
Gonadal Mosaicism as a Rare Inheritance Pattern in Recessive Genodermatoses: Report of Two Cases with Pseudoxanthoma Elasticum and Literature Review

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
- All article types describing original research were included (no reviews);
- Neither the date of publication nor the journal played a role in the selection;
- Only articles describing humans were considered;
- Only articles written in English were considered;
- Only articles of which the full text was available were included in the analysis.
3. Results
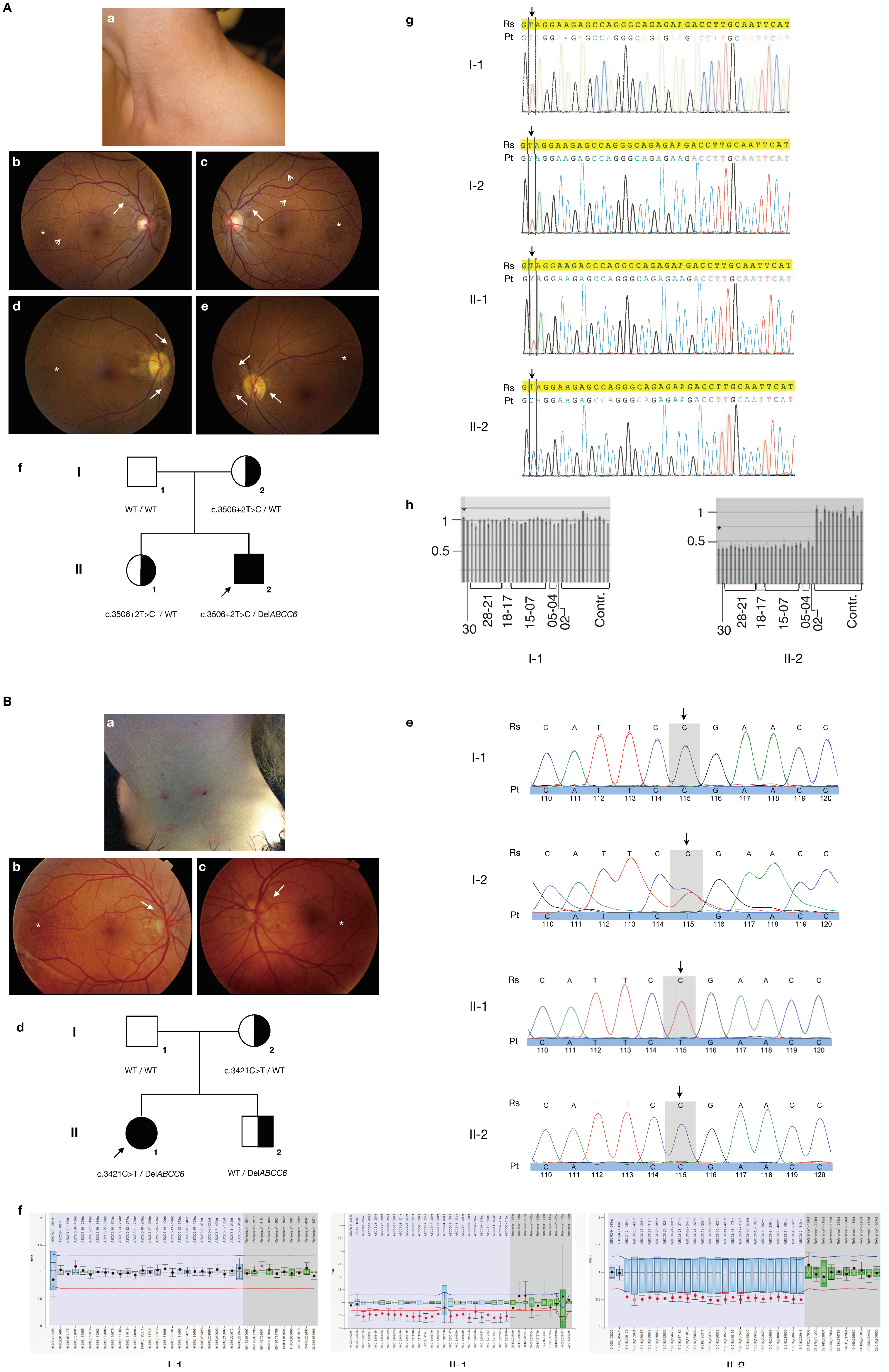
3.1. Case Reports
3.2. Literature Review

{kind=link}
{kind=link}
| Disease | Gene | Variant Type | Parental Origin | N | Somatic Mosaicism | Semen Analysis | Paternity | Reference |
|---|---|---|---|---|---|---|---|---|
| Cockayne syndrome | ERCC6 | Gene deletion | Maternal | 1 | NA | - | Yes | [21] |
| Cutis laxa | GORAB | Gene deletion | Paternal | 1 | NA | NA | Yes | [22] |
| Dystrophic EB | COL7A1 | Nonsense | Paternal | 1 | NA | NA | Yes | [19] |
| COL7A1 | Nonsense | Paternal | 1 | No | NA | Yes | [20] | |
| COL7A1 | Gene deletion | Maternal | 1 | NA | - | Yes | [25] | |
| EB with late-onset muscular dystrophy | PLEC1 | Nonsense | Paternal | 2 | NA | NA | Yes | [18] |
| PLEC1 | Nonsense | Paternal | 1 | NA | NA | Yes | [26] | |
| Hermansky–Pudlak syndrome | HSP1 | Nonsense | Paternal | 1 | NA | NA | Yes | [27] |
| Hypohydrotic ectodermal dysplasia | EDAR | Missense | Paternal | 1 | NA | NA | Yes | [28] |
| Junctional EB | LAMB3 | Nonsense | Paternal | 1 | NA | Positive | Yes | [29] |
| Oculocutaneous albinism | TYRP1 | Nonsense | Paternal | 1 | NA | NA | Yes | [30] |
| Pseudoxanthoma elasticum | ABCC6 | Gene deletion | Paternal | 2 | No | NA | Yes | Current report |
| ABCC6 | Nonsense | Maternal | 1 | NA | - | - | [23] | |
| Vici syndrome | EPG5 | Exon 1 duplication | Paternal | 1 | NA | NA | Yes | [24] |
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Cserhalmi-Friedman, P.B.; Garzon, M.C.; Guzman, E.; Martinez-Mir, A.; Chung, W.K.; Anyane-Yeboa, K.; Christiano, A.M. Maternal germline mosaicism in dominant dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2001, 117, 1327–1328. [Google Scholar] [CrossRef] [PubMed]
- Kirchman, T.T.; Levy, M.L.; Lewis, R.A.; Kanzler, M.H.; Nelson, D.L.; Scheuerle, A.E. Gonadal mosaicism for incontinentia pigmenti in a healthy male. J. Med. Genet. 1995, 32, 887–890. [Google Scholar] [CrossRef]
- Sbidian, E.; Feldmann, D.; Bengoa, J.; Fraitag, S.; Abadie, V.; De Prost, Y.; Bodemer, C.; Hadj-Rabia, S. Germline mosaicism in keratitis-ichthyosis-deafness syndrome: Pre-natal diagnosis in a familial lethal form. Clin. Genet. 2010, 77, 587–592. [Google Scholar] [CrossRef]
