
Molecular Analysis of High-Grade Serous Ovarian Carcinoma Exhibiting Low-Grade Serous Carcinoma and Serous Borderline Tumor

 ,
,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Information
2.2. Immunohistochemistry
2.3. DNA Extraction and Next-Generation Sequencing (NGS)
2.4. DNA Methylation Analysis
3. Results
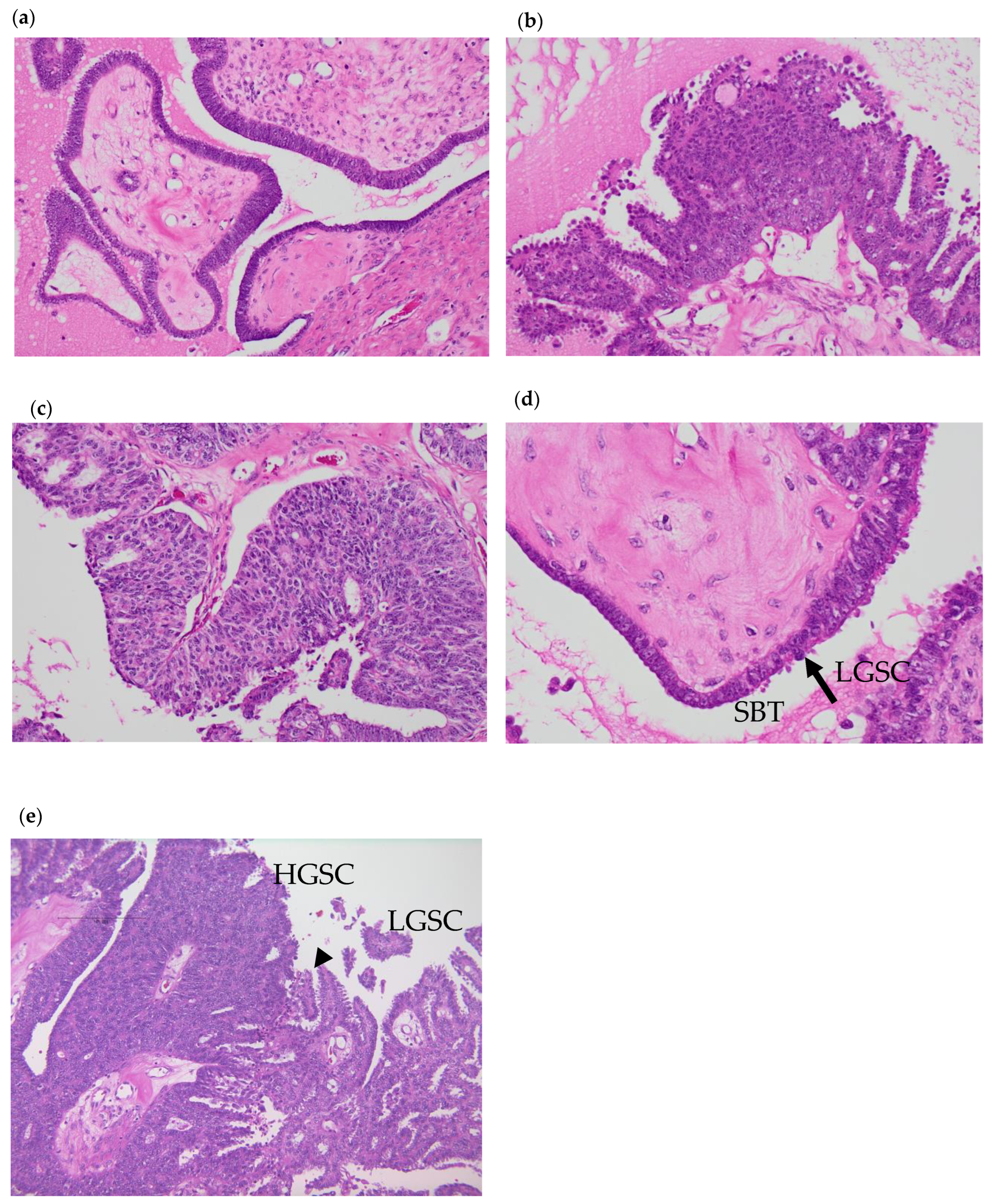
3.1. Pathological Analysis
