
Unraveling the Mechanisms of S100A8/A9 in Myocardial Injury and Dysfunction


Abstract
1. Introduction
2. Origin and Functions of S100A8/A9
3. The Role of S100A8/A9 in Cardiac Injury
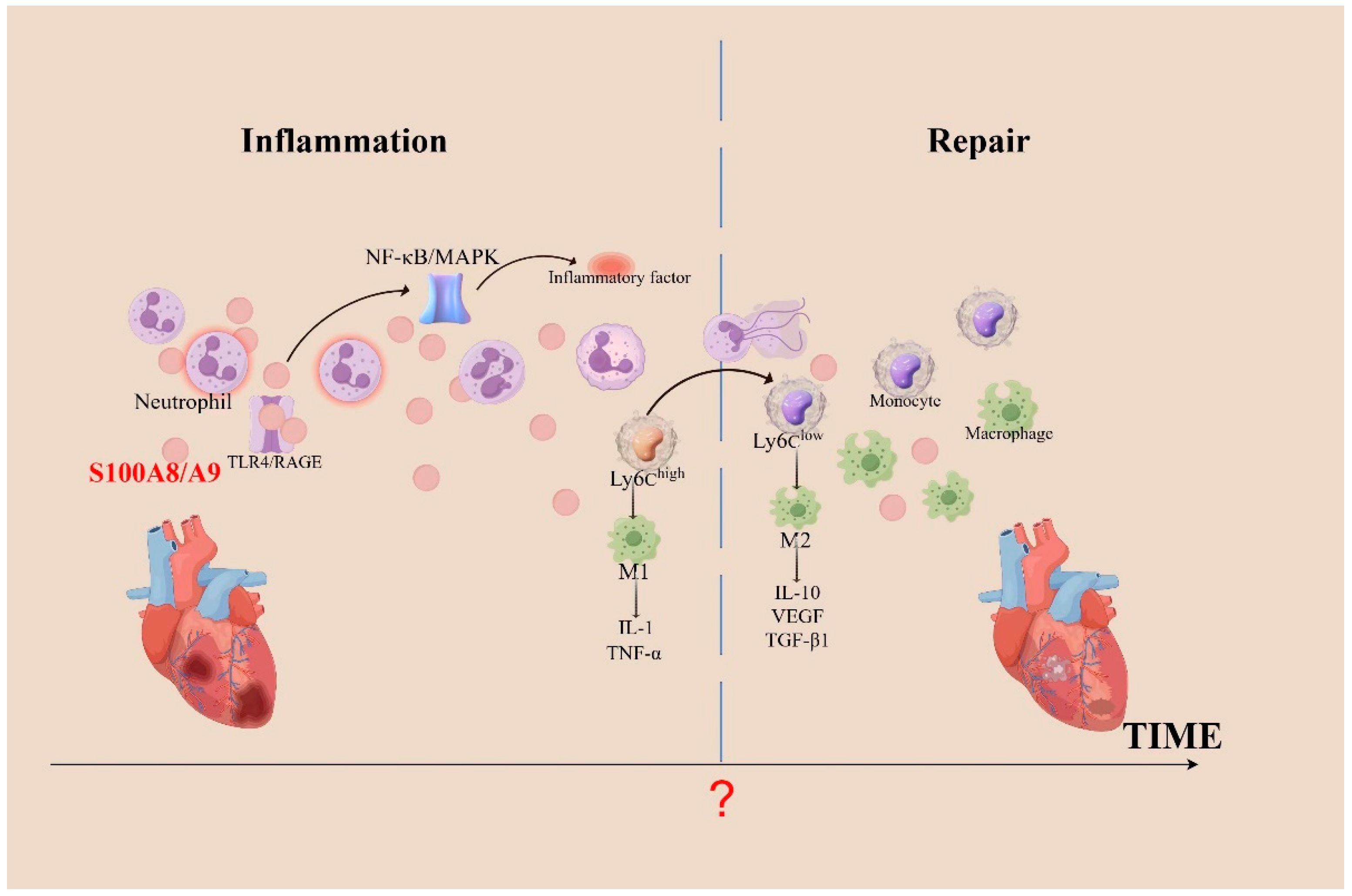
3.1. Regulating the Progression of Inflammation
3.1.1. Acting as a Mediator of Inflammation
3.1.2. Possessing Anti-Inflammatory Properties
3.2. Causing Mitochondrial Dysfunction
3.3. Modulating Myocardial Fibrosis Progression
3.4. Regulating Autophagy and Apoptosis
4. S100A8/A9: A Biomarker for Cardiovascular Disease
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Z.; Yang, Y.; Wang, X.; Yang, N.; He, L.; Wang, J.; Ping, F.; Xu, L.; Zhang, H.; Li, W.; et al. Comparative analysis of atherosclerotic cardiovascular disease burden between ages 20–54 and over 55 years: Insights from the Global Burden of Disease Study 2019. BMC Med. 2024, 22, 303. [Google Scholar] [CrossRef] [PubMed]
- Sreejit, G.; Latif, A.A.; Murphy, A.J.; Nagareddy, P.R. Emerging roles of neutrophil-borne S100A8/A9 in cardiovascular inflammation. Pharmacol. Res. 2020, 161, 105212. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Xie, Q.; Hu, T.; Yao, Q.; Zhao, J.; Wu, Q.; Tang, Q. S100A8/A9 in Myocardial Infarction: A Promising Biomarker and Therapeutic Target. Front. Cell Dev. Biol. 2020, 8, 603902. [Google Scholar] [CrossRef]
- Averill, M.M.; Kerkhoff, C.; Bornfeldt, K.E. S100A8 and S100A9 in cardiovascular biology and disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Pruenster, M.; Vogl, T.; Roth, J.; Sperandio, M. S100A8/A9: From basic science to clinical application. Pharmacol. Ther. 2016, 167, 120–131. [Google Scholar] [CrossRef]
- Moore, B.W. A soluble protein characteristic of the nervous system. Biochem. Biophys. Res. Commun. 1965, 19, 739–744. [Google Scholar] [CrossRef]
- Ceron, J.J.; Ortin-Bustillo, A.; Lopez-Martinez, M.J.; Martinez-Subiela, S.; Eckersall, P.D.; Tecles, F.; Tvarijonaviciute, A.; Munoz-Prieto, A. S-100 Proteins: Basics and Applications as Biomarkers in Animals with Special Focus on Calgranulins (S100A8, A9, and A12). Biology 2023, 12, 881. [Google Scholar] [CrossRef]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 proteins in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef]
- Sobiak, B.; Graczyk-Jarzynka, A.; Lesniak, W. Comparison of DNA Methylation and Expression Pattern of S100 and Other Epidermal Differentiation Complex Genes in Differentiating Keratinocytes. J. Cell Biochem. 2016, 117, 1092–1098. [Google Scholar] [CrossRef]
- Chen, Y.; Ouyang, Y.; Li, Z.; Wang, X.; Ma, J. S100A8 and S100A9 in Cancer. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188891. [Google Scholar] [CrossRef]
- Ahn, G.O.; Seita, J.; Hong, B.J.; Kim, Y.E.; Bok, S.; Lee, C.J.; Kim, K.S.; Lee, J.C.; Leeper, N.J.; Cooke, J.P.; et al. Transcriptional activation of hypoxia-inducible factor-1 (HIF-1) in myeloid cells promotes angiogenesis through VEGF and S100A8. Proc. Natl. Acad. Sci. USA 2014, 111, 2698–2703. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Inagaki, Y.; Kido, J.; Nagata, T. Advanced glycation end products increase expression of S100A8 and A9 via RAGE-MAPK in rat dental pulp cells. Oral. Dis. 2015, 21, 328–334. [Google Scholar] [CrossRef]
- Yonekawa, K.; Neidhart, M.; Altwegg, L.A.; Wyss, C.A.; Corti, R.; Vogl, T.; Grigorian, M.; Gay, S.; Luscher, T.F.; Maier, W. Myeloid related proteins activate Toll-like receptor 4 in human acute coronary syndromes. Atherosclerosis 2011, 218, 486–492. [Google Scholar] [CrossRef]
