. Biol. 2023, 45, 2609–2628. [Google Scholar] [CrossRef]
- Dermitzakis, I.; Theotokis, P.; Evangelidis, P.; Delilampou, E.; Evangelidis, N.; Chatzisavvidou, A.; Avramidou, E.; Manthou, M.E. CNS Border-Associated Macrophages: Ontogeny and Potential Implication in Disease. Curr. Issues Mol. Biol. 2023, 45, 4285–4300. [Google Scholar] [CrossRef]
- Fuchs, E. Epithelial Skin Biology: Three Decades of Developmental Biology, a Hundred Questions Answered and a Thousand New Ones to Address. Curr. Top. Dev. Biol. 2016, 116, 357–374. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Duan, E. Epidermal Development in Mammals: Key Regulators, Signals from beneath, and Stem Cells. Int. J. Mol. Sci. 2013, 14, 10869–10895. [Google Scholar] [CrossRef]
- Inoue, T.; Kuwano, T.; Uehara, Y.; Yano, M.; Oya, N.; Takada, N.; Tanaka, S.; Ueda, Y.; Hachiya, A.; Takahashi, Y.; et al. Non-Invasive Human Skin Transcriptome Analysis Using MRNA in Skin Surface Lipids. Commun. Biol. 2022, 5, 215. [Google Scholar] [CrossRef]
- Lopez-Pajares, V.; Yan, K.; Zarnegar, B.J.; Jameson, K.L.; Khavari, P.A. Genetic Pathways in Disorders of Epidermal Differentiation. Trends Genet. 2013, 29, 31–40. [Google Scholar] [CrossRef]
- Moss, C. Genetic Skin Disorders. Semin. Neonatol. 2000, 5, 311–320. [Google Scholar] [CrossRef]
- Mariath, L.M.; Santin, J.T.; Schuler-Faccini, L.; Kiszewski, A.E. Inherited Epidermolysis Bullosa: Update on the Clinical and Genetic Aspects. An. Bras. Dermatol. 2020, 95, 551–569. [Google Scholar] [CrossRef]
- Naik, P.P.; Desai, M.B. Basal Cell Carcinoma: A Narrative Review on Contemporary Diagnosis and Management. Oncol. Ther. 2022, 10, 317–335. [Google Scholar] [CrossRef]
- Fania, L.; Didona, D.; Di Pietro, F.R.; Verkhovskaia, S.; Morese, R.; Paolino, G.; Donati, M.; Ricci, F.; Coco, V.; Ricci, F.; et al. Cutaneous Squamous Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2021, 9, 171. [Google Scholar] [CrossRef]
- Fuchs, E. Scratching the Surface of Skin Development. Nature 2007, 445, 834–842. [Google Scholar] [CrossRef]
- Keller, R.; Davidson, L.A.; Shook, D.R. How We Are Shaped: The Biomechanics of Gastrulation. Differ. Orig. Artic. 2003, 71, 171–205. [Google Scholar] [CrossRef]
- Stern, C.D. Neural Induction: Old Problem, New Findings, yet More Questions. Development 2005, 132, 2007–2021. [Google Scholar] [CrossRef]
- Forni, M.F.; Trombetta-Lima, M.; Sogayar, M.C. Stem Cells in Embryonic Skin Development. Biol. Res. 2012, 45, 215–222. [Google Scholar] [CrossRef]
- Bleuming, S.A.; He, X.C.; Kodach, L.L.; Hardwick, J.C.; Koopman, F.A.; Ten Kate, F.J.; van Deventer, S.J.H.; Hommes, D.W.; Peppelenbosch, M.P.; Offerhaus, G.J.; et al. Bone Morphogenetic Protein Signaling Suppresses Tumorigenesis at Gastric Epithelial Transition Zones in Mice. Cancer Res. 2007, 67, 8149–8155. [Google Scholar] [CrossRef]
- Bottcher, R.T.; Niehrs, C. Fibroblast Growth Factor Signaling during Early Vertebrate Development. Endocr. Rev. 2005, 26, 63–77. [Google Scholar] [CrossRef]
- Haegel, H.; Larue, L.; Ohsugi, M.; Fedorov, L.; Herrenknecht, K.; Kemler, R. Lack of Beta-Catenin Affects Mouse Development at Gastrulation. Development 1995, 121, 3529–3537. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Graff, J.M. Embryonic Patterning: To BMP or Not to BMP, That Is the Question. Cell 1997, 89, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Troy, T.-C.; Turksen, K. Commitment of Embryonic Stem Cells to an Epidermal Cell Fate and Differentiation in Vitro. Dev. Dyn. 2005, 232, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.; Tainsky, M.; Fuchs, E. Programming Gene Expression in Developing Epidermis. Development 1994, 120, 2369–2383. [Google Scholar] [CrossRef]
- Sun, T.-T.; Eichner, R.; Nelson, W.G.; Scheffer Tseng, C.G.; Weiss, R.A.; Jarvinen, M.; Woodcock-Mitchell, J. Keratin Classes: Molecular Markers for Different Types of Epithelial Differentiation. J. Investig. Dermatol. 1983, 81, S109–S115. [Google Scholar] [CrossRef]
- Wu, Y.J.; Parker, L.M.; Binder, N.E.; Beckett, M.A.; Sinard, J.H.; Griffiths, C.T.; Rheinwald, J.G. The Mesothelial Keratins: A New Family of Cytoskeletal Proteins Identified in Cultured Mesothelial Cells and Nonkeratinizing Epithelia. Cell 1982, 31, 693–703. [Google Scholar] [CrossRef]
- Fuchs, E.; Green, H. Changes in Keratin Gene Expression during Terminal Differentiation of the Keratinocyte. Cell 1980, 19, 1033–1042. [Google Scholar] [CrossRef]
- Crum, C.P.; McKeon, F.D. P63 in Epithelial Survival, Germ Cell Surveillance, and Neoplasia. Annu. Rev. Pathol. 2010, 5, 349–371. [Google Scholar] [CrossRef]
- Koster, M.I.; Dai, D.; Roop, D.R. Conflicting Roles for P63 in Skin Development and Carcinogenesis. Cell Cycle Georget. Tex 2007, 6, 269–273. [Google Scholar] [CrossRef]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. P63 Is Essential for Regenerative Proliferation in Limb, Craniofacial and Epithelial Development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Koster, M.I.; Kim, S.; Huang, J.; Williams, T.; Roop, D.R. TAp63α Induces AP-2γ as an Early Event in Epidermal Morphogenesis. Dev. Biol. 2006, 289, 253–261. [Google Scholar] [CrossRef]
- Romano, R.-A.; Birkaya, B.; Sinha, S. A Functional Enhancer of Keratin14 Is a Direct Transcriptional Target of ΔNp63. J. Investig. Dermatol. 2007, 127, 1175–1186. [Google Scholar] [CrossRef]
- Ihrie, R.A.; Marques, M.R.; Nguyen, B.T.; Horner, J.S.; Papazoglu, C.; Bronson, R.T.; Mills, A.A.; Attardi, L.D. Perp Is a P63-Regulated Gene Essential for Epithelial Integrity. Cell 2005, 120, 843–856. [Google Scholar] [CrossRef]
- McGowan, K.M.; Coulombe, P.A. Onset of Keratin 17 Expression Coincides with the Definition of Major Epithelial Lineages during Skin Development. J. Cell Biol. 1998, 143, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Richardson, R.J.; Hammond, N.L.; Coulombe, P.A.; Saloranta, C.; Nousiainen, H.O.; Salonen, R.; Berry, A.; Hanley, N.; Headon, D.; Karikoski, R.; et al. Periderm Prevents Pathological Epithelial Adhesions during Embryogenesis. J. Clin. Investig. 2014, 124, 3891–3900. [Google Scholar] [CrossRef] [PubMed]
- M’boneko, V.; Merker, H.-J. Development and Morphology of the Periderm of Mouse Embryos (Days 9–12 of Gestation). Cells Tissues Organs 1988, 133, 325–336. [Google Scholar] [CrossRef]
- Hammond, N.L.; Dixon, J.; Dixon, M.J. Periderm: Life-Cycle and Function during Orofacial and Epidermal Development. Semin. Cell Dev. Biol. 2019, 91, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Polakowska, R.R.; Piacentini, M.; Bartlett, R.; Goldsmith, L.A.; Haake, A.R. Apoptosis in Human Skin Development: Morphogenesis, Periderm, and Stem Cells. Dev. Dyn. 1994, 199, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.; Annusver, K.; Czarnewski, P.; Dalessandri, T.; Kalk, C.; Levra Levron, C.; Campamà Sanz, N.; Kastriti, M.E.; Mikkola, M.L.; Rendl, M.; et al. Molecular and Spatial Landmarks of Early Mouse Skin Development. Dev. Cell 2023, 58, 2140–2162.e5. [Google Scholar] [CrossRef]
- Watt, F.M.; Green, H. Stratification and Terminal Differentiation of Cultured Epidermal Cells. Nature 1982, 295, 434–436. [Google Scholar] [CrossRef] [PubMed]