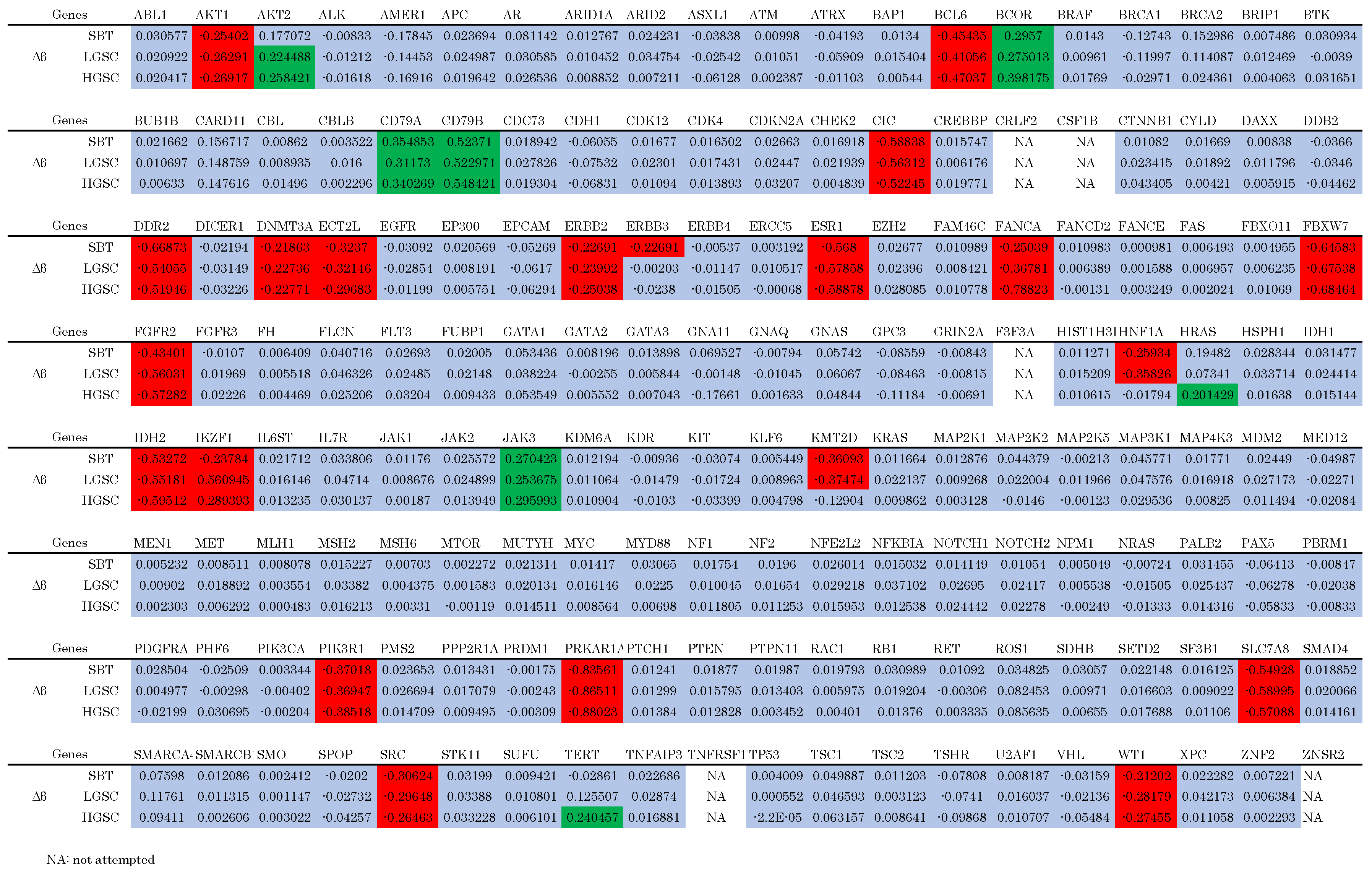
3.2. Genomic Analysis

3.3. Epigenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cancer Statistics. Cancer Information Service, National Cancer Center, Japan (National Cancer Registry, Ministry of Health, Labour and Welfare). Available online: https://ganjoho.jp/reg_stat/statistics/data/dl/en.html (accessed on 6 October 2022).