- Narumi, K.; Miyakawa, R.; Ueda, R.; Hashimoto, H.; Yamamoto, Y.; Yoshida, T.; Aoki, K. Proinflammatory Proteins S100A8/S100A9 Activate NK Cells via Interaction with RAGE. J. Immunol. 2015, 194, 5539–5548. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Zhang, C.; Wang, Y.; Cui, W.; Li, H.; Du, J. S100a8/a9 released by CD11b+Gr1+ neutrophils activates cardiac fibroblasts to initiate angiotensin II-Induced cardiac inflammation and injury. Hypertension 2014, 63, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Sreejit, G.; Nooti, S.K.; Jaggers, R.M.; Athmanathan, B.; Ho Park, K.; Al-Sharea, A.; Johnson, J.; Dahdah, A.; Lee, M.K.S.; Ma, J.; et al. Retention of the NLRP3 Inflammasome-Primed Neutrophils in the Bone Marrow Is Essential for Myocardial Infarction-Induced Granulopoiesis. Circulation 2022, 145, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.R.; Park, B.S.; Kim, S.Y.; Gu, A.; Kim, D.H.; Lee, J.S.; Kim, I.S. Cytokine secreted by S100A9 via TLR4 in monocytes delays neutrophil apoptosis by inhibition of caspase 9/3 pathway. Cytokine 2016, 86, 53–63. [Google Scholar] [CrossRef]
- Sreejit, G.; Abdel-Latif, A.; Athmanathan, B.; Annabathula, R.; Dhyani, A.; Noothi, S.K.; Quaife-Ryan, G.A.; Al-Sharea, A.; Pernes, G.; Dragoljevic, D.; et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation 2020, 141, 1080–1094. [Google Scholar] [CrossRef]
- Vogl, T.; Ludwig, S.; Goebeler, M.; Strey, A.; Thorey, I.S.; Reichelt, R.; Foell, D.; Gerke, V.; Manitz, M.P.; Nacken, W.; et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood 2004, 104, 4260–4268. [Google Scholar] [CrossRef] [PubMed]
- Stephan, J.R.; Yu, F.; Costello, R.M.; Bleier, B.S.; Nolan, E.M. Oxidative Post-translational Modifications Accelerate Proteolytic Degradation of Calprotectin. J. Am. Chem. Soc. 2018, 140, 17444–17455. [Google Scholar] [CrossRef]
- Chakraborty, D.; Zenker, S.; Rossaint, J.; Holscher, A.; Pohlen, M.; Zarbock, A.; Roth, J.; Vogl, T. Alarmin S100A8 Activates Alveolar Epithelial Cells in the Context of Acute Lung Injury in a TLR4-Dependent Manner. Front. Immunol. 2017, 8, 1493. [Google Scholar] [CrossRef]
- Ma, L.; Sun, P.; Zhang, J.C.; Zhang, Q.; Yao, S.L. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int. J. Mol. Med. 2017, 40, 31–38. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Gerhardt, L.M.; Tan, T.C.; Winter, C.; Holderried, T.A.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef]
- Marinkovic, G.; Larsen, H.G.; Yndigegn, T.; Szabo, I.A.; Mares, R.G.; de Camp, L.; Weiland, M.; Tomas, L.; Goncalves, I.; Nilsson, J.; et al. Inhibition of pro-inflammatory myeloid cell responses by short-term S100A9 blockade improves cardiac function after myocardial infarction. Eur. Heart J. 2019, 40, 2713–2723. [Google Scholar] [CrossRef]
- Zhang, W.; Lavine, K.J.; Epelman, S.; Evans, S.A.; Weinheimer, C.J.; Barger, P.M.; Mann, D.L. Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J. Am. Heart Assoc. 2015, 4, e001993. [Google Scholar] [CrossRef]
- Riva, M.; Kallberg, E.; Bjork, P.; Hancz, D.; Vogl, T.; Roth, J.; Ivars, F.; Leanderson, T. Induction of nuclear factor-kappaB responses by the S100A9 protein is Toll-like receptor-4-dependent. Immunology 2012, 137, 172–182. [Google Scholar] [CrossRef]
- Robinson, M.J.; Tessier, P.; Poulsom, R.; Hogg, N. The S100 family heterodimer; MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J. Biol. Chem. 2002, 277, 3658–3665. [Google Scholar] [CrossRef]
- Sager, H.B.; Kessler, T.; Schunkert, H. Monocytes and macrophages in cardiac injury and repair. J. Thorac. Dis. 2017, 9, S30–S35. [Google Scholar] [CrossRef]
- Pruenster, M.; Kurz, A.R.; Chung, K.J.; Cao-Ehlker, X.; Bieber, S.; Nussbaum, C.F.; Bierschenk, S.; Eggersmann, T.K.; Rohwedder, I.; Heinig, K.; et al. Extracellular MRP8/14 is a regulator of beta2 integrin-dependent neutrophil slow rolling and adhesion. Nat. Commun. 2015, 6, 6915. [Google Scholar] [CrossRef]