- Bottillo, I.; Torrente, I.; Lanari, V.; Pinna, V.; Giustini, S.; Divona, L.; De Luca, A.; Dallapiccola, B. Germline mosaicism in neurofibromatosis type 1 due to a paternally derived multi-exon deletion. Am. J. Med. Genet. Part A 2010, 152A, 1467–1473. [Google Scholar] [CrossRef]
- Bergen, A.A.; Plomp, A.S.; Schuurman, E.J.; Terry, S.F.; Breuning, M.H.; Dauwerse, H.G.; Swart, J.; Kool, M.; Van Soest, S.; Baas, F.; et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 228–231. [Google Scholar] [CrossRef]
- Le Saux, O.; Urban, Z.; Tschuch, C.; Csiszar, K.; Bacchelli, B.; Quaglino, D.; Pasquali-Ronchetti, I.; Pope, F.M.; Richards, A.; Terry, S.F.; et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, Y.; Baujat, G.; Botschen, U.; Wittkampf, T.; du Moulin, M.; Stella, J.; Le Merrer, M.; Guest, G.; Lambot, K.; Tazarourte-Pinturier, M.-F.; et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am. J. Hum. Genet. 2012, 90, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Pfau, K.; Lengyel, I.; Norel, J.O.-V.; van Leeuwen, R.; Risseeuw, S.; Leftheriotis, G.; Scholl, H.P.; Feltgen, N.; Holz, F.G.; Pfau, M. Pseudoxanthoma elasticum—Genetics, pathophysiology, and clinical presentation. Prog. Retin. Eye Res. 2024, 102, 101274. [Google Scholar] [CrossRef]
- Ghaoui, N.; Abou-Rahal, J.; Nasser, N.; Kurban, M.; Abbas, O. Pseudoxanthoma Elasticum-like Changes: Associations- and Underlying Mechanisms. Skinmed 2024, 22, 172–177. [Google Scholar]
- Stumpf, M.J.; Schahab, N.; Nickenig, G.; Skowasch, D.; Schaefer, C.A. Therapy of Pseudoxanthoma Elasticum: Current Knowledge and Future Perspectives. Biomedicines 2021, 9, 1895. [Google Scholar] [CrossRef]
- Verschuere, S.; Van Gils, M.; Nollet, L.; Vanakker, O.M. From membrane to mineralization: The curious case of the ABCC6 transporter. FEBS Lett. 2020, 594, 4109–4133. [Google Scholar] [CrossRef]
- Terry, S.F.; Uitto, J. Pseudoxanthoma Elasticum. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 4 June 2020. [Google Scholar]
- Legrand, A.; Pujol, C.; Durand, C.M.; Mesnil, A.; Rubera, I.; Duranton, C.; Zuily, S.; Sousa, A.B.; Renaud, M.; Boucher, J.; et al. Pseudoxanthoma elasticum overlaps hereditary spastic paraplegia type 56. J. Intern. Med. 2021, 289, 709–725. [Google Scholar] [CrossRef]
- Li, Q.; Schurgers, L.J.; Smith, A.C.; Tsokos, M.; Uitto, J.; Cowen, E.W. Co-existent pseudoxanthoma elasticum and vitamin K-dependent coagulation factor deficiency: Compound heterozygosity for mutations in the GGCX gene. Am. J. Pathol. 2009, 174, 534–540. [Google Scholar] [CrossRef]
- Stuppia, L.; Antonucci, I.; Palka, G.; Gatta, V. Use of the MLPA assay in the molecular diagnosis of gene copy number alterations in human genetic diseases. Int. J. Mol. Sci. 2012, 13, 3245–3276. [Google Scholar] [CrossRef]