- Nagase, S. Patient Annual Report for 2022. Acta Obstet. Gynaecol. Jpn. 2022, 74, 2345–2402. [Google Scholar]
- Kurman, R.J.; Carcangiu, M.L.; Young, R.H.; Herrington, C.S. (Eds.) WHO Classification of Tumours of Female Reproductive Organs. In World Health Organization Classification of Tumours, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2014. [Google Scholar]
- Shih, I.-M.; Kurman, R.J. Ovarian Tumorigenesis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.-M.; Kurman, R.J. Molecular Pathogenesis of Ovarian Borderline Tumors: New Insights and Old Challenges. Clin. Cancer Res. 2005, 11, 7273–7279. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.-M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733. [Google Scholar] [CrossRef]
- Shih, I.-M.; Wang, Y.; Wang, T.-L. The Origin of Ovarian Cancer Species and Precancerous Landscape. Am. J. Pathol. 2021, 191, 26–39. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Cancer Genes and the Pathways They Control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Gull, N.; Jones, M.R.; Peng, P.-C.; Coetzee, S.G.; Silva, T.C.; Plummer, J.T.; Reyes, A.L.P.; Davis, B.D.; Chen, S.S.; Lawrenson, K.; et al. DNA Methylation and Transcriptomic Features Are Preserved throughout Disease Recurrence and Chemoresistance in High Grade Serous Ovarian Cancers. J. Exp. Clin. Cancer Res. 2022, 41, 232. [Google Scholar] [CrossRef]
- Shih, I.-M.; Chen, L.; Wang, C.C.; Gu, J.; Davidson, B.; Cope, L.; Kurman, R.J.; Xuan, J.; Wang, T.-L. Distinct DNA Methylation Profiles in Ovarian Serous Neoplasms and Their Implications in Ovarian Carcinogenesis. Am. J. Obstet. Gynecol. 2010, 203, 584.e1–584.e22. [Google Scholar] [CrossRef]
- Zarei, S.; Wang, Y.; Jenkins, S.M.; Voss, J.S.; Kerr, S.E.; Bell, D.A. Clinicopathologic, Immunohistochemical, and Molecular Characteristics of Ovarian Serous Carcinoma With Mixed Morphologic Features of High-Grade and Low-Grade Serous Carcinoma. Am. J. Surg. Pathol. 2020, 44, 316–328. [Google Scholar] [CrossRef]
- Kurose, S.; Nakayama, K.; Razia, S.; Ishikawa, M.; Ishibashi, T.; Yamashita, H.; Sato, S.; Sakiyama, A.; Yoshioka, S.; Kobayashi, M.; et al. Whole-Exome Sequencing of Rare Site Endometriosis-Associated Cancer. Diseases 2021, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Aimono, E.; Tanishima, S.; Imai, M.; Nagatsuma, A.K.; Hayashi, H.; Yoshimura, Y.; Nakayama, K.; Kyo, S.; Nishihara, H. Intratumoral Genomic Heterogeneity May Hinder Precision Medicine Strategies in Patients with Serous Ovarian Carcinoma. Diagnostics 2020, 10, 200. [Google Scholar] [CrossRef]
- Sawada, K.; Nakayama, K.; Nakamura, K.; Yoshimura, Y.; Razia, S.; Ishikawa, M.; Yamashita, H.; Ishibashi, T.; Sato, S.; Kyo, S. Clinical Outcomes of Genotype-Matched Therapy for Recurrent Gynecological Cancers: A Single Institutional Experience. Healthcare 2021, 9, 1395. [Google Scholar] [CrossRef] [PubMed]