- Flynn, M.C.; Kraakman, M.J.; Tikellis, C.; Lee, M.K.S.; Hanssen, N.M.J.; Kammoun, H.L.; Pickering, R.J.; Dragoljevic, D.; Al-Sharea, A.; Barrett, T.J.; et al. Transient Intermittent Hyperglycemia Accelerates Atherosclerosis by Promoting Myelopoiesis. Circ. Res. 2020, 127, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tao, T.; Wang, H.; Zhao, H.; Lu, L.; Wu, F. Arterial Thrombosis Is Accompanied by Elevated Mitogen-Activated Protein Kinase (MAPK) and Cyclooxygenase-2 (COX-2) Expression via Toll-like Receptor 4 (TLR-4) Activation by S100A8/A9. Med. Sci. Monit. 2018, 24, 7673–7681. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Raftery, M.J.; Goyette, J.; Hsu, K.; Geczy, C.L. Oxidative modifications of S100 proteins: Functional regulation by redox. J. Leukoc. Biol. 2009, 86, 577–587. [Google Scholar] [CrossRef]
- Jia, J.; Arif, A.; Terenzi, F.; Willard, B.; Plow, E.F.; Hazen, S.L.; Fox, P.L. Target-selective protein S-nitrosylation by sequence motif recognition. Cell 2014, 159, 623–634. [Google Scholar] [CrossRef]
- Otsuka, K.; Terasaki, F.; Ikemoto, M.; Fujita, S.; Tsukada, B.; Katashima, T.; Kanzaki, Y.; Sohmiya, K.; Kono, T.; Toko, H.; et al. Suppression of inflammation in rat autoimmune myocarditis by S100A8/A9 through modulation of the proinflammatory cytokine network. Eur. J. Heart Fail. 2009, 11, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.J.; Yeh, Y.T.; Peng, F.S.; Li, A.H.; Wu, S.C. S100A8/A9 Enhances Immunomodulatory and Tissue-Repairing Properties of Human Amniotic Mesenchymal Stem Cells in Myocardial Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2021, 22, 11175. [Google Scholar] [CrossRef]
- Sun, K.; Li, Y.Y.; Jin, J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal Transduct. Target. Ther. 2021, 6, 79. [Google Scholar] [CrossRef]
- Dehn, S.; Thorp, E.B. Myeloid receptor CD36 is required for early phagocytosis of myocardial infarcts and induction of Nr4a1-dependent mechanisms of cardiac repair. FASEB J. 2018, 32, 254–264. [Google Scholar] [CrossRef]
- Zhou, Y.; Nomigni, M.T.; Gaigneaux, A.; Tolle, F.; Wright, H.L.; Bueb, J.L.; Brechard, S. miRNA-132-5p mediates a negative feedback regulation of IL-8 secretion through S100A8/A9 downregulation in neutrophil-like HL-60 cells. Front. Immunol. 2023, 14, 1274378. [Google Scholar] [CrossRef]
- Wu, M.; Xu, L.; Wang, Y.; Zhou, N.; Zhen, F.; Zhang, Y.; Qu, X.; Fan, H.; Liu, S.; Chen, Y.; et al. S100A8/A9 induces microglia activation and promotes the apoptosis of oligodendrocyte precursor cells by activating the NF-kappaB signaling pathway. Brain Res. Bull. 2018, 143, 234–245. [Google Scholar] [CrossRef]
- Schenten, V.; Plancon, S.; Jung, N.; Hann, J.; Bueb, J.L.; Brechard, S.; Tschirhart, E.J.; Tolle, F. Secretion of the Phosphorylated Form of S100A9 from Neutrophils Is Essential for the Proinflammatory Functions of Extracellular S100A8/A9. Front. Immunol. 2018, 9, 447. [Google Scholar] [CrossRef]
- Russo, A.; Schurmann, H.; Brandt, M.; Scholz, K.; Matos, A.L.L.; Grill, D.; Revenstorff, J.; Rembrink, M.; von Wulffen, M.; Fischer-Riepe, L.; et al. Alarming and Calming: Opposing Roles of S100A8/S100A9 Dimers and Tetramers on Monocytes. Adv. Sci. 2022, 9, e2201505. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yan, Z.; Fan, Y.; Fan, X.; Li, A.; Qi, Z.; Zhang, J. Cardiac repair after myocardial infarction: A two-sided role of inflammation-mediated. Front. Cardiovasc. Med. 2022, 9, 1077290. [Google Scholar] [CrossRef]
- Roger, A.J.; Munoz-Gomez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef]
- Harapas, C.R.; Idiiatullina, E.; Al-Azab, M.; Hrovat-Schaale, K.; Reygaerts, T.; Steiner, A.; Laohamonthonkul, P.; Davidson, S.; Yu, C.H.; Booty, L.; et al. Organellar homeostasis and innate immune sensing. Nat. Rev. Immunol. 2022, 22, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [PubMed]
- Li, Y.; Chen, B.; Yang, X.; Zhang, C.; Jiao, Y.; Li, P.; Liu, Y.; Li, Z.; Qiao, B.; Lau, W.B.; et al. S100a8/a9 Signaling Causes Mitochondrial Dysfunction and Cardiomyocyte Death in Response to Ischemic/Reperfusion Injury. Circulation 2019, 140, 751–764. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, Y.T.; Teng, F.; Li, H.H.; Guo, S.B. S100a8/a9 contributes to sepsis-induced cardiomyopathy by activating ERK1/2-Drp1-mediated mitochondrial fission and respiratory dysfunction. Int. Immunopharmacol. 2023, 115, 109716. [Google Scholar] [CrossRef]
