
miRNA Sequencing Analysis in Maize Roots Treated with Neutral and Alkaline Salts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Maize Seedling Stress Treatment
2.2. miRNA Sequencing Analysis
2.3. RNA Extraction, Quantification and Quantitative Real Time PCR (qPCR)
2.4. Computational Prediction of miRNA Targets
2.5. Ion Content Determination
3. Results
3.1. Phenotypic Identification of Maize Roots under Neutral Salt and Alkaline Stress
3.2. miRNA Sequencing Results and Data Analysis in Maize Roots under Different Salt Stress Conditions
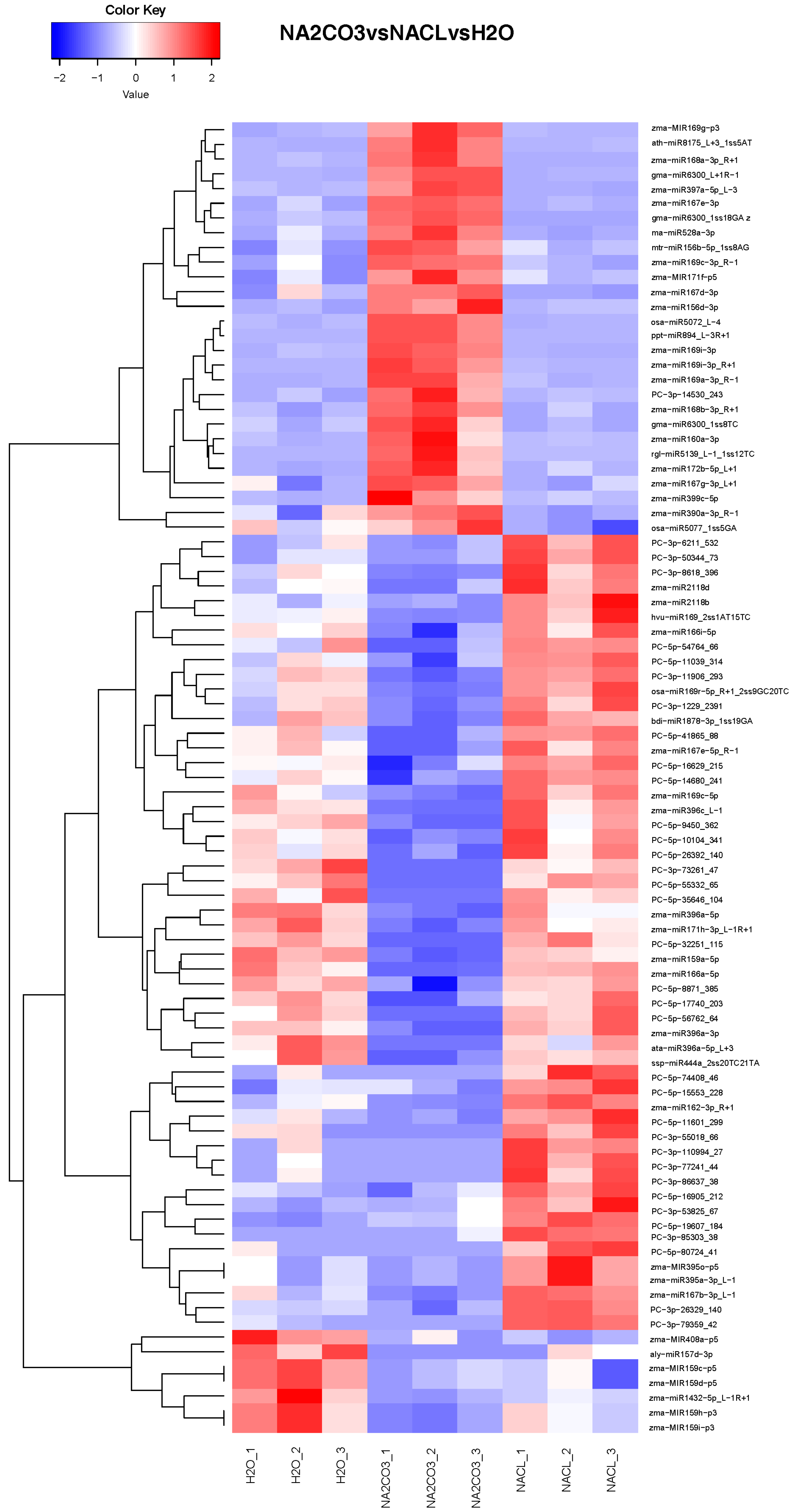
3.3. Differential miRNA Expression Analysis
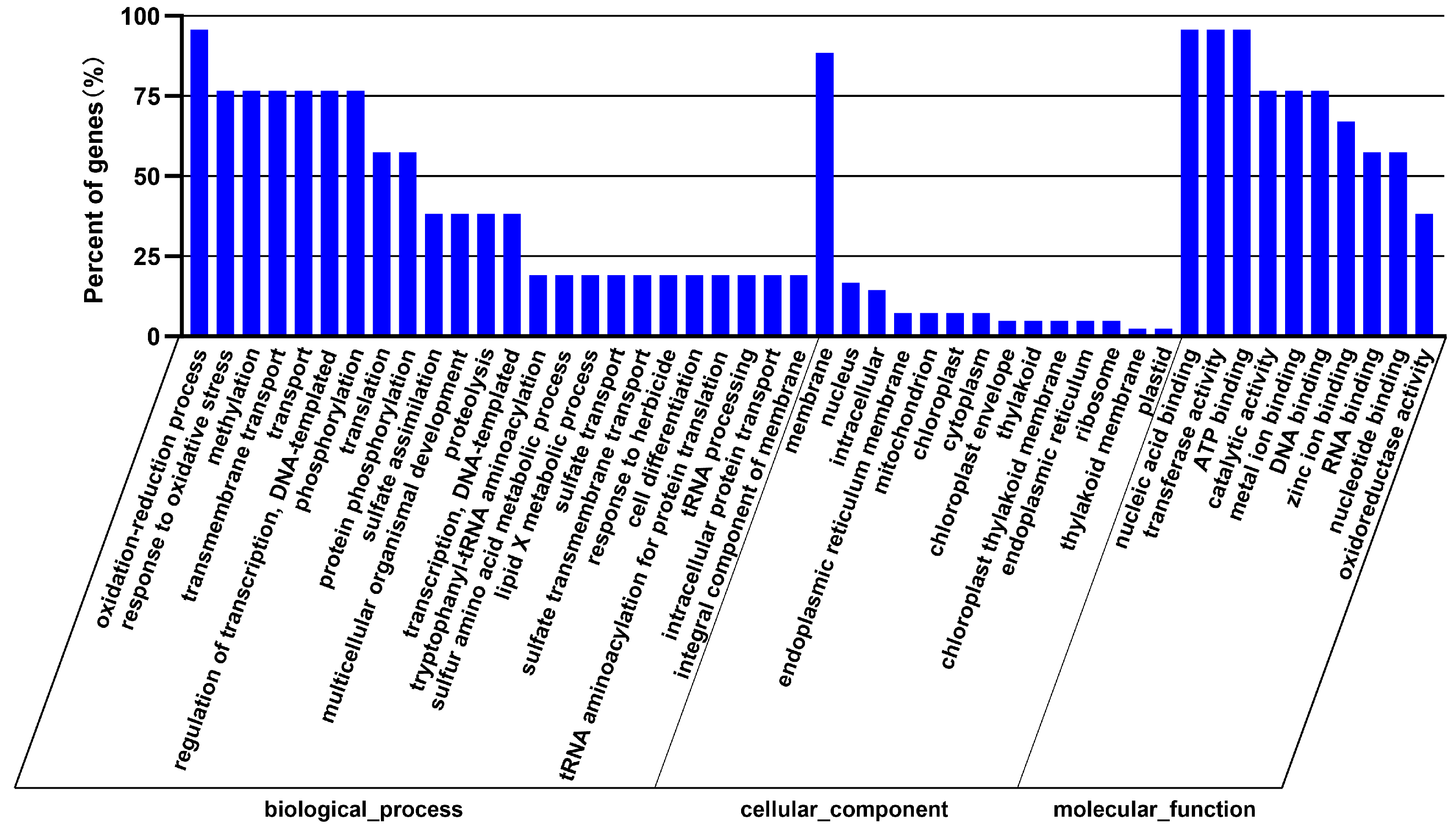
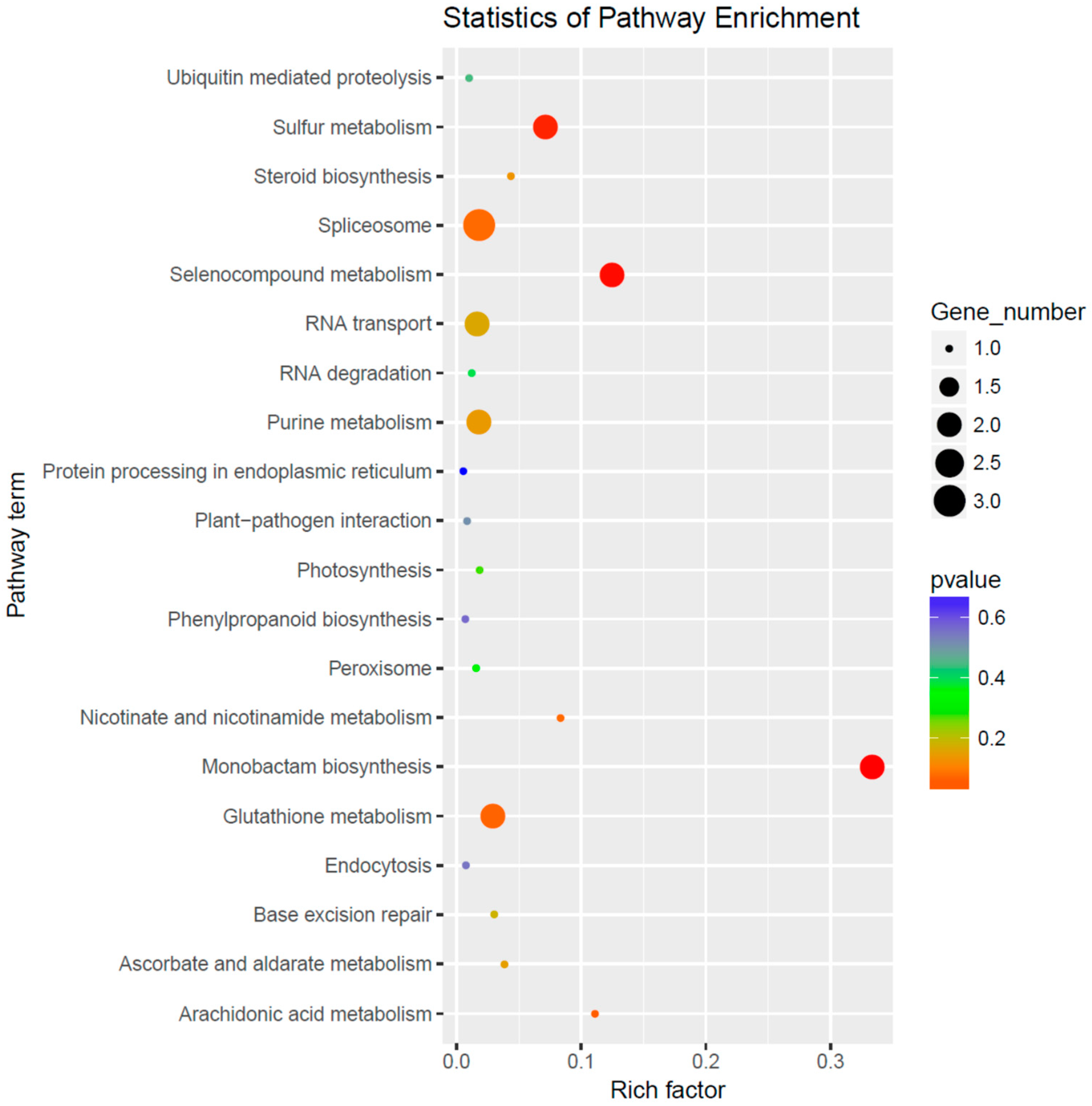
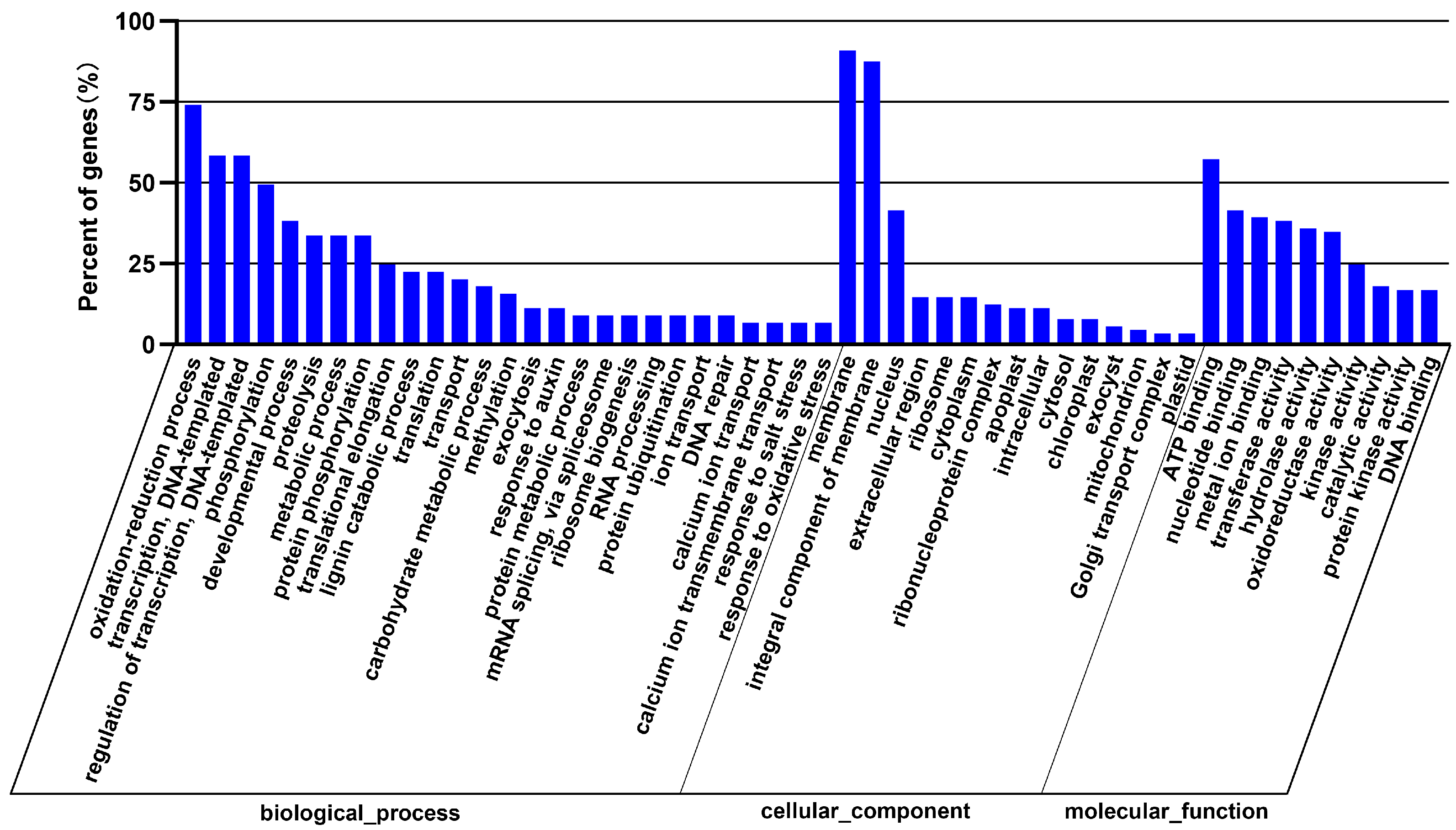
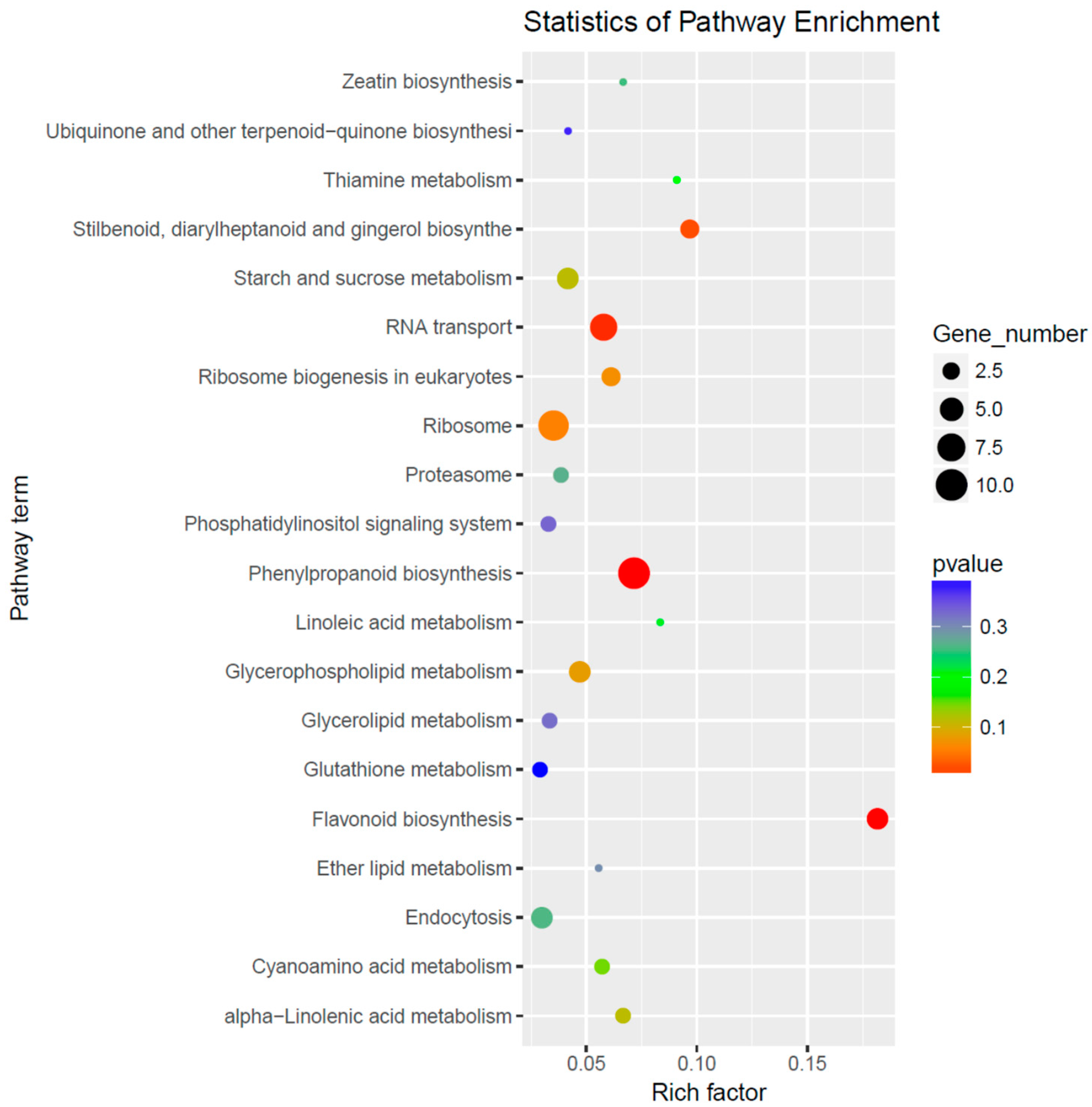
3.4. Prediction and Bioinformatics Analysis of Differentially Expressed miRNA Target Genes
3.5. Analysis of Differentially Expressed miRNAs Shared by Different Comparison Groups
3.6. qPCR Validation of Sequencing Results
3.7. The Changes in the Content of Different Ions in Maize Roots under Neutral and Alkaline Salt Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, L.M.; Liu, X.G.; Qu, X.N.; Yu, Y.; Han, S.P.; Dou, Y.; Xu, Y.Y.; Jing, H.C.; Hao, D.Y. Early transcriptomic adaptation to Na2CO3 stress altered the expression of a quarter of the total genes in the maize genome and exhibited shared and distinctive profiles with NaCl and high pH stresses. J. Integr. Plant Biol. 2013, 55, 1147–1165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wei, L.; Wang, Z.; Wang, T. Physiological and molecular features of Puccinellia tenuiflora tolerating salt and alkaline-salt stress. J. Integr. Plant Biol. 2013, 55, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chong, J.; Li, C.; Kim, C.; Shi, D.; Wang, D. Osmotic adjustment and ion balance traits of an alkali resistant halophyte Kochia sieversiana during adaptation to salt and alkali conditions. Plant Soil 2007, 294, 263–276. [Google Scholar] [CrossRef]
- Zhang, H.; Han, B.; Wang, T.; Chen, S.; Li, H.Y.; Zhang, Y.H.; Dai, S.J. Mechanisms of plant salt response: Insights from proteomics. J. Proteome Res. 2012, 11, 49–67. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef]
- Zhu, J.K. Plant salt tolerance. Trends Plant Sci. 2001, 6, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Rengasamy, P. Soil processes affecting crop production in salt-affected soils. Funct. Plant Biol. 2010, 37, 613–620. [Google Scholar] [CrossRef]
- Parida, A.K.; Das, A.B. Salt tolerance and salinity effects on plants: A review. Ecotoxicol. Environ. Saf. 2005, 60, 324–349. [Google Scholar] [CrossRef]
- Badger, M.R.; Price, G.D. The role of carbonic anhydrase in photosynthesis. Annu. Rev. Plant Biol. 1994, 45, 369–392. [Google Scholar] [CrossRef]
- Miller, G.A.D.; Suzuki, N.; Ciftci-Yilmaz, S.; Mittler, R.O.N. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ. 2010, 33, 453–467. [Google Scholar] [CrossRef]
- Sun, G.R.; Peng, Y.Z.; Shao, H.B.; Chu, L.Y.; Zhao, X.N.; Min, H.Y. Does Puccinellia tenuiflora have the ability of salt exudation? Colloids Surf. B Biointerfaces 2005, 46, 197–203. [Google Scholar]
- Yu, J.; Chen, S.; Wang, T.; Sun, G.; Dai, S. Comparative proteomic analysis of Puccinellia tenuiflora leaves under Na2CO3 stress. Int. J. Mol. Sci. 2013, 14, 1740–1762. [Google Scholar] [CrossRef]
