
Immune Characteristics and Immunotherapy of HIV-Associated Lymphoma


Abstract

1. Introduction
2. The Pathogenesis of HIV-Associated Lymphoma
2.1. Direct Carcinogenesis
2.2. Indirect Carcinogenesis
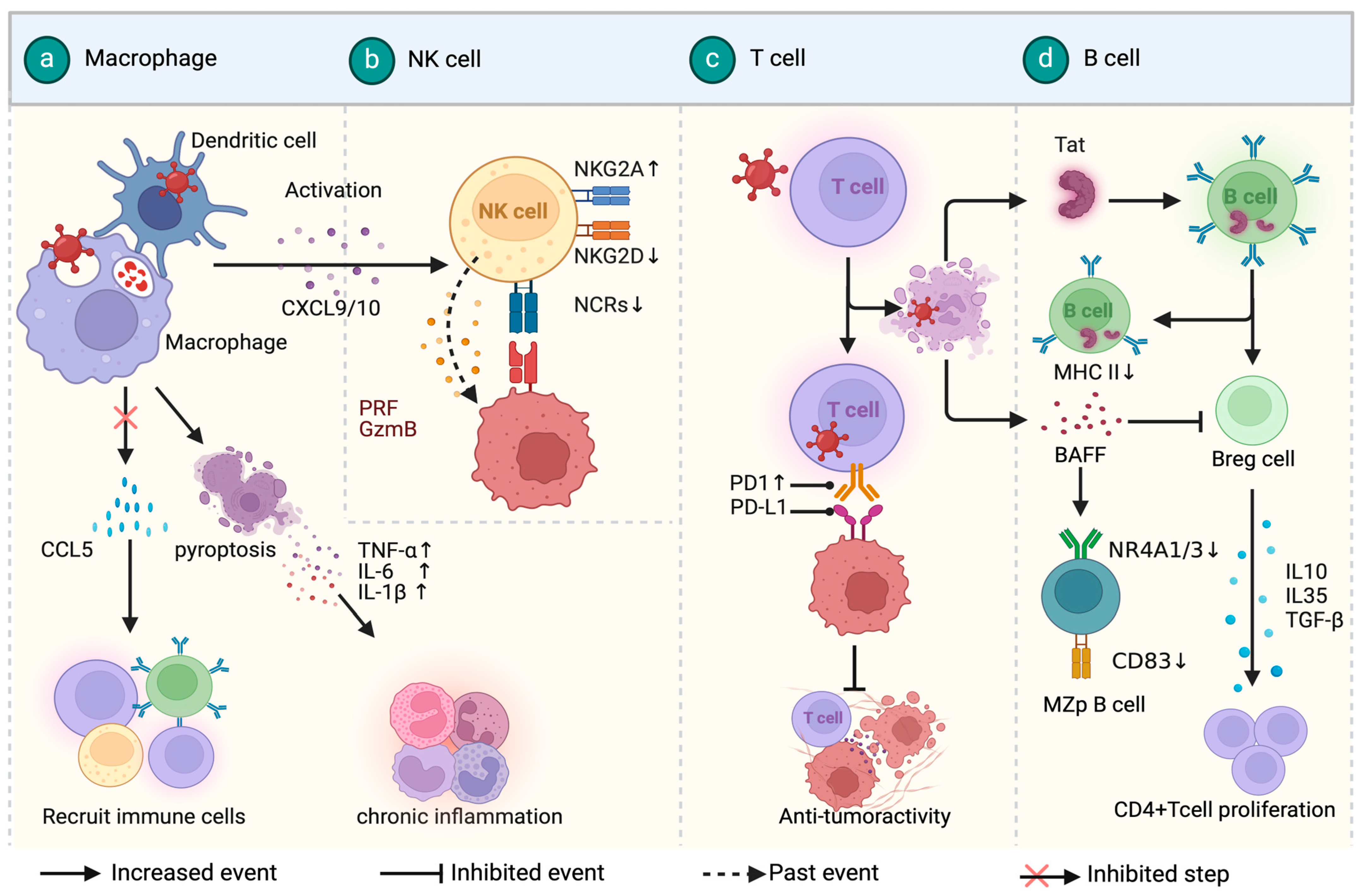
3. Immune Microenvironment in HIV-Associated Lymphoma
3.1. HIV-Associated Diffuse Large B-Cell Lymphoma
3.2. HIV-Associated Burkitt Lymphoma
3.3. HIV-Associated Hodgkin Lymphoma
3.4. Other Rare HIV-Associated Lymphomas
4. Immunotherapy of HIV-Associated Lymphomas
4.1. Monoclonal Antibody Therapies
4.2. Immune Checkpoint Inhibitors
4.3. Adoptive Cellular Immunotherapies

{kind=link}
{kind=link}
{kind=link}
| Drug | Research Type | Lymphoma Subtype | Treatment Options | Outcomes | Reference |
|---|---|---|---|---|---|
| Nivolumab (Nivo) | Case report | Relapsed/refractory classical Hodgkin lymphoma | Nivo 3 mg/kg q 2 wk | Partial remission was achieved after 5 months (10 doses) of Navulizumab treatment | [91] |
| Nivolumab (Nivo) | Prospective cohort study | Relapsed/refractory classical Hodgkin lymphoma | Nivo 3 mg/kg q 2 wk | Incidence of infectious complications was 10% with the median time of onset—98 days; OS at 1 year after first Nivo administration was 96.5% | [92] |
| Nivolumab Ipilimumab (Ipi) | A phase I study (NCT02408861) | Classical Hodgkin lymphoma | Nivo 3 mg/kg q 2 wk. Nivo 240 mg q 2 wk + Ipi 1 mg/kg q 6 wk | NA | [86] |
| Pembrolizumab (Pemb) | Retrospective study | Diffuse large B-cell lymphoma; primary effusion lymphoma; plasmablastic lymphoma | Pemb 200 mg q 3 wk. Pemb 200 mg q 4 wk + pomalidomide 4 mg q.d. | PFS was 4.1 months; OS was 14.7 months; there were four irAEs, all CTCAEv5 grade 2–3; no irAEs occurred in patients receiving the combination of pemb and pomalidomide. | [93] |
| Pembrolizumab (Pemb) | A phase I study (NCT02595866) | Non- Hodgkin lymphoma | Pemb 200 mg q 3 wk | Partial response in 2 participants with NHL, 1 participant with DLBCL, 1 participant with primary effusion lymphoma | [94] |
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carbone, A.; Vaccher, E.; Gloghini, A. Hematologic cancers in individuals infected by HIV. Blood 2022, 139, 995–1012. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Isaguliants, M.; Bayurova, E.; Avdoshina, D.; Kondrashova, A.; Chiodi, F.; Palefsky, J.M. Oncogenic Effects of HIV-1 Proteins, Mechanisms Behind. Cancers 2021, 13, 305. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, Z.; Zeng, D.; Song, S.; Yang, Y.; Wang, A.; Xu, J.; Guo, W.; Wu, M.; Shi, Y.; et al. High expression of HIV-1 matrix protein p17 in both lymphoma and lymph node tissues of AIDS patients. Pathol. Res. Pract. 2022, 237, 154061. [Google Scholar] [CrossRef]
- Carroll, V.A.; Lafferty, M.K.; Marchionni, L.; Bryant, J.L.; Gallo, R.C.; Garzino-Demo, A. Expression of HIV-1 matrix protein p17 and association with B-cell lymphoma in HIV-1 transgenic mice. Proc. Natl. Acad. Sci. USA 2016, 113, 13168–13173. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, A.; Caccuri, F.; Filippini, F.; Ravelli, C.; Caruso, A. Binding to PI(4,5)P(2) is indispensable for secretion of B-cell clonogenic HIV-1 matrix protein p17 variants. J. Biol. Chem. 2021, 297, 100934. [Google Scholar] [CrossRef]
- Giagulli, C.; Caccuri, F.; Zorzan, S.; Bugatti, A.; Zani, A.; Filippini, F.; Manocha, E.; D’Ursi, P.; Orro, A.; Dolcetti, R.; et al. B-cell clonogenic activity of HIV-1 p17 variants is driven by PAR1-mediated EGF transactivation. Cancer Gene Ther. 2021, 28, 649–666. [Google Scholar] [CrossRef]