- Benitah, S.A.; Frye, M. Stem Cells in Ectodermal Development. J. Mol. Med. 2012, 90, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.H.; Watt, F.M. Separation of Human Epidermal Stem Cells from Transit Amplifying Cells on the Basis of Differences in Integrin Function and Expression. Cell 1993, 73, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.R.; Hyman, A.A. Asymmetric Cell Division in C. Elegans: Cortical Polarity and Spindle Positioning. Annu. Rev. Cell Dev. Biol. 2004, 20, 427–453. [Google Scholar]
- Lechler, T.; Fuchs, E. Asymmetric Cell Divisions Promote Stratification and Differentiation of Mammalian Skin. Nature 2005, 437, 275–280. [Google Scholar] [CrossRef]
- Roegiers, F.; Jan, Y.N. Asymmetric Cell Division. Curr. Opin. Cell Biol. 2004, 16, 195–205. [Google Scholar] [CrossRef]
- Koster, M.I.; Dai, D.; Marinari, B.; Sano, Y.; Costanzo, A.; Karin, M.; Roop, D.R. P63 Induces Key Target Genes Required for Epidermal Morphogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 3255–3260. [Google Scholar] [CrossRef] [PubMed]
- Candi, E.; Schmidt, R.; Melino, G. The Cornified Envelope: A Model of Cell Death in the Skin. Nat. Rev. Mol. Cell Biol. 2005, 6, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Lin, M.-H.; Tian, X.; Cheng, H.-T.; Gridley, T.; Shen, J.; Kopan, R. γ-Secretase Functions through Notch Signaling to Maintain Skin Appendages but Is Not Required for Their Patterning or Initial Morphogenesis. Dev. Cell 2004, 7, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Lowry, W.E.; Pasolli, H.A.; Fuchs, E. Canonical Notch Signaling Functions as a Commitment Switch in the Epidermal Lineage. Genes Dev. 2006, 20, 3022–3035. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-J.; Liu, Y.; Dai, Z.-M.; Zhang, X.; Yang, X.; Li, Y.; Qiu, M.; Fu, J.; Hsu, W.; Chen, Y.; et al. BMP-FGF Signaling Axis Mediates Wnt-Induced Epidermal Stratification in Developing Mammalian Skin. PLoS Genet. 2014, 10, e1004687. [Google Scholar] [CrossRef]
- Nair, M.; Teng, A.; Bilanchone, V.; Agrawal, A.; Li, B.; Dai, X. Ovol1 Regulates the Growth Arrest of Embryonic Epidermal Progenitor Cells and Represses C-Myc Transcription. J. Cell Biol. 2006, 173, 253–264. [Google Scholar] [CrossRef]
- Richardson, R.J.; Dixon, J.; Malhotra, S.; Hardman, M.J.; Knowles, L.; Boot-Handford, R.P.; Shore, P.; Whitmarsh, A.; Dixon, M.J. Irf6 Is a Key Determinant of the Keratinocyte Proliferation-Differentiation Switch. Nat. Genet. 2006, 38, 1329–1334. [Google Scholar] [CrossRef]
- Li, Q.; Lu, Q.; Estepa, G.; Verma, I.M. Identification of 14-3-3σ Mutation Causing Cutaneous Abnormality in Repeated-Epilation Mutant Mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 15977–15982. [Google Scholar] [CrossRef]
- Lin, Z.; Jin, S.; Chen, J.; Li, Z.; Lin, Z.; Tang, L.; Nie, Q.; Andersen, B. Murine Interfollicular Epidermal Differentiation Is Gradualistic with GRHL3 Controlling Progression from Stem to Transition Cell States. Nat. Commun. 2020, 11, 5434. [Google Scholar] [CrossRef]
- Miroshnikova, Y.A.; Le, H.Q.; Schneider, D.; Thalheim, T.; Rübsam, M.; Bremicker, N.; Polleux, J.; Kamprad, N.; Tarantola, M.; Wang, I.; et al. Adhesion Forces and Cortical Tension Couple Cell Proliferation and Differentiation to Drive Epidermal Stratification. Nat. Cell Biol. 2018, 20, 69–80. [Google Scholar] [CrossRef]
- Sen, G.L.; Boxer, L.D.; Webster, D.E.; Bussat, R.T.; Qu, K.; Zarnegar, B.J.; Johnston, D.; Siprashvili, Z.; Khavari, P.A. ZNF750 Is a P63 Target Gene That Induces KLF4 to Drive Terminal Epidermal Differentiation. Dev. Cell 2012, 22, 669–677. [Google Scholar] [CrossRef]
- Hennings, H.; Kruszewski, F.H.; Yuspa, S.H.; Tucker, R.W. Intracellular Calcium Alterations in Response to Increased External Calcium in Normal and Neoplastic Keratinocytes. Carcinogenesis 1989, 10, 777–780. [Google Scholar] [CrossRef]
- Matoltsy, A.G.; Balsamo, C.A. A Study of the Components of the Cornified Epithelium of Human Skin. J. Cell Biol. 1955, 1, 339–360. [Google Scholar] [CrossRef]
- McLafferty, E.; Hendry, C.; Alistair, F. The Integumentary System: Anatomy, Physiology and Function of Skin. Nurs. Stand. 2012, 27, 35–42. [Google Scholar] [CrossRef]
- Hardy, M.H. The Secret Life of the Hair Follicle. Trends Genet. 1992, 8, 55–61. [Google Scholar] [CrossRef]
- Park, B.-Y.; Saint-Jeannet, J.-P. Induction and Segregation of the Vertebrate Cranial Placodes; Developmental Biology; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Paus, R.; Müller-Röver, S.; Van Der Veen, C.; Maurer, M.; Eichmüller, S.; Ling, G.; Hofmann, U.; Foitzik, K.; Mecklenburg, L.; Handjiski, B. A Comprehensive Guide for the Recognition and Classification of Distinct Stages of Hair Follicle Morphogenesis. J. Investig. Dermatol. 1999, 113, 523–532. [Google Scholar] [CrossRef]
- Olivera-Martinez, I.; Thélu, J.; Dhouailly, D. Molecular Mechanisms Controlling Dorsal Dermis Generation from the Somitic Dermomyotome. Int. J. Dev. Biol. 2004, 48, 93–101. [Google Scholar] [CrossRef]
- Wang, X.; Tredget, E.E.; Wu, Y. Dynamic Signals for Hair Follicle Development and Regeneration. Stem Cells Dev. 2012, 21, 7–18. [Google Scholar] [CrossRef]
- Blanpain, C.; Fuchs, E. Epidermal Stem Cells of the Skin. Annu. Rev. Cell Dev. Biol. 2006, 22, 339–373. [Google Scholar] [CrossRef] [PubMed]
- Sennett, R.; Rendl, M. Mesenchymal-Epithelial Interactions during Hair Follicle Morphogenesis and Cycling. Semin. Cell Dev. Biol. 2012, 23, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tomann, P.; Andl, T.; Gallant, N.M.; Huelsken, J.; Jerchow, B.; Birchmeier, W.; Paus, R.; Piccolo, S.; Mikkola, M.L.; et al. Reciprocal Requirements for EDA/EDAR/NF-KappaB and Wnt/Beta-Catenin Signaling Pathways in Hair Follicle Induction. Dev. Cell 2009, 17, 49–61. [Google Scholar] [CrossRef]
- Chen, D.; Jarrell, A.; Guo, C.; Lang, R.; Atit, R. Dermal β-Catenin Activity in Response to Epidermal Wnt Ligands Is Required for Fibroblast Proliferation and Hair Follicle Initiation. Development 2012, 139, 1522–1533. [Google Scholar] [CrossRef]
- Huelsken, J.; Vogel, R.; Erdmann, B.; Cotsarelis, G.; Birchmeier, W. Beta-Catenin Controls Hair Follicle Morphogenesis and Stem Cell Differentiation in the Skin. Cell 2001, 105, 533–545. [Google Scholar] [CrossRef]
- Andl, T.; Reddy, S.T.; Gaddapara, T.; Millar, S.E. WNT Signals Are Required for the Initiation of Hair Follicle Development. Dev. Cell 2002, 2, 643–653. [Google Scholar] [CrossRef]
- Pincha, N.; Marangoni, P.; Haque, A.; Klein, O.D. Parallels in Signaling between Development and Regeneration in Ectodermal Organs. Curr. Top. Dev. Biol. 2022, 149, 373–419. [Google Scholar] [CrossRef]
- Rishikaysh, P.; Dev, K.; Diaz, D.; Qureshi, W.M.S.; Filip, S.; Mokry, J. Signaling Involved in Hair Follicle Morphogenesis and Development. Int. J. Mol. Sci. 2014, 15, 1647–1670. [Google Scholar] [CrossRef]
- Noramly, S.; Morgan, B.A. BMPs Mediate Lateral Inhibition at Successive Stages in Feather Tract Development. Development 1998, 125, 3775–3787. [Google Scholar] [CrossRef]
- Pummila, M.; Fliniaux, I.; Jaatinen, R.; James, M.J.; Laurikkala, J.; Schneider, P.; Thesleff, I.; Mikkola, M.L. Ectodysplasin Has a Dual Role in Ectodermal Organogenesis: Inhibition of Bmp Activity and Induction of Shh Expression. Development 2007, 134, 117–125. [Google Scholar] [CrossRef]