- Kobayashi, S.; Abe, N.; Yoshida, K.T.; Liu, S.; Takano, T. Molecular cloning and characterization of plasma membrane-and vacuolar-type Na+/H+ antiporters of an alkaline-salt-tolerant monocot, Puccinellia tenuiflora. J. Plant Res. 2012, 125, 587–594. [Google Scholar] [CrossRef]
- Zhong, N.Q.; Han, L.B.; Wu, X.M.; Wang, L.L.; Wang, F.; Ma, Y.H.; Xia, G.X. Ectopic Expression of a Bacterium NhaD-type Na+/H+ Antiporter Leads to Increased Tolerance to Combined Salt/Alkali Stresses. J. Integr. Plant Biol. 2012, 54, 412–421. [Google Scholar] [CrossRef]
- Ma, Z.; Jiang, J.; Hu, Z.; Lyu, T.; Yang, Y.; Jiang, J.; Cao, J. Over-expression of miR158 causes pollen abortion in Brassica campestris ssp. chinensis. Plant Mol. Biol. 2016, 93, 313–326. [Google Scholar] [CrossRef]
- Chen, Z.; Huo, Q.; Yang, H.; Jian, H.; Qu, C.; Lu, K.; Li, J. Joint RNA-Seq and miRNA profiling analyses to reveal molecular mechanisms in regulating thickness of pod canopy in Brassica napus. Genes 2019, 10, 591. [Google Scholar] [CrossRef]
- Xu, T.; Zhang, L.; Yang, Z.; Wei, Y.; Dong, T. Identification and functional characterization of plant miRNA under salt stress shed light on salinity resistance improvement through miRNA manipulation in crops. Front. Plant Sci. 2021, 12, 665439. [Google Scholar] [CrossRef]
- Chen, B.; Ding, Z.; Zhou, X.; Wang, Y.; Huang, F.; Sun, J.; Chen, J.; Han, W. Integrated full-length transcriptome and microRNA sequencing approaches provide insights into salt tolerance in mangrove (Sonneratia apetala Buch.-Ham.). Front. Genet. 2022, 13, 932832. [Google Scholar] [CrossRef]
- Liu, M.; Li, M.; Ruan, J.; Jia, J.; Ge, C.; Cao, W. Analysis of microRNA Expression Profiles in Broiler Muscle Tissues by Feeding Different Levels of Guanidinoacetic Acid. Curr. Issues Mol. Biol. 2024, 46, 3713–3728. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-h.; Ra, W.-j.; Cho, S.; Choi, S.; Soh, B.; Joo, Y.; Lee, K.-W. Method Validation for Determination of Thalliumby Inductively Coupled Plasma Mass Spectrometry and Monitoring of Various Foods in South Korea. Molecules 2021, 26, 6729. [Google Scholar] [CrossRef] [PubMed]