- Alves de Souza Rios, L.; Mapekula, L.; Mdletshe, N.; Chetty, D.; Mowla, S. HIV-1 Transactivator of Transcription (Tat) Co-operates with AP-1 Factors to Enhance c-MYC Transcription. Front. Cell Dev. Biol. 2021, 9, 693706. [Google Scholar] [CrossRef]
- Valyaeva, A.A.; Tikhomirova, M.A.; Potashnikova, D.M.; Bogomazova, A.N.; Snigiryova, G.P.; Penin, A.A.; Logacheva, M.D.; Arifulin, E.A.; Shmakova, A.A.; Germini, D.; et al. Ectopic expression of HIV-1 Tat modifies gene expression in cultured B cells: Implications for the development of B-cell lymphomas in HIV-1-infected patients. PeerJ 2022, 10, e13986. [Google Scholar] [CrossRef]
- Sall, F.B.; El Amine, R.; Markozashvili, D.; Tsfasman, T.; Oksenhendler, E.; Lipinski, M.; Vassetzky, Y.; Germini, D. HIV-1 Tat protein induces aberrant activation of AICDA in human B-lymphocytes from peripheral blood. J. Cell. Physiol. 2019, 234, 15678–15685. [Google Scholar] [CrossRef]
- Akbay, B.; Germini, D.; Bissenbaev, A.K.; Musinova, Y.R.; Sheval, E.V.; Vassetzky, Y.; Dokudovskaya, S. HIV-1 Tat Activates Akt/mTORC1 Pathway and AICDA Expression by Downregulating Its Transcriptional Inhibitors in B Cells. Int. J. Mol. Sci. 2021, 22, 1588. [Google Scholar] [CrossRef] [PubMed]
- Germini, D.; Tsfasman, T.; Klibi, M.; El-Amine, R.; Pichugin, A.; Iarovaia, O.V.; Bilhou-Nabera, C.; Subra, F.; Bou Saada, Y.; Sukhanova, A.; et al. HIV Tat induces a prolonged MYC relocalization next to IGH in circulating B-cells. Leukemia 2017, 31, 2515–2522. [Google Scholar] [CrossRef] [PubMed]
- Mdletshe, N.; Nel, A.; Shires, K.; Mowla, S. HIV Nef enhances the expression of oncogenic c-MYC and activation-induced cytidine deaminase in Burkitt lymphoma cells, promoting genomic instability. Infect. Agents Cancer 2020, 15, 54. [Google Scholar] [CrossRef]
- De Mel, S.; Hue, S.S.; Jeyasekharan, A.D.; Chng, W.J.; Ng, S.B. Molecular pathogenic pathways in extranodal NK/T cell lymphoma. J. Hematol. Oncol. 2019, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Kim, T.S.; Zeng, Y.; Mikolaj, M.; Il Ahn, J.; Alam, M.S.; Monnie, C.M.; Shi, V.; Zhou, M.; Chun, T.W.; et al. Centrosome amplification and aneuploidy driven by the HIV-1-induced Vpr•VprBP•Plk4 complex in CD4+ T cells. Nat. Commun. 2024, 15, 2017. [Google Scholar] [CrossRef]
- Mellors, J.W.; Guo, S.; Naqvi, A.; Brandt, L.D.; Su, L.; Sun, Z.; Joseph, K.W.; Demirov, D.; Halvas, E.K.; Butcher, D.; et al. Insertional activation of STAT3 and LCK by HIV-1 proviruses in T cell lymphomas. Sci. Adv. 2021, 7, eabi8795. [Google Scholar] [CrossRef]
- Armani-Tourret, M.; Bone, B.; Tan, T.S.; Sun, W.; Bellefroid, M.; Struyve, T.; Louella, M.; Yu, X.G.; Lichterfeld, M. Immune targeting of HIV-1 reservoir cells: A path to elimination strategies and cure. Nat. Rev. Microbiol. 2024, 22, 328–344. [Google Scholar] [CrossRef]
- Veenhuis, R.T.; Abreu, C.M.; Shirk, E.N.; Gama, L.; Clements, J.E. HIV replication and latency in monocytes and macrophages. Semin. Immunol. 2021, 51, 101472. [Google Scholar] [CrossRef]
- Sáez-Cirión, A.; Sereti, I. Immunometabolism and HIV-1 pathogenesis: Food for thought. Nat. Rev. Immunol. 2021, 21, 5–19. [Google Scholar] [CrossRef]