- Jakobsson, G.; Papareddy, P.; Andersson, H.; Mulholland, M.; Bhongir, R.; Ljungcrantz, I.; Engelbertsen, D.; Bjorkbacka, H.; Nilsson, J.; Manea, A.; et al. Therapeutic S100A8/A9 blockade inhibits myocardial and systemic inflammation and mitigates sepsis-induced myocardial dysfunction. Crit. Care 2023, 27, 374. [Google Scholar] [CrossRef]
- Sun, S.N.; Ni, S.H.; Li, Y.; Liu, X.; Deng, J.P.; Chen, Z.X.; Li, H.; Feng, W.J.; Huang, Y.S.; Li, D.N.; et al. G-MDSCs promote aging-related cardiac fibrosis by activating myofibroblasts and preventing senescence. Cell Death Dis. 2021, 12, 594. [Google Scholar] [CrossRef]
- Volz, H.C.; Laohachewin, D.; Seidel, C.; Lasitschka, F.; Keilbach, K.; Wienbrandt, A.R.; Andrassy, J.; Bierhaus, A.; Kaya, Z.; Katus, H.A.; et al. S100A8/A9 aggravates post-ischemic heart failure through activation of RAGE-dependent NF-kappaB signaling. Basic. Res. Cardiol. 2012, 107, 250. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wu, B.; Zhao, J.; Zeng, Z.; Xuan, W.; Cao, S.; Huang, X.; Asakura, M.; Xu, D.; Bin, J.; et al. Myocardial Hypertrophic Preconditioning Attenuates Cardiomyocyte Hypertrophy and Slows Progression to Heart Failure Through Upregulation of S100A8/A9. Circulation 2015, 131, 1506–1517; discussion 1517. [Google Scholar] [CrossRef] [PubMed]
- Daseke, M.J., II; Tenkorang, M.A.A.; Chalise, U.; Konfrst, S.R.; Lindsey, M.L. Cardiac fibroblast activation during myocardial infarction wound healing: Fibroblast polarization after MI. Matrix Biol. J. Int. Soc. Matrix Biol. 2020, 91–92, 109–116. [Google Scholar] [CrossRef]
- Mouton, A.J.; Ma, Y.; Gonzalez, O.J.R.; Daseke, M.J.; Flynn, E.R.; Freeman, T.C.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol. 2019, 114, 6. [Google Scholar] [CrossRef]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef] [PubMed]
- Goldblatt, Z.E.; Cirka, H.A.; Billiar, K.L. Mechanical Regulation of Apoptosis in the Cardiovascular System. Ann. Biomed. Eng. 2021, 49, 75–97. [Google Scholar] [CrossRef]
- Dong, Y.; Chen, H.; Gao, J.; Liu, Y.; Li, J.; Wang, J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J. Mol. Cell Cardiol. 2019, 136, 27–41. [Google Scholar] [CrossRef]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.P.; et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef]
- Ghavami, S.; Eshragi, M.; Ande, S.R.; Chazin, W.J.; Klonisch, T.; Halayko, A.J.; McNeill, K.D.; Hashemi, M.; Kerkhoff, C.; Los, M. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res. 2010, 20, 314–331. [Google Scholar] [CrossRef]
- Nakatani, Y.; Yamazaki, M.; Chazin, W.J.; Yui, S. Regulation of S100A8/A9 (calprotectin) binding to tumor cells by zinc ion and its implication for apoptosis-inducing activity. Mediat. Inflamm. 2005, 2005, 280–292. [Google Scholar] [CrossRef]
- Zali, H.; Marashi, S.A.; Rezaei-Tavirani, M.; Toossi, P.; Rahmati-Roodsari, M.; Shokrgozar, M.A. On the mechanism of apoptosis-inducing activity of human calprotectin: Zinc sequestration, induction of a signaling pathway, or something else? Med. Hypotheses 2007, 68, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Bi, Q.Y.; Wang, Z.N.; Han, F.; Liu, L.M.; Song, L.B.; Li, C.Y.; Zhang, A.Q.; Ji, X.M. Qingyihuaji Formula promotes apoptosis and autophagy through inhibition of MAPK/ERK and PI3K/Akt/mTOR signaling pathway on pancreatic cancer in vivo and in vitro. J. Ethnopharmacol. 2023, 307, 116198. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, L.; Zhao, Y.; Jiang, B.; Luo, J.; Shi, D. BOS-93, a novel bromophenol derivative, induces apoptosis and autophagy in human A549 lung cancer cells via PI3K/Akt/mTOR and MAPK signaling pathway. Exp. Ther. Med. 2019, 17, 3848–3858. [Google Scholar] [CrossRef]