- Bitgood, M.J.; McMahon, A.P. Hedgehog and Bmp Genes Are Coexpressed at Many Diverse Sites of Cell-Cell Interaction in the Mouse Embryo. Dev. Biol. 1995, 172, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A.; Botchkareva, N.V.; Roth, W.; Nakamura, M.; Chen, L.H.; Herzog, W.; Lindner, G.; McMahon, J.A.; Peters, C.; Lauster, R.; et al. Noggin Is a Mesenchymally Derived Stimulator of Hair-Follicle Induction. Nat. Cell Biol. 1999, 1, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Feijen, A.; Goumans, M.J.; van den Eijnden-van Raaij, A.J. Expression of Activin Subunits, Activin Receptors and Follistatin in Postimplantation Mouse Embryos Suggests Specific Developmental Functions for Different Activins. Development 1994, 120, 3621–3637. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, A.; Saito, F.; Ohuchi, H.; Noji, S. Differential Expression of Two BMP Antagonists, Gremlin and Follistatin, during Development of the Chick Feather Bud. Mech. Dev. 2001, 100, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Jamora, C.; DasGupta, R.; Kocieniewski, P.; Fuchs, E. Links between Signal Transduction, Transcription and Adhesion in Epithelial Bud Development. Nature 2003, 422, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Matzuk, M.M.; Gerstmayer, B.; Bosio, A.; Lauster, R.; Miyachi, Y.; Werner, S.; Paus, R. Control of Pelage Hair Follicle Development and Cycling by Complex Interactions between Follistatin and Activin. FASEB J. 2003, 17, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Mok, K.-W.; Saxena, N.; Heitman, N.; Grisanti, L.; Srivastava, D.; Muraro, M.J.; Jacob, T.; Sennett, R.; Wang, Z.; Su, Y.; et al. Dermal Condensate Niche Fate Specification Occurs Prior to Formation and Is Placode Progenitor Dependent. Dev. Cell 2019, 48, 32–48.e5. [Google Scholar] [CrossRef]
- Qu, R.; Gupta, K.; Dong, D.; Jiang, Y.; Landa, B.; Saez, C.; Strickland, G.; Levinsohn, J.; Weng, P.-L.; Taketo, M.M.; et al. Decomposing a Deterministic Path to Mesenchymal Niche Formation by Two Intersecting Morphogen Gradients. Dev. Cell 2022, 57, 1053–1067.e5. [Google Scholar] [CrossRef]
- St-Jacques, B.; Dassule, H.R.; Karavanova, I.; Botchkarev, V.A.; Li, J.; Danielian, P.S.; McMahon, J.A.; Lewis, P.M.; Paus, R.; McMahon, A.P. Sonic Hedgehog Signaling Is Essential for Hair Development. Curr. Biol. 1998, 8, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Huh, S.-H.; Närhi, K.; Lindfors, P.H.; Häärä, O.; Yang, L.; Ornitz, D.M.; Mikkola, M.L. Fgf20 Governs Formation of Primary and Secondary Dermal Condensations in Developing Hair Follicles. Genes Dev. 2013, 27, 450–458. [Google Scholar] [CrossRef]
- Karlsson, L.; Bondjers, C.; Betsholtz, C. Roles for PDGF-A and Sonic Hedgehog in Development of Mesenchymal Components of the Hair Follicle. Development 1999, 126, 2611–2621. [Google Scholar] [CrossRef]
- Ouspenskaia, T.; Matos, I.; Mertz, A.F.; Fiore, V.F.; Fuchs, E. WNT-SHH Antagonism Specifies and Expands Stem Cells Prior to Niche Formation. Cell 2016, 164, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Woo, W.-M.; Zhen, H.H.; Oro, A.E. Shh Maintains Dermal Papilla Identity and Hair Morphogenesis via a Noggin-Shh Regulatory Loop. Genes Dev. 2012, 26, 1235–1246. [Google Scholar] [CrossRef]
- Kamp, H.; Geilen, C.C.; Sommer, C.; Blume-Peytavi, U. Regulation of PDGF and PDGF Receptor in Cultured Dermal Papilla Cells and Follicular Keratinocytes of the Human Hair Follicle. Exp. Dermatol. 2003, 12, 662–672. [Google Scholar] [CrossRef]
- Jamora, C.; Lee, P.; Kocieniewski, P.; Azhar, M.; Hosokawa, R.; Chai, Y.; Fuchs, E. A Signaling Pathway Involving TGF-Beta2 and Snail in Hair Follicle Morphogenesis. PLoS Biol. 2005, 3, e11. [Google Scholar] [CrossRef]
- Foitzik, K.; Paus, R.; Doetschman, T.; Dotto, G.P. The TGF-Beta2 Isoform Is Both a Required and Sufficient Inducer of Murine Hair Follicle Morphogenesis. Dev. Biol. 1999, 212, 278–289. [Google Scholar] [CrossRef]
- Kishimoto, J.; Burgeson, R.E.; Morgan, B.A. Wnt Signaling Maintains the Hair-Inducing Activity of the Dermal Papilla. Genes Dev. 2000, 14, 1181–1185. [Google Scholar] [CrossRef]
- Enshell-Seijffers, D.; Lindon, C.; Kashiwagi, M.; Morgan, B.A. Beta-Catenin Activity in the Dermal Papilla Regulates Morphogenesis and Regeneration of Hair. Dev. Cell 2010, 18, 633–642. [Google Scholar] [CrossRef]
- Cui, C.-Y.; Schlessinger, D. Eccrine Sweat Gland Development and Sweat Secretion. Exp. Dermatol. 2015, 24, 644–650. [Google Scholar] [CrossRef]
- Fu, X.; Li, J.; Sun, X.; Sun, T.; Sheng, Z. Epidermal Stem Cells Are the Source of Sweat Glands in Human Fetal Skin: Evidence of Synergetic Development of Stem Cells, Sweat Glands, Growth Factors, and Matrix Metalloproteinases. Wound Repair Regen. 2005, 13, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.P.; Polak, L.; Rocha, A.S.; Pasolli, H.A.; Chen, S.-C.; Sharma, N.; Blanpain, C.; Fuchs, E. Identification of Stem Cell Populations in Sweat Glands and Ducts Reveals Roles in Homeostasis and Wound Repair. Cell 2012, 150, 136–150. [Google Scholar] [CrossRef]
- Sato, K.; Leidal, R.; Sato, F. Morphology and Development of an Apoeccrine Sweat Gland in Human Axillae. Am. J. Physiol. 1987, 252, R166–R180. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Fuchs, E. Sweat Gland Progenitors in Development, Homeostasis, and Wound Repair. Cold Spring Harb. Perspect. Med. 2014, 4, a015222. [Google Scholar] [CrossRef]
- Sato, K.; Kang, W.H.; Saga, K.; Sato, K.T. Biology of Sweat Glands and Their Disorders. I. Normal Sweat Gland Function. J. Am. Acad. Dermatol. 1989, 20, 537–563. [Google Scholar] [CrossRef]
- Moll, I.; Moll, R. Changes of Expression of Intermediate Filament Proteins during Ontogenesis of Eccrine Sweat Glands. J. Investig. Dermatol. 1992, 98, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Dingwall, H.L.; Tomizawa, R.R.; Aharoni, A.; Hu, P.; Qiu, Q.; Kokalari, B.; Martinez, S.M.; Donahue, J.C.; Aldea, D.; Mendoza, M.; et al. Sweat Gland Development Requires an Eccrine Dermal Niche and Couples Two Epidermal Programs. Dev. Cell 2024, 59, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, C.; Hadj-Rabia, S.; Jambou, M.; Mansour, S.; Guigue, P.; Masmoudi, S.; Bal, E.; Chassaing, N.; Vincent, M.-C.; Viot, G.; et al. Only Four Genes (EDA1, EDAR, EDARADD, and WNT10A) Account for 90% of Hypohidrotic/Anhidrotic Ectodermal Dysplasia Cases. Hum. Mutat. 2011, 32, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.-Y.; Yin, M.; Sima, J.; Childress, V.; Michel, M.; Piao, Y.; Schlessinger, D. Involvement of Wnt, Eda and Shh at Defined Stages of Sweat Gland Development. Development 2014, 141, 3752–3760. [Google Scholar] [CrossRef]
- Chassaing, N.; Bourthoumieu, S.; Cossee, M.; Calvas, P.; Vincent, M.-C. Mutations in EDAR Account for One-Quarter of Non-ED1-Related Hypohidrotic Ectodermal Dysplasia. Hum. Mutat. 2006, 27, 255–259. [Google Scholar] [CrossRef]
- Cui, C.-Y.; Schlessinger, D. EDA Signaling and Skin Appendage Development. Cell Cycle Georget. Tex 2006, 5, 2477–2483. [Google Scholar] [CrossRef] [PubMed]
- Laurikkala, J.; Pispa, J.; Jung, H.-S.; Nieminen, P.; Mikkola, M.; Wang, X.; Saarialho-Kere, U.; Galceran, J.; Grosschedl, R.; Thesleff, I. Regulation of Hair Follicle Development by the TNF Signal Ectodysplasin and Its Receptor Edar. Development 2002, 129, 2541–2553. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, M.; Cui, C.-Y.; Piao, Y.; Ko, M.S.H.; Schlessinger, D. Requirement for Shh and Fox Family Genes at Different Stages in Sweat Gland Development. Hum. Mol. Genet. 2009, 18, 1769–1778. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Fuchs, E. Building and Maintaining the Skin. Cold Spring Harb. Perspect. Biol. 2022, 14, a040840. [Google Scholar] [CrossRef]