- Assem, H.; Rambau, P.F.; Lee, S.; Ogilvie, T.; Sienko, A.; Kelemen, L.E.; Köbel, M. High-Grade Endometrioid Carcinoma of the Ovary: A Clinicopathologic Study of 30 Cases. Am. J. Surg. Pathol. 2018, 42, 534. [Google Scholar] [CrossRef] [PubMed]
- Köbel, M.; Kang, E.Y. The Evolution of Ovarian Carcinoma Subclassification. Cancers 2022, 14, 416. [Google Scholar] [CrossRef]
- Babaier, A.; Mal, H.; Alselwi, W.; Ghatage, P. Low-Grade Serous Carcinoma of the Ovary: The Current Status. Diagnostics 2022, 12, 458. [Google Scholar] [CrossRef]
- Vang, R.; Shih, I.-M.; Kurman, R.J. OVARIAN LOW-GRADE AND HIGH-GRADE SEROUS CARCINOMA: Pathogenesis, Clinicopathologic and Molecular Biologic Features, and Diagnostic Problems. Adv. Anat. Pathol. 2009, 16, 267–282. [Google Scholar] [CrossRef]
- Lengyel, E. Ovarian Cancer Development and Metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef]
- Rashid, S.; Arafah, M.A.; Akhtar, M. The Many Faces of Serous Neoplasms and Related Lesions of the Female Pelvis: A Review. Adv. Anat. Pathol. 2022, 29, 154–167. [Google Scholar] [CrossRef]
- Hauptmann, S.; Friedrich, K.; Redline, R.; Avril, S. Ovarian Borderline Tumors in the 2014 WHO Classification: Evolving Concepts and Diagnostic Criteria. Virchows Arch. 2017, 470, 125–142. [Google Scholar] [CrossRef]
- Murali, R.; Selenica, P.; Brown, D.N.; Cheetham, R.K.; Chandramohan, R.; Claros, N.L.; Bouvier, N.; Cheng, D.T.; Soslow, R.A.; Weigelt, B.; et al. Somatic Genetic Alterations in Synchronous and Metachronous Low-Grade Serous Tumours and High-Grade Carcinomas of the Adnexa. Histopathology 2019, 74, 638–650. [Google Scholar] [CrossRef]
- Vang, R.; Levine, D.A.; Soslow, R.A.; Zaloudek, C.; Shih, I.-M.; Kurman, R.J. Molecular Alterations of TP53 Are a Defining Feature of Ovarian High-Grade Serous Carcinoma: A Rereview of Cases Lacking TP53 Mutations in The Cancer Genome Atlas Ovarian Study. Int. J. Gynecol. Pathol. 2016, 35, 48–55. [Google Scholar] [CrossRef]
- Chui, M.H.; Xing, D.; Zeppernick, F.; Wang, Z.Q.; Hannibal, C.G.; Frederiksen, K.; Kjaer, S.K.; Cope, L.; Kurman, R.J.; Shih, I.-M.; et al. Clinicopathologic and Molecular Features of Paired Cases of Metachronous Ovarian Serous Borderline Tumor and Subsequent Serous Carcinoma. Am. J. Surg. Pathol. 2019, 43, 1462–1472. [Google Scholar] [CrossRef]
- Chui, M.H.; Momeni Boroujeni, A.; Mandelker, D.; Ladanyi, M.; Soslow, R.A. Characterization of TP53-Wildtype Tubo-Ovarian High-Grade Serous Carcinomas: Rare Exceptions to the Binary Classification of Ovarian Serous Carcinoma. Mod. Pathol. 2021, 34, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; deFazio, A.; et al. Driver Mutations in TP53 Are Ubiquitous in High Grade Serous Carcinoma of the Ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Saiki, A.Y.; Caenepeel, S. The Role of MDM2 Amplification and Overexpression in Tumorigenesis. Cold Spring Harb. Perspect. Med. 2016, 6, a026336. [Google Scholar] [CrossRef]