- Yi, W.; Zhu, R.; Hou, X.; Wu, F.; Feng, R. Integrated Analysis Reveals S100a8/a9 Regulates Autophagy and Apoptosis through the MAPK and PI3K-AKT Signaling Pathway in the Early Stage of Myocardial Infarction. Cells 2022, 11, 1911. [Google Scholar] [CrossRef]
- Zhu, H.; He, M.; Wang, Y.L.; Zhang, Y.; Dong, J.; Chen, B.Y.; Li, Y.L.; Zhou, L.J.; Du, L.J.; Liu, Y.; et al. Low-intensity pulsed ultrasound alleviates doxorubicin-induced cardiotoxicity via inhibition of S100a8/a9-mediated cardiac recruitment of neutrophils. Bioeng. Transl. Med. 2023, 8, e10570. [Google Scholar] [CrossRef]
- Shan, H.; Yu, Y.; Zhao, R. The Impact of miR-206-3p Targeting S100A9 on Hypoxia/Reoxygenation-Induced Cardiomyocyte Injury. Chin. J. Integr. Med. Cardio Cerebrovasc. Dis. 2021, 19, 4066–4071+4076. [Google Scholar]
- Shi, S.; Yi, J.L. S100A8/A9 promotes MMP-9 expression in the fibroblasts from cardiac rupture after myocardial infarction by inducing macrophages secreting TNFalpha. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3925–3935. [Google Scholar] [PubMed]
- Sakuma, M.; Tanaka, A.; Kotooka, N.; Hikichi, Y.; Toyoda, S.; Abe, S.; Taguchi, I.; Node, K.; Simon, D.I.; Inoue, T. Myeloid-related protein-8/14 in acute coronary syndrome. Int. J. Cardiol. 2017, 249, 25–31. [Google Scholar] [CrossRef]
- Langley, S.R.; Willeit, K.; Didangelos, A.; Matic, L.P.; Skroblin, P.; Barallobre-Barreiro, J.; Lengquist, M.; Rungger, G.; Kapustin, A.; Kedenko, L.; et al. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J. Clin. Investig. 2017, 127, 1546–1560. [Google Scholar] [CrossRef]
- Saenz-Pipaon, G.; Ravassa, S.; Larsen, K.L.; Martinez-Aguilar, E.; Orbe, J.; Rodriguez, J.A.; Fernandez-Alonso, L.; Gonzalez, A.; Martin-Ventura, J.L.; Paramo, J.A.; et al. Lipocalin-2 and Calprotectin Potential Prognosis Biomarkers in Peripheral Arterial Disease. Eur. J. Vasc. Endovasc. Surg. 2022, 63, 648–656. [Google Scholar] [CrossRef]
- Chalise, U.; Becirovic-Agic, M.; Daseke, M.J., 2nd; Konfrst, S.R.; Rodriguez-Paar, J.R.; Feng, D.; Salomon, J.D.; Anderson, D.R.; Cook, L.M.; Lindsey, M.L. S100A9 is a functional effector of infarct wall thinning after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H145–H155. [Google Scholar] [CrossRef] [PubMed]
- Saenz-Pipaon, G.; Martin, P.S.; Planell, N.; Maillo, A.; Ravassa, S.; Vilas-Zornoza, A.; Martinez-Aguilar, E.; Rodriguez, J.A.; Alameda, D.; Lara-Astiaso, D.; et al. Functional and transcriptomic analysis of extracellular vesicles identifies calprotectin as a new prognostic marker in peripheral arterial disease (PAD). J. Extracell. Vesicles 2020, 9, 1729646. [Google Scholar] [CrossRef] [PubMed]
- Muller, I.; Vogl, T.; Kuhl, U.; Krannich, A.; Banks, A.; Trippel, T.; Noutsias, M.; Maisel, A.S.; van Linthout, S.; Tschope, C. Serum alarmin S100A8/S100A9 levels and its potential role as biomarker in myocarditis. ESC Heart Fail. 2020, 7, 1442–1451. [Google Scholar] [CrossRef]
- Goel, R.; Nair, A.; Kabeerdoss, J.; Mohan, H.; Jeyaseelan, V.; Joseph, G.; Danda, D. Study of serial serum myeloid-related protein 8/14 as a sensitive biomarker in Takayasu arteritis: A single centre study. Rheumatol. Int. 2018, 38, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, C.; Wang, L.; Feng, X.; Chen, Y.; Zhang, R.; Cheng, Y.; Liu, Z.; Chen, Q. Identification of S100A8/A9 involved in thromboangiitis obliterans dev elopment using tandem mass tags-labeled quantitative proteomics analysis. Cell. Signal. 2024, 120, 111199. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Xie, N.; Qin, L.; Xia, K.; Fang, F.; Yang, T. Correlation of serum calprotectin level with the range of coronary lesion in patients with acute coronary syndrome. J. Cent. S. Univ. Med. Sci. 2014, 39, 912–916. [Google Scholar]
- Santilli, F.; Paloscia, L.; Liani, R.; Di Nicola, M.; Di Marco, M.; Lattanzio, S.; La Barba, S.; Pascale, S.; Mascellanti, M.; Davi, G. Circulating myeloid-related protein-8/14 is related to thromboxane-dependent platelet activation in patients with acute coronary syndrome, with and without ongoing low-dose aspirin treatment. J. Am. Heart Assoc. 2014, 3, e000903. [Google Scholar] [CrossRef]