- Ruco, L.P.; Uccini, S.; Baroni, C.D. The Langerhans’ Cells. Allergy 1989, 44 (Suppl. S9), 27–30. [Google Scholar] [CrossRef]
- Vega-Lopez, G.A.; Cerrizuela, S.; Aybar, M.J. Trunk Neural Crest Cells: Formation, Migration and Beyond. Int. J. Dev. Biol. 2017, 61, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Sommer, L. Generation of Melanocytes from Neural Crest Cells. Pigment Cell Melanoma Res. 2011, 24, 411–421. [Google Scholar] [CrossRef]
- Vandamme, N.; Berx, G. From Neural Crest Cells to Melanocytes: Cellular Plasticity during Development and Beyond. Cell. Mol. Life Sci. 2019, 76, 1919–1934. [Google Scholar] [CrossRef] [PubMed]
- Chatzi, D.; Kyriakoudi, S.A.; Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Theotokis, P. Clinical and Genetic Correlation in Neurocristopathies: Bridging a Precision Medicine Gap. J. Clin. Med. 2024, 13, 2223. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.Z.; Evrard, M.; Ng, L.G. Lights, Camera, and Action: Vertebrate Skin Sets the Stage for Immune Cell Interaction with Arthropod-Vectored Pathogens. Front. Immunol. 2013, 4, 286. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.H. Ontogeny and Function of Murine Epidermal Langerhans Cells. Nat. Immunol. 2017, 18, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto-Hill, S.; Friesen, L.; Park, S.; Im, S.; Kaplan, M.H.; Kim, C.H. RARα Supports the Development of Langerhans Cells and Langerin-Expressing Conventional Dendritic Cells. Nat. Commun. 2018, 9, 3896. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Tacke, F.; Angeli, V.; Bogunovic, M.; Loubeau, M.; Dai, X.-M.; Stanley, E.R.; Randolph, G.J.; Merad, M. Langerhans Cells Arise from Monocytes in Vivo. Nat. Immunol. 2006, 7, 265–273. [Google Scholar] [CrossRef]
- Hoeffel, G.; Wang, Y.; Greter, M.; See, P.; Teo, P.; Malleret, B.; Leboeuf, M.; Low, D.; Oller, G.; Almeida, F.; et al. Adult Langerhans Cells Derive Predominantly from Embryonic Fetal Liver Monocytes with a Minor Contribution of Yolk Sac-Derived Macrophages. J. Exp. Med. 2012, 209, 1167–1181. [Google Scholar] [CrossRef]
- Foster, C.A.; Holbrook, K.A.; Farr, A.G. Ontogeny of Langerhans Cells in Human Embryonic and Fetal Skin: Expression of HLA-DR and OKT-6 Determinants. J. Investig. Dermatol. 1986, 86, 240–243. [Google Scholar] [CrossRef]
- Collin, M.; Milne, P. Langerhans Cell Origin and Regulation. Curr. Opin. Hematol. 2016, 23, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Fainaru, O.; Woolf, E.; Lotem, J.; Yarmus, M.; Brenner, O.; Goldenberg, D.; Negreanu, V.; Bernstein, Y.; Levanon, D.; Jung, S.; et al. Runx3 Regulates Mouse TGF-Beta-Mediated Dendritic Cell Function and Its Absence Results in Airway Inflammation. EMBO J. 2004, 23, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Hacker, C.; Kirsch, R.D.; Ju, X.-S.; Hieronymus, T.; Gust, T.C.; Kuhl, C.; Jorgas, T.; Kurz, S.M.; Rose-John, S.; Yokota, Y.; et al. Transcriptional Profiling Identifies Id2 Function in Dendritic Cell Development. Nat. Immunol. 2003, 4, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Greter, M.; Lelios, I.; Pelczar, P.; Hoeffel, G.; Price, J.; Leboeuf, M.; Kündig, T.M.; Frei, K.; Ginhoux, F.; Merad, M.; et al. Stroma-Derived Interleukin-34 Controls the Development and Maintenance of Langerhans Cells and the Maintenance of Microglia. Immunity 2012, 37, 1050–1060. [Google Scholar] [CrossRef]
- Romani, N.; Holzmann, S.; Tripp, C.H.; Koch, F.; Stoitzner, P. Langerhans Cells—Dendritic Cells of the Epidermis. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2003, 111, 725–740. [Google Scholar] [CrossRef]
- Moll, R.; Moll, I.; Franke, W.W. Identification of Merkel Cells in Human Skin by Specific Cytokeratin Antibodies: Changes of Cell Density and Distribution in Fetal and Adult Plantar Epidermis. Differ. Res. Biol. Divers. 1984, 28, 136–154. [Google Scholar] [CrossRef]
- Perdigoto, C.N.; Dauber, K.L.; Bar, C.; Tsai, P.-C.; Valdes, V.J.; Cohen, I.; Santoriello, F.J.; Zhao, D.; Zheng, D.; Hsu, Y.-C.; et al. Polycomb-Mediated Repression and Sonic Hedgehog Signaling Interact to Regulate Merkel Cell Specification during Skin Development. PLoS Genet. 2016, 12, e1006151. [Google Scholar] [CrossRef] [PubMed]
- Kiran, N.K.; Tilak Raj, T.N.; Mukunda, K.S.; Rajashekar Reddy, V. Nevoid Basal Cell Carcinoma Syndrome (Gorlin-Goltz Syndrome). Contemp. Clin. Dent. 2012, 3, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L. Nevoid Basal Cell Carcinoma Syndrome (Gorlin Syndrome). Orphanet J. Rare Dis. 2008, 3, 32. [Google Scholar] [CrossRef]
- Şereflican, B.; Tuman, B.; Şereflican, M.; Halıcıoğlu, S.; Özyalvaçlı, G.; Bayrak, S. Gorlin-Goltz Syndrome. Turk Pediatri Ars. 2017, 52, 173–177. [Google Scholar] [CrossRef]
- Göppner, D.; Leverkus, M. Basal Cell Carcinoma: From the Molecular Understanding of the Pathogenesis to Targeted Therapy of Progressive Disease. J. Ski. Cancer 2011, 2011, 650258. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Miyashita, T. Gorlin Syndrome (Nevoid Basal Cell Carcinoma Syndrome): Update and Literature Review. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 2014, 56, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Bare, J.W.; Lebo, R.V.; Epstein, E.H. Loss of Heterozygosity at Chromosome 1q22 in Basal Cell Carcinomas and Exclusion of the Basal Cell Nevus Syndrome Gene from This Site. Cancer Res. 1992, 52, 1494–1498. [Google Scholar]
- Sidransky, D. Is Human Patched the Gatekeeper of Common Skin Cancers? Nat. Genet. 1996, 14, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; Dutta, A.; Sarkar, S.; Nag, S.S.; Biswas, S.K.; Mandal, P. Focal Dermal Hypoplasia (Goltz Syndrome): A Cross-Sectional Study from Eastern India. Indian J. Dermatol. 2017, 62, 498–504. [Google Scholar] [CrossRef]
- Ellenbogen, E. Embryology of the Skin. In Skin and the Heart; Salavastru, C., Murrell, D.F., Otton, J., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 3–9. ISBN 978-3-030-54779-0. [Google Scholar]
- van der Geer, S.; Ostertag, J.U.; Krekels, G.A.M. Treatment of Basal Cell Carcinomas in Patients with Nevoid Basal Cell Carcinoma Syndrome. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Prodinger, C.; Reichelt, J.; Bauer, J.W.; Laimer, M. Epidermolysis Bullosa: Advances in Research and Treatment. Exp. Dermatol. 2019, 28, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, P.A.; Kerns, M.L.; Fuchs, E. Epidermolysis Bullosa Simplex: A Paradigm for Disorders of Tissue Fragility. J. Clin. Investig. 2009, 119, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Morasso, M.I.; Tomic-Canic, M. Epidermal Stem Cells: The Cradle of Epidermal Determination, Differentiation and Wound Healing. Biol. Cell 2005, 97, 173–183. [Google Scholar] [CrossRef]
- Kim, S.; Coulombe, P.A. Intermediate Filament Scaffolds Fulfill Mechanical, Organizational, and Signaling Functions in the Cytoplasm. Genes Dev. 2007, 21, 1581–1597. [Google Scholar] [CrossRef]
- Morley, S.M.; Dundas, S.R.; James, J.L.; Gupta, T.; Brown, R.A.; Sexton, C.J.; Navsaria, H.A.; Leigh, I.M.; Lane, E.B. Temperature Sensitivity of the Keratin Cytoskeleton and Delayed Spreading of Keratinocyte Lines Derived from EBS Patients. J. Cell Sci. 1995, 108 Pt 11, 3463–3471. [Google Scholar] [CrossRef]