- Singh, H.; Jadhav, S.; Arif Khan, A.; Aggarwal, S.K.; Choudhari, R.; Verma, S.; Aggarwal, S.; Gupta, V.; Singh, A.; Nain, S.; et al. APOBEC3, TRIM5α, and BST2 polymorphisms in healthy individuals of various populations with special references to its impact on HIV transmission. Microb. Pathog. 2022, 162, 105326. [Google Scholar] [CrossRef]
- Singh, H.; Samani, D.; Ghate, M.V.; Gangakhedkar, R.R. Impact of cellular restriction gene (TRIM5α, BST-2) polymorphisms on the acquisition of HIV-1 and disease progression. J. Gene Med. 2018, 20, e3004. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Lê-Bury, G.; Niedergang, F. Defective Phagocytic Properties of HIV-Infected Macrophages: How Might They Be Implicated in the Development of Invasive Salmonella Typhimurium? Front. Immunol. 2018, 9, 531. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, G.; Yao, Z.Q.; Moorman, J.P.; Ning, S. MicroRNA regulation of viral immunity, latency, and carcinogenesis of selected tumor viruses and HIV. Rev. Med. Virol. 2015, 25, 320–341. [Google Scholar] [CrossRef] [PubMed]
- Adu-Gyamfi, C.G.; Savulescu, D.; George, J.A.; Suchard, M.S. Indoleamine 2, 3-Dioxygenase-Mediated Tryptophan Catabolism: A Leading Star or Supporting Act in the Tuberculosis and HIV Pas-de-Deux? Front. Cell. Infect. Microbiol. 2019, 9, 372. [Google Scholar] [CrossRef]
- Planès, R.; Bahraoui, E. HIV-1 Tat protein induces the production of IDO in human monocyte derived-dendritic cells through a direct mechanism: Effect on T cells proliferation. PLoS ONE 2013, 8, e74551. [Google Scholar] [CrossRef]
- Favre, D.; Mold, J.; Hunt, P.W.; Kanwar, B.; Loke, P.; Seu, L.; Barbour, J.D.; Lowe, M.M.; Jayawardene, A.; Aweeka, F.; et al. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci. Transl. Med. 2010, 2, 32ra36. [Google Scholar] [CrossRef] [PubMed]
- Terness, P.; Bauer, T.M.; Röse, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: Mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef]
- Judge, C.J.; Kostadinova, L.; Sherman, K.E.; Butt, A.A.; Falck-Ytter, Y.; Funderburg, N.T.; Landay, A.L.; Lederman, M.M.; Sieg, S.F.; Sandberg, J.K.; et al. CD56(bright) NK IL-7Rα expression negatively associates with HCV level, and IL-7-induced NK function is impaired during HCV and HIV infections. J. Leukoc. Biol. 2017, 102, 171–184. [Google Scholar] [CrossRef]
- Lucar, O.; Reeves, R.K.; Jost, S. A Natural Impact: NK Cells at the Intersection of Cancer and HIV Disease. Front. Immunol. 2019, 10, 1850. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, Y.; Lu, J.; Chen, Y.F.; Hu, H.Y.; Xu, X.Q.; Fu, G.F. Changes in NK Cell Subsets and Receptor Expressions in HIV-1 Infected Chronic Patients and HIV Controllers. Front. Immunol. 2021, 12, 792775. [Google Scholar] [CrossRef] [PubMed]
- Fromentin, R.; Bakeman, W.; Lawani, M.B.; Khoury, G.; Hartogensis, W.; DaFonseca, S.; Killian, M.; Epling, L.; Hoh, R.; Sinclair, E.; et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS Pathog. 2016, 12, e1005761. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, R.; Gordon, A.; Zemp, F.J.; Kumar, M.; Sarkar, S.; Luchman, H.A.; Bellail, A.C.; Hao, C.; Mahoney, D.J.; Dunn, J.F.; et al. PD-1 independent of PD-L1 ligation promotes glioblastoma growth through the NFκB pathway. Sci. Adv. 2021, 7, eabh2148. [Google Scholar] [CrossRef] [PubMed]
- Demers, K.R.; Makedonas, G.; Buggert, M.; Eller, M.A.; Ratcliffe, S.J.; Goonetilleke, N.; Li, C.K.; Eller, L.A.; Rono, K.; Maganga, L.; et al. Temporal Dynamics of CD8+ T Cell Effector Responses during Primary HIV Infection. PLoS Pathog. 2016, 12, e1005805. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhang, Z.N.; Yin, L.B.; Fu, Y.J.; Jiang, Y.J.; Shang, H. Reduced eIF3d accelerates HIV disease progression by attenuating CD8+ T cell function. J. Transl. Med. 2019, 17, 167. [Google Scholar] [CrossRef]