- Jonasson, L.; Larsen, H.G.; Lundberg, A.K.; Gullstrand, B.; Bengtsson, A.A.; Schiopu, A. Stress-induced release of the S100A8/A9 alarmin is elevated in coronary artery disease patients with impaired cortisol response. Sci. Rep. 2017, 7, 17545. [Google Scholar] [CrossRef]
- Yu, S.; Li, M.; Li, Z.; Xu, P.; Yao, Z.; Qian, S.; Qian, F.; Gao, D.; Wang, H. Positive correlations between plasma BPI level and MPO-DNA and S100A8/A9 in myocardial infarction. Platelets 2022, 33, 603–611. [Google Scholar]
- Katashima, T.; Naruko, T.; Terasaki, F.; Fujita, M.; Otsuka, K.; Murakami, S.; Sato, A.; Hiroe, M.; Ikura, Y.; Ueda, M.; et al. Enhanced expression of the S100A8/A9 complex in acute myocardial infarction patients. Circ. J. 2010, 74, 741–748. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Sreejit, G.; Abo-Aly, M.; Jaggers, R.M.; Chelvarajan, L.; Johnson, J.; Pernes, G.; Athmanathan, B.; Abdel-Latif, A.; Murphy, A.J. NETosis Is Required for S100A8/A9-Induced Granulopoiesis After Myocardial Infarction. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2805–2807. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guan, M.; Zhang, X.; Ma, T.; Wu, M.; Li, Y.; Chen, X.; Zheng, Y. The Association Between S100A8/A9 and the Development of Very Late Stent Thrombosis in Patients With Acute Myocardial Infarction. Clin. Appl. Thromb. Hemost. Off. J. Int. Acad. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620943295. [Google Scholar] [CrossRef] [PubMed]
- Engelberger, R.P.; Limacher, A.; Kucher, N.; Baumann, F.; Silbernagel, G.; Benghozi, R.; Do, D.D.; Willenberg, T.; Baumgartner, I. Biological variation of established and novel biomarkers for atherosclerosis: Results from a prospective, parallel-group cohort study. Clin. Chim. Acta 2015, 447, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, G.; Dorr, M.; Sappa, P.K.; Ameling, S.; Dhople, V.; Steil, L.; Klingel, K.; Empen, K.; Beug, D.; Volker, U.; et al. Endomyocardial proteomic signature corresponding to the response of patients with dilated cardiomyopathy to immunoadsorption therapy. J. Proteom. 2017, 150, 121–129. [Google Scholar] [CrossRef]
- Gupta, A.; Qaisar, R.; Halwani, R.; Kannan, M.; Ahmad, F. TFPI and FXIII negatively and S100A8/A9 and Cystatin C positively correlate with D-dimer in COVID-19. Exp. Biol. Med. 2022, 247, 1570–1576. [Google Scholar] [CrossRef]
- Chapuis, N.; Ibrahimi, N.; Belmondo, T.; Goulvestre, C.; Berger, A.E.; Mariaggi, A.A.; Andrieu, M.; Chenevier-Gobeaux, C.; Bayle, A.; Campos, L.; et al. Dynamics of circulating calprotectin accurately predict the outcome of moderate COVID-19 patients. EBioMedicine 2022, 80, 104077. [Google Scholar] [CrossRef]
- Lofblad, L.; Hov, G.G.; Asberg, A.; Videm, V. Calprotectin and CRP as biomarkers of cardiovascular disease risk in patients with chronic kidney disease: A follow-up study at 5 and 10 years. Scand. J. Clin. Lab. Investig. 2023, 83, 258–263. [Google Scholar] [CrossRef]
- Lood, C.; Tyden, H.; Gullstrand, B.; Jonsen, A.; Kallberg, E.; Morgelin, M.; Kahn, R.; Gunnarsson, I.; Leanderson, T.; Ivars, F.; et al. Platelet-Derived S100A8/A9 and Cardiovascular Disease in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2016, 68, 1970–1980. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, B.; Pi, Q.; Zhou, G.; Cheng, Z.; Qu, C.; Wang, X.; Kong, L.; Luo, S.; Du, D.; et al. Deficiency of S100A8/A9 attenuates pulmonary microvascular leakage in septic mice. Respir. Res. 2023, 24, 288. [Google Scholar] [CrossRef]
- Wang, Q.; Long, G.; Luo, H.; Zhu, X.; Han, Y.; Shang, Y.; Zhang, D.; Gong, R. S100A8/A9: An emerging player in sepsis and sepsis-induced organ injury. Biomed. Pharmacother. 2023, 168, 115674. [Google Scholar] [CrossRef]