- Gjertson, D.W.; Brenner, C.H.; Baur, M.P.; Carracedo, A.; Guidet, F.; Luque, J.A.; Lessig, R.; Mayr, W.R.; Pascali, V.L.; Prinz, M.; et al. Recommendations on biostatistics in paternity testing. Forensic Sci. Int. Genet. 2007, 1, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Costrop, L.M.; Vanakker, O.O.; Van Laer, L.; Le Saux, O.; Martin, L.; Chassaing, N.; Guerra, D.; Pasquali-Ronchetti, I.; Coucke, P.J.; De Paepe, A. Novel deletions causing pseudoxanthoma elasticum underscore the genomic instability of the ABCC6 region. J. Hum. Genet. 2010, 55, 112–117. [Google Scholar] [CrossRef]
- Rouan, F.; Pulkkinen, L.; LaForgia, S.; Hyde, P.; Richard, G.; Uitto, J.; Meneguzzi, G.; Kim, D.U. Epidermolysis bullosa: Novel and de novo premature termination codon and deletion mutations in the plectin gene predict late-onset muscular dystrophy. J. Investig. Dermatol. 2000, 114, 381–387. [Google Scholar] [CrossRef]
- Cuadrado-Corrales, N.; Sánchez-Jimeno, C.; García, M.; Ayuso, C.; De Lucas, R.; Vicario, J.; Conti, C.; Zambruno, G.; Escamez, M.; Del Rio, M. A recurrent nonsense mutation occurring as a de novo event in a patient with recessive dystrophic epidermolysis bullosa. Dermatology 2011, 223, 219–221. [Google Scholar] [CrossRef]
- Kern, J.S.; Kohlhase, J.; Bruckner-Tuderman, L.; Has, C. Expanding the COL7A1 mutation database: Novel and recurrent mutations and unusual genotype-phenotype constellations in 41 patients with dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2006, 126, 1006–1012. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, J.; Ye, J.; Gong, Z.; Gu, X. Maternal origin of a de novo microdeletion spanning the ERCC6 gene in a classic form of the Cockayne syndrome. Eur. J. Med. Genet. 2011, 54, e389–e393. [Google Scholar] [CrossRef] [PubMed]
- Al-Bughaili, M.; Neuhann, T.M.; Flöttmann, R.; Mundlos, S.; Spielmann, M.; Kornak, U.; Fischer-Zirnsak, B. A de novo 1q23.3-q24.2 deletion combined with a GORAB missense mutation causes a distinctive phenotype with cutis laxa. J. Hum. Genet. 2017, 62, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Ringpfeil, F.; McGuigan, K.; Fuchsel, L.; Kozic, H.; Larralde, M.; Lebwohl, M.; Uitto, J. Pseudoxanthoma elasticum is a recessive disease characterized by compound heterozygosity. J. Investig. Dermatol. 2006, 126, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Hirasawa, K.; Takeshita, A.; Nakatsukasa, H.; Yamamoto-Shimojima, K.; Imaizumi, T.; Nagata, S.; Yamamoto, T. Novel compound heterozygous EPG5 mutations consisted with a missense mutation and a microduplication in the exon 1 region identified in a Japanese patient with Vici syndrome. Am. J. Med. Genet. Part A 2018, 176, 2803–2807. [Google Scholar] [CrossRef]