- Yoshioka, N. Roles of Dystonin Isoforms in the Maintenance of Neural, Muscle, and Cutaneous Tissues. Anat. Sci. Int. 2024, 99, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.; Türk, M.; Harter, P.N.; Mittelbronn, M.; Kornblum, C.; Norwood, F.; Jungbluth, H.; Thiel, C.T.; Schlötzer-Schrehardt, U.; Schröder, R. Downstream Effects of Plectin Mutations in Epidermolysis Bullosa Simplex with Muscular Dystrophy. Acta Neuropathol. Commun. 2016, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Monteleon, C.L.; Lee, I.Y.; Ridky, T.W. Exophilin-5 Supports Lysosome-Mediated Trafficking Required for Epidermal Differentiation. J. Investig. Dermatol. 2019, 139, 2219–2222.e6. [Google Scholar] [CrossRef]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus Reclassification of Inherited Epidermolysis Bullosa and Other Disorders with Skin Fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef]
- Lin, Z.; Li, S.; Feng, C.; Yang, S.; Wang, H.; Ma, D.; Zhang, J.; Gou, M.; Bu, D.; Zhang, T.; et al. Stabilizing Mutations of KLHL24 Ubiquitin Ligase Cause Loss of Keratin 14 and Human Skin Fragility. Nat. Genet. 2016, 48, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.W.; Liu, L.; Hsu, C.-K.; Aristodemou, S.; Ozoemena, L.; Ogboli, M.; Moss, C.; Martinez, A.E.; Mellerio, J.E.; McGrath, J.A. Mutations in KLHL24 Add to the Molecular Heterogeneity of Epidermolysis Bullosa Simplex. J. Investig. Dermatol. 2017, 137, 1378–1380. [Google Scholar] [CrossRef] [PubMed]
- Bolling, M.C.; Jonkman, M.F. KLHL24: Beyond Skin Fragility. J. Investig. Dermatol. 2019, 139, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Walko, G.; Castañón, M.J.; Wiche, G. Molecular Architecture and Function of the Hemidesmosome. Cell Tissue Res. 2015, 360, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.H.; Rouhani, M.J.; Maughan, E.F.; Orr, J.C.; Kolluri, K.K.; Pearce, D.R.; Haughey, E.K.; Sutton, L.; Flatau, S.; Balboa, P.L.; et al. Lentiviral Expression of Wild-Type LAMA3A Restores Cell Adhesion in Airway Basal Cells from Children with Epidermolysis Bullosa. Mol. Ther. 2024, 32, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Kiritsi, D.; Has, C.; Bruckner-Tuderman, L. Laminin 332 in Junctional Epidermolysis Bullosa. Cell Adhes. Migr. 2013, 7, 135–141. [Google Scholar] [CrossRef]
- Schumann, H.; Kiritsi, D.; Pigors, M.; Hausser, I.; Kohlhase, J.; Peters, J.; Ott, H.; Hyla-Klekot, L.; Gacka, E.; Sieron, A.L.; et al. Phenotypic Spectrum of Epidermolysis Bullosa Associated with A6β4 Integrin Mutations. Br. J. Dermatol. 2013, 169, 115–124. [Google Scholar] [CrossRef]
- Ma, S.; Li, X.; Cao, R.; Zhan, G.; Fu, X.; Xiao, R.; Yang, Z. Developmentally Regulated Expression of Integrin Alpha-6 Distinguishes Neural Crest Derivatives in the Skin. Front. Cell Dev. Biol. 2023, 11, 1140554. [Google Scholar] [CrossRef] [PubMed]
- Poyyakkara, A.; Raji, G.R.; Padmaja, K.P.; Ramachandran, V.; Changmai, U.; Edatt, L.; Punathil, R.; Kumar, V.B.S. Integrin Β4 Induced Epithelial-to-Mesenchymal Transition Involves MiR-383 Mediated Regulation of GATA6 Levels. Mol. Biol. Rep. 2023, 50, 8623–8637. [Google Scholar] [CrossRef] [PubMed]
- Mariath, L.M.; Santin, J.T.; Frantz, J.A.; Doriqui, M.J.R.; Schuler-Faccini, L.; Kiszewski, A.E. Genotype-Phenotype Correlations on Epidermolysis Bullosa with Congenital Absence of Skin: A Comprehensive Review. Clin. Genet. 2021, 99, 29–41. [Google Scholar] [CrossRef]
- Zhang, H.; Cui, X.; Cao, A.; Li, X.; Li, L. ITGA3 Interacts with VASP to Regulate Stemness and Epithelial-Mesenchymal Transition of Breast Cancer Cells. Gene 2020, 734, 144396. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Balasubramanian, M.; Humphreys, N.; Waruiru, C.; Brauner, M.; Kohlhase, J.; O’Reilly, R.; Has, C. Intronic ITGA3 Mutation Impacts Splicing Regulation and Causes Interstitial Lung Disease, Nephrotic Syndrome, and Epidermolysis Bullosa. J. Investig. Dermatol. 2016, 136, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.A. Recently Identified Forms of Epidermolysis Bullosa. Ann. Dermatol. 2015, 27, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kitahata, H.; Kosumi, H.; Watanabe, M.; Fujimura, Y.; Takashima, S.; Osada, S.-I.; Hirose, T.; Nishie, W.; Nagayama, M.; et al. Collagen XVII Deficiency Alters Epidermal Patterning. Lab. Investig. 2022, 102, 581–588. [Google Scholar] [CrossRef]
- Tanimura, S.; Tadokoro, Y.; Inomata, K.; Binh, N.T.; Nishie, W.; Yamazaki, S.; Nakauchi, H.; Tanaka, Y.; McMillan, J.R.; Sawamura, D.; et al. Hair Follicle Stem Cells Provide a Functional Niche for Melanocyte Stem Cells. Cell Stem Cell 2011, 8, 177–187. [Google Scholar] [CrossRef]
- Liu, N.; Matsumura, H.; Kato, T.; Ichinose, S.; Takada, A.; Namiki, T.; Asakawa, K.; Morinaga, H.; Mohri, Y.; De Arcangelis, A.; et al. Stem Cell Competition Orchestrates Skin Homeostasis and Ageing. Nature 2019, 568, 344–350. [Google Scholar] [CrossRef]
- Watanabe, M.; Natsuga, K.; Nishie, W.; Kobayashi, Y.; Donati, G.; Suzuki, S.; Fujimura, Y.; Tsukiyama, T.; Ujiie, H.; Shinkuma, S.; et al. Type XVII Collagen Coordinates Proliferation in the Interfollicular Epidermis. eLife 2017, 6, e26635. [Google Scholar] [CrossRef]
- Has, C.; Kern, J.S. Collagen XVII. Dermatol. Clin. 2010, 28, 61–66. [Google Scholar] [CrossRef]
- McGrath, J.A.; Gatalica, B.; Christiano, A.M.; Li, K.; Owaribe, K.; McMillan, J.R.; Eady, R.A.; Uitto, J. Mutations in the 180-KD Bullous Pemphigoid Antigen (BPAG2), a Hemidesmosomal Transmembrane Collagen (COL17A1), in Generalized Atrophic Benign Epidermolysis Bullosa. Nat. Genet. 1995, 11, 83–86. [Google Scholar] [CrossRef]
- Roig-Rosello, E.; Rousselle, P. The Human Epidermal Basement Membrane: A Shaped and Cell Instructive Platform That Aging Slowly Alters. Biomolecules 2020, 10, 1607. [Google Scholar] [CrossRef]
- Conradt, G.; Hausser, I.; Nyström, A. Epidermal or Dermal Collagen VII Is Sufficient for Skin Integrity: Insights to Anchoring Fibril Homeostasis. J. Investig. Dermatol. 2024, 144, 1301–1310.e7. [Google Scholar] [CrossRef]
- Burgeson, R.E. Type VII Collagen, Anchoring Fibrils, and Epidermolysis Bullosa. J. Investig. Dermatol. 1993, 101, 252–255. [Google Scholar] [CrossRef]
- Chung, H.J.; Uitto, J. Type VII Collagen: The Anchoring Fibril Protein at Fault in Dystrophic Epidermolysis Bullosa. Dermatol. Clin. 2010, 28, 93–105. [Google Scholar] [CrossRef]
- Baardman, R.; Bremer, J.; Diercks, G.F.H.; Jan, S.Z.; Lemmink, H.H.; Bolling, M.C.; Van den Akker, P.C. Single Glycine Deletion in COL7A1 Acting as Glycine Substitution in Dystrophic Epidermolysis Bullosa. J. Eur. Acad. Dermatol. Venereol. 2021, 35, e597–e600. [Google Scholar] [CrossRef]
- So, J.Y.; Nazaroff, J.; Yenamandra, V.K.; Gorell, E.S.; Harris, N.; Fulchand, S.; Eid, E.; Dolorito, J.A.; Marinkovich, M.P.; Tang, J.Y. Functional Genotype-Phenotype Associations in Recessive Dystrophic Epidermolysis Bullosa. J. Am. Acad. Dermatol. 2024. [CrossRef]
- Fine, J.-D.; Bruckner-Tuderman, L.; Eady, R.A.J.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited Epidermolysis Bullosa: Updated Recommendations on Diagnosis and Classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Lai-Cheong, J.E.; Tanaka, A.; Hawche, G.; Emanuel, P.; Maari, C.; Taskesen, M.; Akdeniz, S.; Liu, L.; McGrath, J.A. Kindler Syndrome: A Focal Adhesion Genodermatosis. Br. J. Dermatol. 2009, 160, 233–242. [Google Scholar] [CrossRef]
- Shimizu, H.; Sato, M.; Ban, M.; Kitajima, Y.; Ishizaki, S.; Harada, T.; Bruckner-Tuderman, L.; Fine, J.D.; Burgeson, R.; Kon, A.; et al. Immunohistochemical, Ultrastructural, and Molecular Features of Kindler Syndrome Distinguish It from Dystrophic Epidermolysis Bullosa. Arch. Dermatol. 1997, 133, 1111–1117. [Google Scholar] [CrossRef]