- Kurmanbayeva, A.; Brychkova, G.; Bekturova, A.; Khozin, I.; Sagi, M. Determination of Total Sulfur, Sulfate, Sulfite, Thiosulfate, and Sulfolipids in Plants. Methods Mol. Biol. 2017, 1670, 255–265. [Google Scholar]
- Xu, J.; Li, H.D.; Chen, L.Q.; Wang, Y.; Liu, L.L.; He, L.; Wu, W.H. A protein kinase, interacting with two calcineurin B-like proteins, regulates K+ transporter AKT1 in Arabidopsis. Cell 2006, 125, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- AbdElgawad, H.; Peshev, D.; Zinta, G.; Van den Ende, W.; Janssens, I.A.; Asard, H. Climate extreme effects on the chemical composition of temperate grassland species under ambient and elevated CO2: A comparison of fructan and non-fructan accumulators. PLoS ONE 2014, 9, e92044. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, J.; Zhang, X.; Jia, Y. MicroRNA-mediated regulation of plant responses to abiotic stress. J. Integr. Plant Biol. 2012, 54, 885–897. [Google Scholar]
- Satendra, K.M.; Surekha, A.; Sailaja, B.; Sheshu, M.M.; Voleti, S.R. MicroRNAs and their role in salt stress response in plants. Genes Genom. 2013, 35, 377–388. [Google Scholar]
- Zhang, H.; Liu, X.; Yang, X.; Wu, H.; Zhu, J.; Zhang, H. MicroRNA–mRNA Integrated Analysis Reveals Roles for MicroRNAs in A Typical Halophyte, Reaumuria soongorica, During Seed Germination Under Salt Stress. Plants 2020, 9, 803. [Google Scholar]