- Lee, M.; Xu, G.; Wang, K.; Wang, H.; Zhang, J.; Tang, Z.; Lin, Z.; Yang, Y. Recessive dystrophic epidermolysis bullosa caused by a de novo interstitial deletion spanning COL7A1 and a hemizygous splicing mutation in trans. Clin. Exp. Dermatol. 2016, 41, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Yiu, E.M.; Klausegger, A.; Waddell, L.B.; Grasern, N.; Lloyd, L.; Tran, K.; North, K.N.; Bauer, J.W.; McKelvie, P.; Chow, C.; et al. Epidermolysis bullosa with late-onset muscular dystrophy and plectin deficiency. Muscle Nerve 2011, 44, 135–141. [Google Scholar] [CrossRef]
- González-Conejero, R.; Rivera, J.; Escolar, G.; Zuazu-Jausoro, I.; Vicente, V.; Corral, J. Molecular, ultrastructural and functional characterization of a Spanish family with Hermansky-Pudlak syndrome: Role of insC974 in platelet function and clinical relevance. Br. J. Haematol. 2003, 123, 132–138. [Google Scholar] [CrossRef]
- Bashyam, M.; Chaudhary, A.; Reddy, E.; Reddy, V.; Acharya, V.; Nagarajaram, H.; Devi, A.; Bashyam, L.; Dalal, A.; Gupta, N.; et al. A founder ectodysplasin A receptor (EDAR) mutation results in a high frequency of the autosomal recessive form of hypohidrotic ectodermal dysplasia in India. Br. J. Dermatol. 2012, 166, 819–829. [Google Scholar] [CrossRef]
- Cserhalmi-Friedman, P.B.; Anyane-Yeboa, K.; Christiano, A.M. Paternal germline mosaicism in Herlitz junctional epidermolysis bullosa. Exp. Dermatol. 2002, 11, 468–470. [Google Scholar] [CrossRef]
- Rooryck, C.; Roudaut, C.; Robine, E.; Müsebeck, J.; Arveiler, B. Oculocutaneous albinism with TYRP1 gene mutations in a Caucasian patient. Pigment. Cell Res. 2006, 19, 239–242. [Google Scholar] [CrossRef]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef]
- Gu, W.; Zhang, F.; Lupski, J.R. Mechanisms for human genomic rearrangements. Pathogenetics 2008, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, N.; Martin, L.; Bourthoumieu, S.; Calvas, P.; Hovnanian, A. Contribution of ABCC6 genomic rearrangements to the diagnosis of pseudoxanthoma elasticum in French patients. Hum. Mutat. 2007, 28, 1046. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Beck, K.; Sachsinger, C.; Silvestri, C.; Treiber, C.; Göring, H.H.; Johnson, E.W.; De Paepe, A.; Pope, F.M.; Pasquali-Ronchetti, I.; et al. A spectrum of ABCC6 mutations is responsible for pseudoxanthoma elasticum. Am. J. Hum. Genet. 2001, 69, 749–764. [Google Scholar] [CrossRef]
- Girard, S.L.; Bourassa, C.V.; Perreault, L.-P.L.; Legault, M.-A.; Barhdadi, A.; Ambalavanan, A.; Brendgen, M.; Vitaro, F.; Noreau, A.; Dionne, G.; et al. Paternal age explains a major portion of de novo germline mutation rate variability in healthy individuals. PLoS ONE 2016, 11, e0164212. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Francioli, L.C.; Hormozdiari, F.; Marschall, T.; Hehir-Kwa, J.Y.; Abdellaoui, A.; Lameijer, E.-W.; Moed, M.H.; Koval, V.; Renkens, I.; et al. Characteristics of de novo structural changes in the human genome. Genome Res. 2015, 26, 792–801. [Google Scholar] [CrossRef]