- Muller, I.; Vogl, T.; Pappritz, K.; Miteva, K.; Savvatis, K.; Rohde, D.; Most, P.; Lassner, D.; Pieske, B.; Kuhl, U.; et al. Pathogenic Role of the Damage-Associated Molecular Patterns S100A8 and S100A9 in Coxsackievirus B3-Induced Myocarditis. Circ. Heart Fail. 2017, 10, e004125. [Google Scholar] [CrossRef] [PubMed]
- Kraakman, M.J.; Lee, M.K.; Al-Sharea, A.; Dragoljevic, D.; Barrett, T.J.; Montenont, E.; Basu, D.; Heywood, S.; Kammoun, H.L.; Flynn, M.; et al. Neutrophil-derived S100 calcium-binding proteins A8/A9 promote reticulated thrombocytosis and atherogenesis in diabetes. J. Clin. Investig. 2017, 127, 2133–2147. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Pestrak, M.J.; Wu, Q.; Ahumada, O.S.; Dellos-Nolan, S.; Saljoughian, N.; Shukla, R.K.; Mitchem, C.F.; Nagareddy, P.R.; Ganesan, L.P.; et al. Pseudomonas aeruginosa pulmonary infection results in S100A8/A9-dependent cardiac dysfunction. PLoS Pathog. 2023, 19, e1011573. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, J.; Wu, M.; Kang, L.; Xu, B. The effector cells and cellular mediators of immune system involved in cardiac inflammation and fibrosis after myocardial infarction. J. Cell Physiol. 2020, 235, 8996–9004. [Google Scholar] [CrossRef]
- Ghavami, S.; Rashedi, I.; Dattilo, B.M.; Eshraghi, M.; Chazin, W.J.; Hashemi, M.; Wesselborg, S.; Kerkhoff, C.; Los, M. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J. Leukoc. Biol. 2008, 83, 1484–1492. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Murata, H.; Aoyama, Y.; Hibino, T.; Putranto, E.W.; Ruma, I.M.; Inoue, Y.; Sakaguchi, Y.; Yamamoto, K.; Kinoshita, R.; et al. DNAX-activating protein 10 (DAP10) membrane adaptor associates with receptor for advanced glycation end products (RAGE) and modulates the RAGE-triggered signaling pathway in human keratinocytes. J. Biol. Chem. 2014, 289, 23389–23402. [Google Scholar] [CrossRef]
- Kwon, C.H.; Moon, H.J.; Park, H.J.; Choi, J.H.; Park, D.Y. S100A8 and S100A9 promotes invasion and migration through p38 mitogen-activated protein kinase-dependent NF-kappaB activation in gastric cancer cells. Mol. Cells 2013, 35, 226–234. [Google Scholar] [CrossRef]
- Kologrivova, I.; Shtatolkina, M.; Suslova, T.; Ryabov, V. Cells of the Immune System in Cardiac Remodeling: Main Players in Resolution of Inflammation and Repair After Myocardial Infarction. Front. Immunol. 2021, 12, 664457. [Google Scholar] [CrossRef]
- Tan, Y.; Bao, X.; Li, Y.; Song, G.; Lu, H.; Sun, X.; Gu, R.; Kang, L.; Xu, B. Colchicine Attenuates Microvascular Obstruction after Myocardial Ischemia-Reperfusion Injury by Inhibiting the Proliferation of Neutrophil in Bone Marrow. Cardiovasc. Drugs Ther. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Tousif, S.; Singh, A.P.; Umbarkar, P.; Galindo, C.; Wheeler, N.; Cora, A.T.; Zhang, Q.; Prabhu, S.D.; Lal, H. Ponatinib Drives Cardiotoxicity by S100A8/A9-NLRP3-IL-1beta Mediated Inflammation. Circ. Res. 2023, 132, 267–289. [Google Scholar] [CrossRef]
- Chen, Y.C.; Smith, M.; Ying, Y.L.; Makridakis, M.; Noonan, J.; Kanellakis, P.; Rai, A.; Salim, A.; Murphy, A.; Bobik, A.; et al. Quantitative proteomic landscape of unstable atherosclerosis identifies molecular signatures and therapeutic targets for plaque stabilization. Commun. Biol. 2023, 6, 265. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhao, Y.; Su, M.; Sun, L.; Chen, M.; Zhang, Z.; Ilyas, I.; Wang, Z.; Little, P.J.; Wang, L.; et al. A proteome-wide screen identifies the calcium binding proteins, S100A8/S100A9, as clinically relevant therapeutic targets in aortic dissection. Pharmacol. Res. 2024, 199, 107029. [Google Scholar] [CrossRef]
- Cai, X.; Hong, L.; Liu, Y.; Huang, X.; Lai, H.; Shao, L. Salmonella pathogenicity island 1 knockdown confers protection against myocardial fibrosis and inflammation in uremic cardiomyopathy via down-regulation of S100 Calcium Binding Protein A8/A9 transcription. Ren. Fail. 2022, 44, 1819–1832. [Google Scholar] [CrossRef]