- Wan, J.; Meng, S.; Wang, Q.; Zhao, J.; Qiu, X.; Wang, L.; Li, J.; Lin, Y.; Mu, L.; Dang, K.; et al. Suppression of microRNA168 enhances salt tolerance in rice (Oryza sativa L.). BMC Plant Biol. 2022, 22, 563. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Fingering too many proteins. Cell 1988, 56, 675. [Google Scholar] [CrossRef] [PubMed]
- Bland, J.M.; Altman, D.G. Multiple significance tests: The Bonferroni method. BMJ 1995, 310, 170. [Google Scholar] [CrossRef] [PubMed]
- Takatsuji, H. Zinc-finger transcription factors in plants. Cell. Mol. Life Sci. 1998, 54, 582–596. [Google Scholar] [CrossRef]
- Merk, H.L.; Yarnes, S.C.; Van Deynze, A. Trait diversity and potential for selection indices based on variation among regionally adapted processing tomato germplasm. J. Am. Soc. Hort. Sci. 2012, 137, 427–437. [Google Scholar] [CrossRef]
- Welch, D.; Hassan, H.; Blilou, I.; Immink, R.; Heidstra, R. Arabidopsis JACKDAW and MAGPIE zinc finger proteins delimit asymmetric cell division and stabilize tissue boundaries by restricting SHORT-ROOT action. Genes Dev. 2007, 21, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Ryu, J.Y.; Baek, K.; Park, C.M. High temperature attenuates the gravitropism of inflorescence stems by inducing SHOOT GRAVITROPISM 5 alternative splicing in Arabidopsis. New Phytol. 2016, 209, 265–279. [Google Scholar] [CrossRef]
- Omidbakhshfard, M.A.; Proost, S.; Fujikura, U.; Mueller-Roeber, B. Growth-regulating factors (GRFs): A small transcription factor family with important functions in plant biology. Mol. Plant 2015, 8, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Choi, D.; Kende, H. The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant J. 2003, 36, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Hewezi, T.; Baum, T.J. Complex feedback regulations govern the expression of miRNA396 and its GRF target genes. Plant Signal. Behav. 2012, 7, 749–751. [Google Scholar] [CrossRef]