- Shmakova, A.; Hugot, C.; Kozhevnikova, Y.; Schwager Karpukhina, A.; Tsimailo, I.; Gérard, L.; Boutboul, D.; Oksenhendler, E.; Szewczyk-Roszczenko, O.; Roszczenko, P.; et al. Chronic HIV-1 Tat action induces HLA-DR downregulation in B cells: A mechanism for lymphoma immune escape in people living with HIV. J. Med. Virol. 2024, 96, e29423. [Google Scholar] [CrossRef] [PubMed]
- Doyon-Laliberté, K.; Aranguren, M.; Poudrier, J.; Roger, M. Marginal Zone B-Cell Populations and Their Regulatory Potential in the Context of HIV and Other Chronic Inflammatory Conditions. Int. J. Mol. Sci. 2022, 23, 3372. [Google Scholar] [CrossRef] [PubMed]
- Doyon-Laliberté, K.; Aranguren, M.; Byrns, M.; Chagnon-Choquet, J.; Paniconi, M.; Routy, J.P.; Tremblay, C.; Quintal, M.C.; Brassard, N.; Kaufmann, D.E.; et al. Excess BAFF Alters NR4As Expression Levels and Breg Function of Human Precursor-like Marginal Zone B-Cells in the Context of HIV-1 Infection. Int. J. Mol. Sci. 2022, 23, 15142. [Google Scholar] [CrossRef]
- Münz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef]
- Lurain, K.; Ramaswami, R.; Yarchoan, R. The role of viruses in HIV-associated lymphomas. Semin. Hematol. 2022, 59, 183–191. [Google Scholar] [CrossRef]
- McHugh, D.; Myburgh, R.; Caduff, N.; Spohn, M.; Kok, Y.L.; Keller, C.W.; Murer, A.; Chatterjee, B.; Rühl, J.; Engelmann, C.; et al. EBV renders B cells susceptible to HIV-1 in humanized mice. Life Sci. Alliance 2020, 3, e202000640. [Google Scholar] [CrossRef]
- Katsuya, H.; Cook, L.B.M.; Rowan, A.G.; Melamed, A.; Turpin, J.; Ito, J.; Islam, S.; Miyazato, P.; Jek Yang Tan, B.; Matsuo, M.; et al. Clonality of HIV-1- and HTLV-1-Infected Cells in Naturally Coinfected Individuals. J. Infect. Dis. 2022, 225, 317–326. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, X.; Wang, X. Targeting the tumor microenvironment in B-cell lymphoma: Challenges and opportunities. J. Hematol. Oncol. 2021, 14, 125. [Google Scholar] [CrossRef]
- Taylor, J.G.; Liapis, K.; Gribben, J.G. The role of the tumor microenvironment in HIV-associated lymphomas. Biomark. Med. 2015, 9, 473–482. [Google Scholar] [CrossRef]
- Hernández-Ramírez, R.U.; Qin, L.; Lin, H.; Leyden, W.; Neugebauer, R.S.; Althoff, K.N.; Achenbach, C.J.; Hessol, N.A.; D’Souza, G.; Gebo, K.A.; et al. Association of immunosuppression and HIV viraemia with non-Hodgkin lymphoma risk overall and by subtype in people living with HIV in Canada and the USA: A multicentre cohort study. Lancet HIV 2019, 6, e240–e249. [Google Scholar] [CrossRef]
- Gopal, S.; Patel, M.R.; Yanik, E.L.; Cole, S.R.; Achenbach, C.J.; Napravnik, S.; Burkholder, G.A.; Reid, E.G.; Rodriguez, B.; Deeks, S.G.; et al. Temporal trends in presentation and survival for HIV-associated lymphoma in the antiretroviral therapy era. J. Natl. Cancer Inst. 2013, 105, 1221–1229. [Google Scholar] [CrossRef]
- Chapman, J.R.; Bouska, A.C.; Zhang, W.; Alderuccio, J.P.; Lossos, I.S.; Rimsza, L.M.; Maguire, A.; Yi, S.; Chan, W.C.; Vega, F.; et al. EBV-positive HIV-associated diffuse large B cell lymphomas are characterized by JAK/STAT (STAT3) pathway mutations and unique clinicopathologic features. Br. J. Haematol. 2021, 194, 870–878. [Google Scholar] [CrossRef]