- Marinkovic, G.; Koenis, D.S.; de Camp, L.; Jablonowski, R.; Graber, N.; de Waard, V.; de Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ. Res. 2020, 127, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. S100A8/A9 as a therapeutic target in myocardial infarction: Cellular mechanisms, molecular interactions, and translational challenges. Eur. Heart J. 2019, 40, 2724–2726. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Major Methods | Diseases | Biomarker Effects |
|---|---|---|
| Laboratory | ||
| Immunohistochemistry | Acute coronary syndrome (ACS) | Associated with thrombus formation [69] |
| Proteomic comparison Immunohistochemistry and qPCR | Coronary artery disease (CAD) | Improved risk prediction and diagnostics [70] Useful for risk stratification in advanced cases [71] |
| Neutrophil proteome and echocardiography assessments | Myocardial infarction (MI) | A functional biomarker for infarct wall thinning [72] |
| Functional and transcriptomic analyses of extracellular vesicles | Peripheral arterial disease (PAD) | Associated with increased amputation risk [73] |
| Specific ELISAs | Lymphocytic myocarditis (MC) | Served as an additional diagnostic tool [74] |
| Commercial ELISA | Takayasu arteritis (TA) | Prognostic implications [75] |
| Quantitative proteomic analysis | Thromboangiitis obliterans (TAO) | Implicated in TAO development [76] |
| Clinical | ||
| Short-term prognostic value of S100A8/A0 serum levels was determined Plasma MRP-8/14 and urinary 11-dehydro-TXB2 levels in patients were evaluated | Acute coronary syndrome (ACS) | Correlated with the number of coronary lesion branches [77] A target to test different antiplatelet strategies in ACS [78] |
| The association between S100A8/A9 release and anti-inflammatory glucocorticoid secretion parameters was studied | Coronary artery disease (CAD) | May be associated with dysregulated cortisol secretion [79] |
| The correlation between plasma BPI levels and circulating inflammatory biomarker levels was explored Serum S100A8/A9 levels were serially measured Various markers of NETosis were measured Serum concentrations of S100A8/A9 and high-sensitivity C-reactive protein (hs-CRP) were assessed and compared | Myocardial infarction (MI) | Positively correlated with plasma BPI levels [80] Implicated in the pathophysiology of AMI [3,81] Reducing S100A8/A9 release could reduce post-MI inflammation [82] Related to the development of very late stent thrombosis (VLST) [83] |
| Blood samples were collected for biomarker calculation | Peripheral arterial disease (PAD) | Both higher mean levels and a favorable variation profile [84] |
| Differences in the myocardial proteome were evaluated | Dilated cardiomyopathy (DCM) | Developed as a classifier [85] |
| The distribution of platelet S100A8/A9 protein, etc., was detected | Cardiovascular complications of other diseases | Strongly and positively correlated with myoglobin, etc. [86,87,88,89,90,91,92,93,94] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Wang, Y.; Ning, K.; Bao, Y. Unraveling the Mechanisms of S100A8/A9 in Myocardial Injury and Dysfunction. Curr. Issues Mol. Biol. 2024, 46, 9707-9720. https://doi.org/10.3390/cimb46090577
Xu Y, Wang Y, Ning K, Bao Y. Unraveling the Mechanisms of S100A8/A9 in Myocardial Injury and Dysfunction. Current Issues in Molecular Biology. 2024; 46(9):9707-9720. https://doi.org/10.3390/cimb46090577
Chicago/Turabian StyleXu, Yuanbo, Yixuan Wang, Ke Ning, and Yimin Bao. 2024. "Unraveling the Mechanisms of S100A8/A9 in Myocardial Injury and Dysfunction" Current Issues in Molecular Biology 46, no. 9: 9707-9720. https://doi.org/10.3390/cimb46090577
APA StyleXu, Y., Wang, Y., Ning, K., & Bao, Y. (2024). Unraveling the Mechanisms of S100A8/A9 in Myocardial Injury and Dysfunction. Current Issues in Molecular Biology, 46(9), 9707-9720. https://doi.org/10.3390/cimb46090577