- Bao, M.; Bian, H.; Zha, Y.; Li, F.; Sun, Y.; Bai, B.; Chen, Z.; Wang, J.; Zhu, M.; Han, N. miR396a-mediated basic helix-loop-helix transcription factor bHLH74 repression acts as a regulator for root growth in Arabidopsis seedlings. Plant Cell Physiol. 2014, 55, 1343–1353. [Google Scholar] [CrossRef]
- Debernardi, J.M.; Mecchia, M.A.; Vercruyssen, L.; Smaczniak, C.; Kaufmann, K.; Inzé, D.; Rodriguez, R.E.; Palatnik, J.F. Post-transcriptional control of GRF transcription factors by microRNA miR396 and GIF co-activator affects leaf size and longevity. Plant J. 2014, 79, 413–426. [Google Scholar] [CrossRef]
- Liu, H.; Guo, S.; Xu, Y.; Li, C.; Zhang, Z.; Zhang, D.; Xu, S.; Zhang, C.; Chong, K. OsmiR396d-regulated OsGRFs function in floral organogenesis in rice through binding to their targets OsJMJ706 and OsCR4. Plant Physiol. 2014, 165, 160–174. [Google Scholar] [CrossRef]
- Pajoro, A.; Madrigal, P.; Muiño, J.M.; Matus, J.T.; Jin, J.; Mecchia, M.A.; Debernardi, J.M.; Palatnik, J.F.; Balazadeh, S.; Arif, M.; et al. Dynamics of chromatin accessibility and gene regulation by MADS-domain transcription factors in flower development. Genome Biol. 2014, 15, R41. [Google Scholar] [CrossRef]
- Liang, G.; He, H.; Li, Y.; Yu, D. Molecular mechanism of microRNA396 mediating pistil development in Arabidopsis. Plant Physiol. 2015, 170, 2490–2504. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Zhao, J.; Li, Z.; Hu, Q.; Yuan, N.; Zhou, M.; Xia, X.; Noorai, R.; Saski, C.; Li, S.; et al. MicroRNA396-mediated alteration in plant development and salinity stress response in creeping bentgrass. Horticulturae 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yao, S.; Kim, S.C.; Wang, X. Lipid phosphorylation by a diacylglycerol kinase suppresses ABA biosynthesis to regulate plant stress responses. Mol. Plant 2024, 17, 342–358. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, G.; Sutoh, K.; Zhu, J.K.; Zhang, W. Identification of cold-inducible microRNAs in plants by transcriptome analysis. Biochim. Biophys. Acta Gene Regul. Mech. 2008, 1779, 780–788. [Google Scholar]
- Frazier, T.P.; Sun, G.L.; Burklew, C.E.; Zhang, B.H. Salt and drought stresses induce the aberrant expression of microRNA genes in tobacco. Mol. Biotechnol. 2011, 49, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Zhang, N.; Ma, C.Y.; Qu, Y.; Si, H.J.; Wang, D. Prediction and verification of microRNAs related to proline accumulation under drought stress in potato. Comput. Biol. Chem. 2013, 46, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Candar-Cakir, B.; Arican, E.; Zhang, B.H. Small RNA and degradome deep sequencing reveals drought-and tissue-specific microRNAs and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol. J. 2016, 14, 1727–1746. [Google Scholar] [CrossRef] [PubMed]
- Sahito, Z.A.; Wang, L.; Sun, Z.; Yan, Q.; Zhang, X.; Jiang, Q.; Ullah, I.; Tong, Y.; Li, X. The miR172c-NNC1 module modulates root plastic development in response to salt in soybean. BMC Plant Biol. 2017, 17, 229. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Wang, Y.; Wang, L.; Yang, L.; Wang, R.; Li, X. miR172b controls the transition to autotrophic development inhibited by ABA in Arabidopsis. PLoS ONE 2013, 8, e64770. [Google Scholar] [CrossRef]
- Liu, X.; Liu, S.; Chen, X.; Prasanna, B.M.; Ni, Z.; Li, X.; He, Y.; Fan, Z.; Zhou, T. Maize miR167-ARF3/30-polyamine oxidase 1 module-regulated H2O2 production confers resistance to maize chlorotic mottle virus. Plant Physiol. 2022, 189, 1065–1082. [Google Scholar] [CrossRef]