- Nowakowski, G.S.; Feldman, T.; Rimsza, L.M.; Westin, J.R.; Witzig, T.E.; Zinzani, P.L. Integrating precision medicine through evaluation of cell of origin in treatment planning for diffuse large B-cell lymphoma. Blood Cancer J. 2019, 9, 48. [Google Scholar] [CrossRef]
- Besson, C.; Lancar, R.; Prevot, S.; Algarte-Genin, M.; Delobel, P.; Bonnet, F.; Meyohas, M.C.; Partisani, M.; Oberic, L.; Gabarre, J.; et al. Outcomes for HIV-associated diffuse large B-cell lymphoma in the modern combined antiretroviral therapy era. Aids 2017, 31, 2493–2501. [Google Scholar] [CrossRef]
- Baptista, M.J.; Tapia, G.; Morgades, M.; Muncunill, J.; Muñoz-Marmol, A.M.; Montoto, S.; Gribben, J.G.; Calaminici, M.; Martinez, A.; Gonzalez-Farre, B.; et al. Using the Lymph2Cx assay for assessing cell-of-origin subtypes of HIV-related diffuse large B-cell lymphoma. Leuk. Lymphoma 2019, 60, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Capello, D.; Scandurra, M.; Poretti, G.; Rancoita, P.M.; Mian, M.; Gloghini, A.; Deambrogi, C.; Martini, M.; Rossi, D.; Greiner, T.C.; et al. Genome wide DNA-profiling of HIV-related B-cell lymphomas. Br. J. Haematol. 2010, 148, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sun, L.; Dai, Y.; Zhang, L.; Yang, K.; Han, X.; Ding, X.; Gao, H.; Zhou, X.; Wang, P. Clinical pathology of primary central nervous system lymphoma in HIV-positive patients—A 41 Chinese patients retrospective study. Ann. Diagn. Pathol. 2023, 63, 152108. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Silverberg, M.J.; Martínez-Maza, O.; Chi, M.; Abrams, D.I.; Haque, R.; Zha, H.D.; McGuire, M.; Xu, L.; Said, J. Epstein-Barr virus infection and expression of B-cell oncogenic markers in HIV-related diffuse large B-cell Lymphoma. Clin. Cancer Res. 2012, 18, 4702–4712. [Google Scholar] [CrossRef] [PubMed]
- Liapis, K.; Clear, A.; Owen, A.; Coutinho, R.; Greaves, P.; Lee, A.M.; Montoto, S.; Calaminici, M.; Gribben, J.G. The microenvironment of AIDS-related diffuse large B-cell lymphoma provides insight into the pathophysiology and indicates possible therapeutic strategies. Blood 2013, 122, 424–433. [Google Scholar] [CrossRef]
- Baptista, M.J.; Tapia, G.; Muñoz-Marmol, A.M.; Muncunill, J.; Garcia, O.; Montoto, S.; Gribben, J.G.; Calaminici, M.; Martinez, A.; Veloza, L.; et al. Genetic and phenotypic characterisation of HIV-associated aggressive B-cell non-Hodgkin lymphomas, which do not occur specifically in this population: Diagnostic and prognostic implications. Histopathology 2022, 81, 826–840. [Google Scholar] [CrossRef]
- Maguire, A.; Chen, X.; Wisner, L.; Malasi, S.; Ramsower, C.; Kendrick, S.; Barrett, M.T.; Glinsmann-Gibson, B.; McGrath, M.; Rimsza, L.M. Enhanced DNA repair and genomic stability identify a novel HIV-related diffuse large B-cell lymphoma signature. Int. J. Cancer 2019, 145, 3078–3088. [Google Scholar] [CrossRef]
- Guech-Ongey, M.; Simard, E.P.; Anderson, W.F.; Engels, E.A.; Bhatia, K.; Devesa, S.S.; Mbulaiteye, S.M. AIDS-related Burkitt lymphoma in the United States: What do age and CD4 lymphocyte patterns tell us about etiology and/or biology? Blood 2010, 116, 5600–5604. [Google Scholar] [CrossRef]
- Sapkota, S.; Shaikh, H. Non-Hodgkin Lymphoma. In StatPearls; StatPearls Publishing Copyright © 2024; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2024. [Google Scholar]