- Heinemann, A.; He, Y.; Zimina, E.; Boerries, M.; Busch, H.; Chmel, N.; Kurz, T.; Bruckner-Tuderman, L.; Has, C. Induction of Phenotype Modifying Cytokines by FERMT1 Mutations. Hum. Mutat. 2011, 32, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, P.; Zhang, W.; Zhou, H.; Guo, E.; Hu, G.; Zhang, L. FERMT1 Contributes to the Migration and Invasion of Nasopharyngeal Carcinoma through Epithelial–Mesenchymal Transition and Cell Cycle Arrest. Cancer Cell Int. 2022, 22, 70. [Google Scholar] [CrossRef] [PubMed]
- Rognoni, E.; Ruppert, R.; Fässler, R. The Kindlin Family: Functions, Signaling Properties and Implications for Human Disease. J. Cell Sci. 2016, 129, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Maier, K.; He, Y.; Wölfle, U.; Esser, P.R.; Brummer, T.; Schempp, C.; Bruckner-Tuderman, L.; Has, C. UV-B-Induced Cutaneous Inflammation and Prospects for Antioxidant Treatment in Kindler Syndrome. Hum. Mol. Genet. 2016, 25, 5339–5352. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Bruckner-Tuderman, L.; Uitto, J. Epidermolysis Bullosa. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014; ISBN 978-0-12-801238-3. [Google Scholar]
- Nishimura, E.K.; Suzuki, M.; Igras, V.; Du, J.; Lonning, S.; Miyachi, Y.; Roes, J.; Beermann, F.; Fisher, D.E. Key Roles for Transforming Growth Factor Beta in Melanocyte Stem Cell Maintenance. Cell Stem Cell 2010, 6, 130–140. [Google Scholar] [CrossRef]
- Piccinni, E.; Di Zenzo, G.; Maurelli, R.; Dellambra, E.; Teson, M.; Has, C.; Zambruno, G.; Castiglia, D. Induction of Senescence Pathways in Kindler Syndrome Primary Keratinocytes. Br. J. Dermatol. 2013, 168, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Nakase, Y.; Hamada, A.; Kitamura, N.; Hata, T.; Toratani, S.; Yamamoto, T.; Okamoto, T. Novel PTCH1 Mutations in Japanese Familial Nevoid Basal Cell Carcinoma Syndrome. Hum. Genome Var. 2020, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Kurata, S.; Okuyama, T.; Osada, M.; Watanabe, T.; Tomimori, Y.; Sato, S.; Iwai, A.; Tsuji, T.; Ikawa, Y.; Katoh, I. P51/P63 Controls Subunit A3 of the Major Epidermis Integrin Anchoring the Stem Cells to the Niche *. J. Biol. Chem. 2004, 279, 50069–50077. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Mei, Y.; Jiang, Y.; Li, H.; Zhao, R.; Sima, J.; Yao, Y. Ectodysplasin A (EDA) Signaling: From Skin Appendage to Multiple Diseases. Int. J. Mol. Sci. 2022, 23, 8911. [Google Scholar] [CrossRef]
- Keller, M.D.; Petersen, M.; Ong, P.; Church, J.; Risma, K.; Burham, J.; Jain, A.; Stiehm, E.R.; Hanson, E.P.; Uzel, G.; et al. Hypohidrotic Ectodermal Dysplasia and Immunodeficiency with Coincident NEMO and EDA Mutations. Front. Immunol. 2011, 2, 61. [Google Scholar] [CrossRef]
- Wohlfart, S.; Hammersen, J.; Schneider, H. Mutational Spectrum in 101 Patients with Hypohidrotic Ectodermal Dysplasia and Breakpoint Mapping in Independent Cases of Rare Genomic Rearrangements. J. Hum. Genet. 2016, 61, 891–897. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 14 June 2024).
- Shchaslyvyi, A.Y.; Antonenko, S.V.; Tesliuk, M.G.; Telegeev, G.D. Current State of Human Gene Therapy: Approved Products and Vectors. Pharmaceuticals 2023, 16, 1416. [Google Scholar] [CrossRef] [PubMed]
- Mavilio, F.; Pellegrini, G.; Ferrari, S.; Di Nunzio, F.; Di Iorio, E.; Recchia, A.; Maruggi, G.; Ferrari, G.; Provasi, E.; Bonini, C.; et al. Correction of Junctional Epidermolysis Bullosa by Transplantation of Genetically Modified Epidermal Stem Cells. Nat. Med. 2006, 12, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Holostem Terapie Avanzate s.r.l. Prospective, Open-Label, Uncontrolled Clinical Trial to Assess the Safety and Efficacy of Autologous Cultured Epidermal Grafts Containing Epidermal Stem Cells Genetically Modified with a Gamma-Retroviral (Rv) Vector Carrying COL17A1 CDNA for Restoration of Epidermis in Patients with Junctional Epidermolysis Bullosa. 2022. Available online: https://clinicaltrials.gov/ (accessed on 29 July 2024).
- Siprashvili, Z.; Nguyen, N.T.; Gorell, E.S.; Loutit, K.; Khuu, P.; Furukawa, L.K.; Lorenz, H.P.; Leung, T.H.; Keene, D.R.; Rieger, K.E.; et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA 2016, 316, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.; Gammon, L.; Liu, L.; Mellerio, J.E.; Dopping-Hepenstal, P.J.C.; Pacy, J.; Elia, G.; Jeffery, R.; Leigh, I.M.; Navsaria, H.; et al. Potential of Fibroblast Cell Therapy for Recessive Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2008, 128, 2179–2189. [Google Scholar] [CrossRef] [PubMed]
- Rashidghamat, E.; McGrath, J.A. Novel and Emerging Therapies in the Treatment of Recessive Dystrophic Epidermolysis Bullosa. Intractable Rare Dis. Res. 2017, 6, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Nyström, A.; Bruckner-Tuderman, L.; Kern, J.S. Cell- and Protein-Based Therapy Approaches for Epidermolysis Bullosa. Methods Mol. Biol. 2013, 961, 425–440. [Google Scholar] [CrossRef]
- Osborn, M.J.; Starker, C.G.; McElroy, A.N.; Webber, B.R.; Riddle, M.J.; Xia, L.; DeFeo, A.P.; Gabriel, R.; Schmidt, M.; von Kalle, C.; et al. TALEN-Based Gene Correction for Epidermolysis Bullosa. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1151–1159. [Google Scholar] [CrossRef]
- Kocher, T.; Peking, P.; Klausegger, A.; Murauer, E.M.; Hofbauer, J.P.; Wally, V.; Lettner, T.; Hainzl, S.; Ablinger, M.; Bauer, J.W.; et al. Cut and Paste: Efficient Homology-Directed Repair of a Dominant Negative KRT14 Mutation via CRISPR/Cas9 Nickases. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2585–2598. [Google Scholar] [CrossRef]
- Hainzl, S.; Peking, P.; Kocher, T.; Murauer, E.M.; Larcher, F.; Del Rio, M.; Duarte, B.; Steiner, M.; Klausegger, A.; Bauer, J.W.; et al. COL7A1 Editing via CRISPR/Cas9 in Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2573–2584. [Google Scholar] [CrossRef]
- Anbouba, G.M.; Carmany, E.P.; Natoli, J.L. The Characterization of Hypodontia, Hypohidrosis, and Hypotrichosis Associated with X-Linked Hypohidrotic Ectodermal Dysplasia: A Systematic Review. Am. J. Med. Genet. Part A 2020, 182, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Huang, C.; Wan, F.; Jiang, C.; Chen, J.; Li, X.; Wang, F.; Wu, J.; Lei, M.; Wu, Y. Structural Insights into Pathogenic Mechanism of Hypohidrotic Ectodermal Dysplasia Caused by Ectodysplasin A Variants. Nat. Commun. 2023, 14, 767. [Google Scholar] [CrossRef] [PubMed]
- Trzeciak, W.H.; Koczorowski, R. Molecular Basis of Hypohidrotic Ectodermal Dysplasia: An Update. J. Appl. Genet. 2016, 57, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Horrell, J.; Snitow, M.; Cui, J.; Gochnauer, H.; Syrett, C.M.; Kallish, S.; Seykora, J.T.; Liu, F.; Gaillard, D.; et al. WNT10A Mutation Causes Ectodermal Dysplasia by Impairing Progenitor Cell Proliferation and KLF4-Mediated Differentiation. Nat. Commun. 2017, 8, 15397. [Google Scholar] [CrossRef] [PubMed]
- Mues, G.; Bonds, J.; Xiang, L.; Vieira, A.R.; Seymen, F.; Klein, O.; D’Souza, R.N. The WNT10A Gene in Ectodermal Dysplasias and Selective Tooth Agenesis. Am. J. Med. Genet. Part A 2014, 164A, 2455–2460. [Google Scholar] [CrossRef] [PubMed]
- Klineberg, I.; Cameron, A.; Whittle, T.; Hobkirk, J.; Bergendal, B.; Maniere, M.-C.; King, N.; Palmer, R.; Hobson, R.; Stanford, C.; et al. Rehabilitation of Children with Ectodermal Dysplasia. Part 1: An International Delphi Study. Int. J. Oral Maxillofac. Implant. 2013, 28, 1090–1100. [Google Scholar] [CrossRef]