- Yamasaki, H.; Cohen, M.F. Biological consilience of hydrogen sulfide and nitric oxide in plants: Gases of primordial earth linking plant, microbial and animal physiologies. Nitric Oxide 2016, 55/56, 91–100. [Google Scholar] [CrossRef]
- Hancock, J.T.; Whiteman, M. Hydrogen sulfide signaling: Interactions with nitric oxide and reactive oxygen species. Ann. N. Y. Acad. Sci. 2016, 1365, 5–14. [Google Scholar] [CrossRef]
- Li, Z.G.; Min, X.; Zhou, Z.H. Hydrogen Sulfide: A Signal Molecule in Plant Cross-Adaptation. Front. Plant Sci. 2016, 7, 1621. [Google Scholar] [CrossRef]
- Corpas, F.J. Hydrogen sulfide: A new warrior against abiotic stress. Trends Plant Sci. 2019, 24, 983–988. [Google Scholar] [CrossRef]
- Hancock, J.H. Hydrogen sulfide and environmental stresses. Environ. Exp. Bot. 2019, 161, 50–56. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Liu, Y.; Wang, Q.; Fei, J.; Liu, X.; Zhang, C.; Yin, Y. miRNA Sequencing Analysis in Maize Roots Treated with Neutral and Alkaline Salts. Curr. Issues Mol. Biol. 2024, 46, 8874-8889. https://doi.org/10.3390/cimb46080524
Chen Z, Liu Y, Wang Q, Fei J, Liu X, Zhang C, Yin Y. miRNA Sequencing Analysis in Maize Roots Treated with Neutral and Alkaline Salts. Current Issues in Molecular Biology. 2024; 46(8):8874-8889. https://doi.org/10.3390/cimb46080524
Chicago/Turabian StyleChen, Ziqi, Yang Liu, Qi Wang, Jianbo Fei, Xiangguo Liu, Chuang Zhang, and Yuejia Yin. 2024. "miRNA Sequencing Analysis in Maize Roots Treated with Neutral and Alkaline Salts" Current Issues in Molecular Biology 46, no. 8: 8874-8889. https://doi.org/10.3390/cimb46080524