- Han, X.; Jemal, A.; Hulland, E.; Simard, E.P.; Nastoupil, L.; Ward, E.; Flowers, C.R. HIV Infection and Survival of Lymphoma Patients in the Era of Highly Active Antiretroviral Therapy. Cancer Epidemiol. Biomark. Prev. 2017, 26, 303–311. [Google Scholar] [CrossRef]
- Mbulaiteye, S.M.; Pullarkat, S.T.; Nathwani, B.N.; Weiss, L.M.; Rao, N.; Emmanuel, B.; Lynch, C.F.; Hernandez, B.; Neppalli, V.; Hawes, D.; et al. Epstein-Barr virus patterns in US Burkitt lymphoma tumors from the SEER residual tissue repository during 1979–2009. Apmis 2014, 122, 5–15. [Google Scholar] [CrossRef]
- Cesarman, E. Pathology of lymphoma in HIV. Curr. Opin. Oncol. 2013, 25, 487–494. [Google Scholar] [CrossRef]
- Ajasin, D.; Eugenin, E.A. HIV-1 Tat: Role in Bystander Toxicity. Front. Cell. Infect. Microbiol. 2020, 10, 61. [Google Scholar] [CrossRef]
- Shmakova, A.; Tsimailo, I.; Kozhevnikova, Y.; Gérard, L.; Boutboul, D.; Oksenhendler, E.; Tuaillon, E.; Rivault, A.; Germini, D.; Vassetzky, Y.; et al. HIV-1 Tat is present in the serum of people living with HIV-1 despite viral suppression. Int. J. Infect. Dis. 2024, 142, 106994. [Google Scholar] [CrossRef]
- Ramorola, B.R.; Goolam-Hoosen, T.; Alves de Souza Rios, L.; Mowla, S. Modulation of Cellular MicroRNA by HIV-1 in Burkitt Lymphoma Cells—A Pathway to Promoting Oncogenesis. Genes 2021, 12, 1302. [Google Scholar] [CrossRef]
- Zelenetz, A.D.; Gordon, L.I.; Abramson, J.S.; Advani, R.H.; Andreadis, B.; Bartlett, N.L.; Budde, L.E.; Caimi, P.F.; Chang, J.E.; Christian, B.; et al. NCCN Guidelines® Insights: B-Cell Lymphomas, Version 6. J. Natl. Compr. Cancer Netw. 2023, 21, 1118–1131. [Google Scholar] [CrossRef]
- Biggar, R.J.; Horm, J.; Goedert, J.J.; Melbye, M. Cancer in a group at risk of acquired immunodeficiency syndrome (AIDS) through 1984. Am. J. Epidemiol. 1987, 126, 578–586. [Google Scholar] [CrossRef]
- Carroll, V.; Garzino-Demo, A. HIV-associated lymphoma in the era of combination antiretroviral therapy: Shifting the immunological landscape. Pathog. Dis. 2015, 73, ftv044. [Google Scholar] [CrossRef]
- Hleyhel, M.; Hleyhel, M.; Bouvier, A.M.; Belot, A.; Tattevin, P.; Pacanowski, J.; Genet, P.; De Castro, N.; Berger, J.L.; Dupont, C.; et al. Risk of non-AIDS-defining cancers among HIV-1-infected individuals in France between 1997 and 2009: Results from a French cohort. Aids 2014, 28, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Kowalkowski, M.A.; Mims, M.A.; Day, R.S.; Du, X.L.; Chan, W.; Chiao, E.Y. Longer duration of combination antiretroviral therapy reduces the risk of Hodgkin lymphoma: A cohort study of HIV-infected male veterans. Cancer Epidemiol. 2014, 38, 386–392. [Google Scholar] [CrossRef]
- Said, J.W. Immunodeficiency-related Hodgkin lymphoma and its mimics. Adv. Anat. Pathol. 2007, 14, 189–194. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Serraino, D.; Spina, M. HIV-associated Hodgkin lymphoma. Curr. Opin. HIV AIDS 2009, 4, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Koulis, A.; Trivedi, P.; Ibrahim, H.; Bower, M.; Naresh, K.N. The role of the microenvironment in human immunodeficiency virus-associated classical Hodgkin lymphoma. Histopathology 2014, 65, 749–756. [Google Scholar] [CrossRef]