- Lee, H.-E.; Chang, I.-K.; Im, M.; Seo, Y.-J.; Lee, J.-H.; Lee, Y. Topical Minoxidil Treatment for Congenital Alopecia in Hypohidrotic Ectodermal Dysplasia. J. Am. Acad. Dermatol. 2013, 68, e139–e140. [Google Scholar] [CrossRef]
- Mineroff, J.; Dowling, J.R.; Golbari, N.M.; Wechter, T.; Jagdeo, J. Hypohidrotic Ectodermal Dysplasia Milia Treatment With Fractional Carbon Dioxide Laser and Laser-Assisted Drug Delivery of Triamcinolone. J. Drugs Dermatol. 2023, 22, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Huttner, K. Future Developments in XLHED Treatment Approaches. Am. J. Med. Genet. Part A 2014, 164A, 2433–2436. [Google Scholar] [CrossRef] [PubMed]
- Körber, I.; Klein, O.D.; Morhart, P.; Faschingbauer, F.; Grange, D.K.; Clarke, A.; Bodemer, C.; Maitz, S.; Huttner, K.; Kirby, N.; et al. Safety and Immunogenicity of Fc-EDA, a Recombinant Ectodysplasin A1 Replacement Protein, in Human Subjects. Br. J. Clin. Pharmacol. 2020, 86, 2063–2069. [Google Scholar] [CrossRef]
- Schneider, H.; Faschingbauer, F.; Schuepbach-Mallepell, S.; Körber, I.; Wohlfart, S.; Dick, A.; Wahlbuhl, M.; Kowalczyk-Quintas, C.; Vigolo, M.; Kirby, N.; et al. Prenatal Correction of X-Linked Hypohidrotic Ectodermal Dysplasia. N. Engl. J. Med. 2018, 378, 1604–1610. [Google Scholar] [CrossRef]
- Adolphe, C.; Xue, A.; Fard, A.T.; Genovesi, L.A.; Yang, J.; Wainwright, B.J. Genetic and Functional Interaction Network Analysis Reveals Global Enrichment of Regulatory T Cell Genes Influencing Basal Cell Carcinoma Susceptibility. Genome Med. 2021, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Choquet, H.; Ashrafzadeh, S.; Kim, Y.; Asgari, M.M.; Jorgenson, E. Genetic and Environmental Factors Underlying Keratinocyte Carcinoma Risk. JCI Insight 2020, 5, e134783. [Google Scholar] [CrossRef] [PubMed]
- Karampinis, E.; Nechalioti, P.-M.; Georgopoulou, K.E.; Goniotakis, G.; Roussaki Schulze, A.V.; Zafiriou, E.; Kouretas, D. Systemic Oxidative Stress Parameters in Skin Cancer Patients and Patients with Benign Lesions. Stresses 2023, 3, 785–812. [Google Scholar] [CrossRef]
- Dourmishev, L.A.; Rusinova, D.; Botev, I. Clinical Variants, Stages, and Management of Basal Cell Carcinoma. Indian Dermatol. Online J. 2013, 4, 12–17. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, B.; Badri, T.; Steele, R.B. Basal Cell Carcinoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Josiah, A.J.; Twilley, D.; Pillai, S.K.; Ray, S.S.; Lall, N. Pathogenesis of Keratinocyte Carcinomas and the Therapeutic Potential of Medicinal Plants and Phytochemicals. Molecules 2021, 26, 1979. [Google Scholar] [CrossRef] [PubMed]
- Perez-Losada, J.; Balmain, A. Stem-Cell Hierarchy in Skin Cancer. Nat. Rev. Cancer 2003, 3, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Y.; Wang, J.; Mancianti, M.-L.; Epstein, E.H. Basal Cell Carcinomas Arise from Hair Follicle Stem Cells in Ptch1+/− Mice. Cancer Cell 2011, 19, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Epstein, E.H. Basal Cell Carcinomas: Attack of the Hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef]
- Briscoe, J.; Thérond, P.P. The Mechanisms of Hedgehog Signalling and Its Roles in Development and Disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog Target Genes: Mechanisms of Carcinogenesis Induced by Aberrant Hedgehog Signaling Activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef]
- Pellegrini, C.; Maturo, M.G.; Di Nardo, L.; Ciciarelli, V.; Gutiérrez García-Rodrigo, C.; Fargnoli, M.C. Understanding the Molecular Genetics of Basal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2485. [Google Scholar] [CrossRef]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened Mutations in Sporadic Basal-Cell Carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef]
- Aszterbaum, M.; Rothman, A.; Johnson, R.L.; Fisher, M.; Xie, J.; Bonifas, J.M.; Zhang, X.; Scott, M.P.; Epstein, E.H. Identification of Mutations in the Human PATCHED Gene in Sporadic Basal Cell Carcinomas and in Patients with the Basal Cell Nevus Syndrome. J. Investig. Dermatol. 1998, 110, 885–888. [Google Scholar] [CrossRef]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic Analysis Identifies New Drivers and Progression Pathways in Skin Basal Cell Carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef]
- Smyth, I.; Narang, M.A.; Evans, T.; Heimann, C.; Nakamura, Y.; Chenevix-Trench, G.; Pietsch, T.; Wicking, C.; Wainwright, B.J. Isolation and Characterization of Human Patched 2 (PTCH2), a Putative Tumour Suppressor Gene Inbasal Cell Carcinoma and Medulloblastoma on Chromosome 1p32. Hum. Mol. Genet. 1999, 8, 291–297. [Google Scholar] [CrossRef]
- Dasgeb, B.; Pajouhanfar, S.; Jazayeri, A.; Schoenberg, E.; Kumar, G.; Fortina, P.; Berger, A.C.; Uitto, J. Novel PTCH1 and Concurrent TP53 Mutations in Four Patients with Numerous Non-Syndromic Basal Cell Carcinomas: The Paradigm of Oncogenic Synergy. Exp. Dermatol. 2022, 31, 736–742. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. P53, Guardian of the Genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Piipponen, M.; Riihilä, P.; Nissinen, L.; Kähäri, V.-M. The Role of P53 in Progression of Cutaneous Squamous Cell Carcinoma. Cancers 2021, 13, 4507. [Google Scholar] [CrossRef]
- Hatton, B.A.; Knoepfler, P.S.; Kenney, A.M.; Rowitch, D.H.; de Alborán, I.M.; Olson, J.M.; Eisenman, R.N. N-Myc Is an Essential Downstream Effector of Shh Signaling during Both Normal and Neoplastic Cerebellar Growth. Cancer Res. 2006, 66, 8655–8661. [Google Scholar] [CrossRef] [PubMed]
- Nowell, C.; Radtke, F. Cutaneous Notch Signaling in Health and Disease. Cold Spring Harb. Perspect. Med. 2013, 3, a017772. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.-T.; Yu, M.; Zloty, D.; Bell, R.H.; Wang, E.; Akhoundsadegh, N.; Leung, G.; Haegert, A.; Carr, N.; Shapiro, J.; et al. Notch Signaling Is Significantly Suppressed in Basal Cell Carcinomas and Activation Induces Basal Cell Carcinoma Cell Apoptosis. Mol. Med. Rep. 2017, 15, 1441–1454. [Google Scholar] [CrossRef]
- Massi, D.; Panelos, J. Notch Signaling and the Developing Skin Epidermis. Adv. Exp. Med. Biol. 2012, 727, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Totaro, A.; Castellan, M.; Battilana, G.; Zanconato, F.; Azzolin, L.; Giulitti, S.; Cordenonsi, M.; Piccolo, S. YAP/TAZ Link Cell Mechanics to Notch Signalling to Control Epidermal Stem Cell Fate. Nat. Commun. 2017, 8, 15206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.-L. The Hippo-YAP Pathway in Organ Size Control and Tumorigenesis: An Updated Version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef]
- Rognoni, E.; Walko, G. The Roles of YAP/TAZ and the Hippo Pathway in Healthy and Diseased Skin. Cells 2019, 8, 411. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.-X.; Brugge, J.S.; Haber, D.A. Transforming Properties of YAP, a Candidate Oncogene on the Chromosome 11q22 Amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.R.; et al. Yap1 Acts Downstream of α-Catenin to Control Epidermal Proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef]
- Griewank, K.G.; Murali, R.; Schilling, B.; Schimming, T.; Möller, I.; Moll, I.; Schwamborn, M.; Sucker, A.; Zimmer, L.; Schadendorf, D.; et al. TERT Promoter Mutations Are Frequent in Cutaneous Basal Cell Carcinoma and Squamous Cell Carcinoma. PLoS ONE 2013, 8, e80354. [Google Scholar] [CrossRef]