- Saleh, A.; Perets, R. Mutated P53 in HGSC—From a Common Mutation to a Target for Therapy. Cancers 2021, 13, 3465. [Google Scholar] [CrossRef] [PubMed]
- Boyd, C.; McCluggage, W.G. Low-Grade Ovarian Serous Neoplasms (Low-Grade Serous Carcinoma and Serous Borderline Tumor) Associated With High-Grade Serous Carcinoma or Undifferentiated Carcinoma: Report of a Series of Cases of an Unusual Phenomenon. Am. J. Surg. Pathol. 2012, 36, 368. [Google Scholar] [CrossRef]
- Inoue, M.; Takenaka, M.; Fukunaga, M.; Isonishi, S. Concurrent High-Grade Serous Carcinoma and Borderline Tumor Demonstrating Different Chemo-Sensitivity. Int. Cancer Conf. J. 2017, 6, 65–69. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| SBT | LGSC | HGSC | |
|---|---|---|---|
| p53 | wild-type | wild-type | wild-type |
| Ki67 | low | low | high |
| WT1 | positive | positive | positive |
| CK7 | positive | positive | positive |
| ER | weak positive | positive | weak positive |
| PR | partially positive | negative | partially positive |
| Mutation | VAF | ||
|---|---|---|---|
| SBT | LGSC | HGSC | |
| KRAS p.G12V | 49.2% | 56.5% | 60.3% |
| NF2 p.W184* | 59.0% | 66.6% | 81.4% |
| Chromosome | Gene | Estimated CN | ||
|---|---|---|---|---|
| SBT | LGSC | HGSC | ||
| chr11 | MEN1 | 1.11 | 0.95 | 1.27 |
| chr12 | MDM2 | 3.2 | 3.66 | 3.3 |
| chr17 | NF1 | 1.27 | 1.37 | 1.25 |
| chr22 | EP300 | 1.09 | 0.95 | 1.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, K.; Nakayama, K.; Razia, S.; Islam, S.H.; Farzana, Z.U.; Sonia, S.B.; Sasamori, H.; Yamashita, H.; Ishibashi, T.; Ishikawa, M.; et al. Molecular Analysis of High-Grade Serous Ovarian Carcinoma Exhibiting Low-Grade Serous Carcinoma and Serous Borderline Tumor. Curr. Issues Mol. Biol. 2024, 46, 9376-9385. https://doi.org/10.3390/cimb46090555
Kanno K, Nakayama K, Razia S, Islam SH, Farzana ZU, Sonia SB, Sasamori H, Yamashita H, Ishibashi T, Ishikawa M, et al. Molecular Analysis of High-Grade Serous Ovarian Carcinoma Exhibiting Low-Grade Serous Carcinoma and Serous Borderline Tumor. Current Issues in Molecular Biology. 2024; 46(9):9376-9385. https://doi.org/10.3390/cimb46090555
Chicago/Turabian StyleKanno, Kosuke, Kentaro Nakayama, Sultana Razia, Sohel Hasibul Islam, Zahan Umme Farzana, Shahataj Begum Sonia, Hiroki Sasamori, Hitomi Yamashita, Tomoka Ishibashi, Masako Ishikawa, and et al. 2024. "Molecular Analysis of High-Grade Serous Ovarian Carcinoma Exhibiting Low-Grade Serous Carcinoma and Serous Borderline Tumor" Current Issues in Molecular Biology 46, no. 9: 9376-9385. https://doi.org/10.3390/cimb46090555
APA StyleKanno, K., Nakayama, K., Razia, S., Islam, S. H., Farzana, Z. U., Sonia, S. B., Sasamori, H., Yamashita, H., Ishibashi, T., Ishikawa, M., Imamura, K., Ishikawa, N., & Kyo, S. (2024). Molecular Analysis of High-Grade Serous Ovarian Carcinoma Exhibiting Low-Grade Serous Carcinoma and Serous Borderline Tumor. Current Issues in Molecular Biology, 46(9), 9376-9385. https://doi.org/10.3390/cimb46090555