- Hartmann, S.; Jakobus, C.; Rengstl, B.; Döring, C.; Newrzela, S.; Brodt, H.R.; Wolf, T.; Hansmann, M.L. Spindle-shaped CD163+ rosetting macrophages replace CD4+ T-cells in HIV-related classical Hodgkin lymphoma. Mod. Pathol. 2013, 26, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Grywalska, E.; Markowicz, J.; Grabarczyk, P.; Pasiarski, M.; Roliński, J. Epstein-Barr virus-associated lymphoproliferative disorders. Postep. Hig. Med. Dosw. 2013, 67, 481–490. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Caruso, A.; De Paoli, P.; Dolcetti, R. The impact of EBV and HIV infection on the microenvironmental niche underlying Hodgkin lymphoma pathogenesis. Int. J. Cancer 2017, 140, 1233–1245. [Google Scholar] [CrossRef]
- De Paoli, P.; Carbone, A. Microenvironmental abnormalities induced by viral cooperation: Impact on lymphomagenesis. Semin. Cancer Biol. 2015, 34, 70–80. [Google Scholar] [CrossRef]
- Caccuri, F.; Giagulli, C.; Bugatti, A.; Benetti, A.; Alessandri, G.; Ribatti, D.; Marsico, S.; Apostoli, P.; Slevin, M.A.; Rusnati, M.; et al. HIV-1 matrix protein p17 promotes angiogenesis via chemokine receptors CXCR1 and CXCR2. Proc. Natl. Acad. Sci. USA 2012, 109, 14580–14585. [Google Scholar] [CrossRef]
- Eliopoulos, A.G.; Stack, M.; Dawson, C.W.; Kaye, K.M.; Hodgkin, L.; Sihota, S.; Rowe, M.; Young, L.S. Epstein-Barr virus-encoded LMP1 and CD40 mediate IL-6 production in epithelial cells via an NF-kappaB pathway involving TNF receptor-associated factors. Oncogene 1997, 14, 2899–2916. [Google Scholar] [CrossRef]
- Re, A.; Cattaneo, C.; Rossi, G. Hiv and Lymphoma: From Epidemiology to Clinical Management. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019004. [Google Scholar] [CrossRef]
- Rezahosseini, O.; Hanaei, S.; Hamadani, M.; Keshavarz-Fathi, M.; Rezaei, N. The promising role of monoclonal antibodies for immunotherapy of the HIV-associated cancer, non-Hodgkin lymphoma. Int. Rev. Immunol. 2018, 37, 165–173. [Google Scholar] [CrossRef]
- Im, A.; Pavletic, S.Z. Immunotherapy in hematologic malignancies: Past, present, and future. J. Hematol. Oncol. 2017, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Schommers, P.; Gillor, D.; Hentrich, M.; Wyen, C.; Wolf, T.; Oette, M.; Zoufaly, A.; Wasmuth, J.C.; Bogner, J.R.; Müller, M.; et al. Incidence and risk factors for relapses in HIV-associated non-Hodgkin lymphoma as observed in the German HIV-related lymphoma cohort study. Haematologica 2018, 103, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, R.; Lurain, K.; Peer, C.J.; Serquiña, A.; Wang, V.; Widell, A.; Goncalves, P.; Steinberg, S.M.; Marshall, V.; George, J.; et al. Tocilizumab in patients with symptomatic Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood 2020, 135, 2316–2319. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, I.; Rossi, G.; Rullo, E.; Ascoli, V. Classic KSHV/HHV-8-positive Primary Effusion Lymphoma (PEL): A Systematic Review and Meta-Analysis of Case Reports. Mediterr. J. Hematol. Infect. Dis. 2022, 14, e2022020. [Google Scholar] [CrossRef]
- Rajdev, L.; Chiao, E.Y.; Lensing, S.; Little, R.F.; Dittmer, D.; Einstein, M.H.; Haigentz, M.; Sparano, J.A.; Mitsuyasu, R.T. AMC 095 (AIDS Malignancy Consortium): A phase I study of ipilimumab (IPI) and nivolumab (NIVO) in advanced HIV associated solid tumors (ST) with expansion cohorts in HIV associated solid tumors and classical Hodgkin lymphoma (cHL). J. Clin. Oncol. 2018, 36, TPS2597. [Google Scholar] [CrossRef]
- Nijland, M.; Veenstra, R.N.; Visser, L.; Xu, C.; Kushekhar, K.; van Imhoff, G.W.; Kluin, P.M.; van den Berg, A.; Diepstra, A. HLA dependent immune escape mechanisms in B-cell lymphomas: Implications for immune checkpoint inhibitor therapy? Oncoimmunology 2017, 6, e1295202. [Google Scholar] [CrossRef]