- Wang, L.; Shi, Y.; Ju, P.; Liu, R.; Yeo, S.P.; Xia, Y.; Owlanj, H.; Feng, Z. Silencing of Diphthamide Synthesis 3 (Dph3) Reduces Metastasis of Murine Melanoma. PLoS ONE 2012, 7, e49988. [Google Scholar] [CrossRef]
- BASSET-SEGUIN, N.; HERMS, F. Update on the Management of Basal Cell Carcinoma. Acta Derm. Venereol. 2020, 100, 5750. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant P53: One Name, Many Proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Ayaz, G.; Yan, H.; Malik, N.; Huang, J. An Updated View of the Roles of P53 in Embryonic Stem Cells. Stem Cells 2022, 40, 883–891. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor Evolution. High Burden and Pervasive Positive Selection of Somatic Mutations in Normal Human Skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [CrossRef]
- Li-Bao, L.; Díaz-Díaz, C.; Raiola, M.; Sierra, R.; Temiño, S.; Moya, F.J.; Rodriguez-Perales, S.; Santos, E.; Giovinazzo, G.; Bleckwehl, T.; et al. Regulation of Myc Transcription by an Enhancer Cluster Dedicated to Pluripotency and Early Embryonic Expression. Nat. Commun. 2024, 15, 3931. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.-F.; et al. Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef]
- Li, J.-C.; Zhao, Y.-H.; Wang, X.-Y.; Yang, Y.; Pan, D.-L.; Qiu, Z.-D.; Su, Y.; Pan, J.-J. Clinical Significance of the Expression of EGFR Signaling Pathway-Related Proteins in Esophageal Squamous Cell Carcinoma. Tumor Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 651–657. [Google Scholar] [CrossRef]
- Weina, K.; Utikal, J. SOX2 and Cancer: Current Research and Its Implications in the Clinic. Clin. Transl. Med. 2014, 3, 19. [Google Scholar] [CrossRef]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 Mutations Occur Early during Cutaneous Squamous Cell Carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem Cells and the Impact of ROS Signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef]
- Sadeqzadeh, E.; de Bock, C.E.; Thorne, R.F. Sleeping Giants: Emerging Roles for the Fat Cadherins in Health and Disease. Med. Res. Rev. 2014, 34, 190–221. [Google Scholar] [CrossRef]
- Stang, A.; Khil, L.; Kajüter, H.; Pandeya, N.; Schmults, C.D.; Ruiz, E.S.; Karia, P.S.; Green, A.C. Incidence and Mortality for Cutaneous Squamous Cell Carcinoma: Comparison across Three Continents. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 6–10. [Google Scholar] [CrossRef]
- Combalia, A.; Carrera, C. Squamous Cell Carcinoma: An Update on Diagnosis and Treatment. Dermatol. Pract. Concept. 2020, 10, e2020066. [Google Scholar] [CrossRef]
- Armstrong, B.K.; Kricker, A. The Epidemiology of UV Induced Skin Cancer. J. Photochem. Photobiol. B 2001, 63, 8–18. [Google Scholar] [CrossRef]
- Brooks, P.J.; Zakhari, S. Acetaldehyde and the Genome: Beyond Nuclear DNA Adducts and Carcinogenesis. Environ. Mol. Mutagen. 2014, 55, 77–91. [Google Scholar] [CrossRef]
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef]
- Owens, D.M.; Romero, M.R.; Gardner, C.; Watt, F.M. Suprabasal Alpha6beta4 Integrin Expression in Epidermis Results in Enhanced Tumourigenesis and Disruption of TGFbeta Signalling. J. Cell Sci. 2003, 116, 3783–3791. [Google Scholar] [CrossRef]
- Morris, R.J. Keratinocyte Stem Cells: Targets for Cutaneous Carcinogens. J. Clin. Investig. 2000, 106, 3–8. [Google Scholar] [CrossRef]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of Genomic Alterations in Cervical Carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef]
- Teven, C.M.; Farina, E.M.; Rivas, J.; Reid, R.R. Fibroblast Growth Factor (FGF) Signaling in Development and Skeletal Diseases. Genes Dis. 2014, 1, 199–213. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef]
- Daniely, Y.; Liao, G.; Dixon, D.; Linnoila, R.I.; Lori, A.; Randell, S.H.; Oren, M.; Jetten, A.M. Critical Role of P63 in the Development of a Normal Esophageal and Tracheobronchial Epithelium. Am. J. Physiol. Cell Physiol. 2004, 287, C171–C181. [Google Scholar] [CrossRef]
- Porter, L.; McCaughan, F. SOX2 and Squamous Cancers. Semin. Cancer Biol. 2020, 67, 154–167. [Google Scholar] [CrossRef]
- Lefort, K.; Mandinova, A.; Ostano, P.; Kolev, V.; Calpini, V.; Kolfschoten, I.; Devgan, V.; Lieb, J.; Raffoul, W.; Hohl, D.; et al. Notch1 Is a P53 Target Gene Involved in Human Keratinocyte Tumor Suppression through Negative Regulation of ROCK1/2 and MRCKalpha Kinases. Genes Dev. 2007, 21, 562–577. [Google Scholar] [CrossRef]
- Demehri, S.; Turkoz, A.; Kopan, R. Epidermal Notch1 Loss Promotes Skin Tumorigenesis by Impacting the Stromal Microenvironment. Cancer Cell 2009, 16, 55–66. [Google Scholar] [CrossRef]
- Mohs, F.E. Chemosurgery for the Microscopically Controlled Excision of Skin Cancer. J. Surg. Oncol. 1971, 3, 257–267. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Skin Disease | Gene | Chromosomal Locus | Ref. |
|---|---|---|---|
| Nevoid basal cell carcinoma syndrome | PTCH1 | 9q22.32 | [151,200] |
| Epidermolysis bullosa simplex | KRT5 | 12q13.13 | [164] |
| KRT14 | 17q21.2 | [164] | |
| DST | 6p12.1 | [161] | |
| PLEC | 8q24.3 | [162] | |
| EXPH5 | 11q22.3 | [163] | |
| KLHL24 | 3q27.1 | [166] | |
| CD151 | 11p15.5 | [168] | |
| Junctional epidermolysis bullosa | LAMA3 | 18q11.2 | [164] |
| LAMB3 | 1q32.2 | [164] | |
| LAMC2 | 1q25.3 | [164] | |
| ITGA6 | 2q31.1 | [171] | |
| ITGB4 | 17q25.1 | [171,173] | |
| ITGA3 | 17q21.33 | [175,201] | |
| COL17A1 | 10q25.1 | [178,181] | |
| Dystrophic epidermolysis bullosa | COL7A1 | 3p21.31 | [185] |
| Kindler epidermolysis bullosa | FERMT1 | 20p12.3 | [193,194] |
| Hypohidrotic ectodermal dysplasia | EDA | Xq13.1 | [202] |
| IKBKG | Xq28 | [203] | |
| WNT10A | 2q35 | [204] | |
| EDAR | 2q13 | [204] | |
| EDARADD | 1q42.3-q43 | [204] |
| Skin Cancer | Gene | Chromosomal Locus | Ref. |
|---|---|---|---|
| Basal cell carcinoma | PTCH1 | 9q22.32 | [240] |
| PTCH2 | 1p34.1 | [243] | |
| SMO | 7q32.1 | [243] | |
| SUFU | 10q24.32 | [243] | |
| TP53 | 17p13.1 | [245] | |
| MYCN | 2p24.3 | [243] | |
| TERT | 5p15.33 | [257] | |
| DPH3 | 3p25.1 | [258] | |
| Squamous cell carcinoma | TP53 | 17p13.1 | [260,261] |
| CDKN2A | 9p21.3 | [262] | |
| CCND1 | 11q13.3 | [263] | |
| MYC | 8q24.21 | [263,264] | |
| FGFR1 | 8p11.23 | [265] | |
| FGFR2 | 10q26.13 | [265] | |
| FGFR3 | 4p16.3 | [265] | |
| EGFR | 7p11.2 | [266] | |
| TP63 | 3q28 | [48] | |
| SOX2 | 3q26.33 | [267] | |
| NOTCH1 | 9q34.3 | [268] | |
| NFE2L2 | 2q31.2 | [269] | |
| FAT1 | 4q35.2 | [270] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dermitzakis, I.; Chatzi, D.; Kyriakoudi, S.A.; Evangelidis, N.; Vakirlis, E.; Meditskou, S.; Theotokis, P.; Manthou, M.E. Skin Development and Disease: A Molecular Perspective. Curr. Issues Mol. Biol. 2024, 46, 8239-8267. https://doi.org/10.3390/cimb46080487
Dermitzakis I, Chatzi D, Kyriakoudi SA, Evangelidis N, Vakirlis E, Meditskou S, Theotokis P, Manthou ME. Skin Development and Disease: A Molecular Perspective. Current Issues in Molecular Biology. 2024; 46(8):8239-8267. https://doi.org/10.3390/cimb46080487
Chicago/Turabian StyleDermitzakis, Iasonas, Despoina Chatzi, Stella Aikaterini Kyriakoudi, Nikolaos Evangelidis, Efstratios Vakirlis, Soultana Meditskou, Paschalis Theotokis, and Maria Eleni Manthou. 2024. "Skin Development and Disease: A Molecular Perspective" Current Issues in Molecular Biology 46, no. 8: 8239-8267. https://doi.org/10.3390/cimb46080487