- Landsburg, D.J.; Koike, A.; Nasta, S.D.; Svoboda, J.; Schuster, S.J.; Wasik, M.A.; Caponetti, G.C. Patterns of immune checkpoint protein expression in MYC-overexpressing aggressive B-cell non-Hodgkin lymphomas. Cancer Immunol. Immunother. 2021, 70, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jia, L.; Zhang, X.; Zhang, T.; Zhang, Y. One arrow for two targets: Potential co-treatment regimens for lymphoma and HIV. Blood Rev. 2022, 55, 100965. [Google Scholar] [CrossRef]
- Hattenhauer, S.T.; Mispelbaum, R.; Hentrich, M.; Boesecke, C.; Monin, M.B. Enabling CAR T-cell therapies for HIV-positive lymphoma patients—A call for action. HIV Med. 2023, 24, 957–964. [Google Scholar] [CrossRef]
- Sandoval-Sus, J.D.; Mogollon-Duffo, F.; Patel, A.; Visweshwar, N.; Laber, D.A.; Kim, R.; Jagal, M.V. Nivolumab as salvage treatment in a patient with HIV-related relapsed/refractory Hodgkin lymphoma and liver failure with encephalopathy. J. Immunother. Cancer 2017, 5, 49. [Google Scholar] [CrossRef]
- Rogacheva, Y.; Popova, M.; Lepik, K.; Kondakova, E.; Zalyalov, Y.; Stelmah, L.; Volkova, A.; Nikolaev, I.; Goloshchapov, O.; Barhatov, I.; et al. INFECTIOUS COMPLICATIONS OF NIVOLUMAB THERAPY IN RELAPSED/REFRACTORY HODGKIN’S LYMPHOMA. Hematol. Oncol. 2019, 37, 502. [Google Scholar] [CrossRef]
- Lurain, K.; Ramaswami, R.; Mangusan, R.; Widell, A.; Ekwede, I.; George, J.; Ambinder, R.; Cheever, M.; Gulley, J.L.; Goncalves, P.H.; et al. Use of pembrolizumab with or without pomalidomide in HIV-associated non-Hodgkin’s lymphoma. J. Immunother. Cancer 2021, 9, e002097. [Google Scholar] [CrossRef] [PubMed]
- Uldrick, T.S.; Gonçalves, P.H.; Abdul-Hay, M.; Claeys, A.J.; Emu, B.; Ernstoff, M.S.; Fling, S.P.; Fong, L.; Kaiser, J.C.; Lacroix, A.M.; et al. Assessment of the Safety of Pembrolizumab in Patients with HIV and Advanced Cancer-A Phase 1 Study. JAMA Oncol. 2019, 5, 1332–1339. [Google Scholar] [CrossRef] [PubMed]

| Subtypes of HIV- Associated Lymphomas | Preferred Regimens | Other Recommended Regimens |
|---|---|---|
| Diffuse large B-cell lymphoma | R-EPOCH | RCHOP |
| Primary effusion lymphoma | R-EPOCH | RCHOP |
| Burkitt lymphoma | CODOX-M/IVAC (modified) or DA-EPOCH-R | R-Hyper CVAD |
| Plasmablastic lymphoma | EPOCH (preferred) | CODOX-M/IVAC (modified) or R-HyperCVAD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Xie, X.; Li, J.; Xiao, Q.; He, S.; Fu, H.; Zhang, X.; Liu, Y. Immune Characteristics and Immunotherapy of HIV-Associated Lymphoma. Curr. Issues Mol. Biol. 2024, 46, 9984-9997. https://doi.org/10.3390/cimb46090596
Liu Y, Xie X, Li J, Xiao Q, He S, Fu H, Zhang X, Liu Y. Immune Characteristics and Immunotherapy of HIV-Associated Lymphoma. Current Issues in Molecular Biology. 2024; 46(9):9984-9997. https://doi.org/10.3390/cimb46090596
Chicago/Turabian StyleLiu, Yi, Xiaoqing Xie, Jun Li, Qing Xiao, Sanxiu He, Huihui Fu, Xiaomei Zhang, and Yao Liu. 2024. "Immune Characteristics and Immunotherapy of HIV-Associated Lymphoma" Current Issues in Molecular Biology 46, no. 9: 9984-9997. https://doi.org/10.3390/cimb46090596
APA StyleLiu, Y., Xie, X., Li, J., Xiao, Q., He, S., Fu, H., Zhang, X., & Liu, Y. (2024). Immune Characteristics and Immunotherapy of HIV-Associated Lymphoma. Current Issues in Molecular Biology, 46(9), 9984-9997. https://doi.org/10.3390/cimb46090596