- Wadhawan, I.; Hai, Y.; Foyouzi Yousefi, N.; Guo, X.; Graham, J.M., Jr.; Rosenfeld, J.A. De novo copy number variants and parental age: Is there an association? Eur. J. Med. Genet. 2020, 63, 103829. [Google Scholar] [CrossRef]
- Hehir-Kwa, J.Y.; Rodríguez-Santiago, B.; Vissers, L.E.; de Leeuw, N.; Pfundt, R.; Buitelaar, J.K.; Perez-Jurado, L.A.; Veltman, J.A. De novo copy number variants associated with intellectual disability have a paternal origin and age bias. J. Med. Genet. 2011, 48, 776–778. [Google Scholar] [CrossRef]
- Roach, J.C.; Glusman, G.; Smit, A.F.; Huff, C.D.; Hubley, R.; Shannon, P.T.; Rowen, L.; Pant, K.P.; Goodman, N.; Bamshad, M.; et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 2010, 328, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Campens, L.; Vanakker, O.M.; Trachet, B.; Segers, P.; Leroy, B.P.; De Zaeytijd, J.; Voet, D.; De Paepe, A.; De Backer, T.; De Backer, J. Characterization of cardiovascular involvement in pseudoxanthoma elasticum families. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2646–2652. [Google Scholar] [CrossRef]
- Nollet, L.; Campens, L.; De Zaeytijd, J.; Leroy, B.; Hemelsoet, D.; Coucke, P.J.; Vanakker, O.M. Clinical and subclinical findings in heterozygous ABCC6 carriers: Results from a Belgian cohort and clinical practice guidelines. J. Med. Genet. 2022, 59, 496–504. [Google Scholar] [CrossRef]
- Martin, L.; Maître, F.; Bonicel, P.; Daudon, P.; Verny, C.; Bonneau, D.; Le Saux, O.; Chassaing, N. Heterozygosity for a single mutation in the ABCC6 gene may closely mimic PXE: Consequences of this phenotype overlap for the definition of PXE. Arch. Dermatol. 2008, 144, 301–306. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dangreau, L.; Hosen, M.J.; De Zaeytijd, J.; Leroy, B.P.; Coucke, P.J.; Vanakker, O.M. Gonadal Mosaicism as a Rare Inheritance Pattern in Recessive Genodermatoses: Report of Two Cases with Pseudoxanthoma Elasticum and Literature Review. Curr. Issues Mol. Biol. 2024, 46, 9998-10007. https://doi.org/10.3390/cimb46090597
Dangreau L, Hosen MJ, De Zaeytijd J, Leroy BP, Coucke PJ, Vanakker OM. Gonadal Mosaicism as a Rare Inheritance Pattern in Recessive Genodermatoses: Report of Two Cases with Pseudoxanthoma Elasticum and Literature Review. Current Issues in Molecular Biology. 2024; 46(9):9998-10007. https://doi.org/10.3390/cimb46090597
Chicago/Turabian StyleDangreau, Lisa, Mohammad J. Hosen, Julie De Zaeytijd, Bart P. Leroy, Paul J. Coucke, and Olivier M. Vanakker. 2024. "Gonadal Mosaicism as a Rare Inheritance Pattern in Recessive Genodermatoses: Report of Two Cases with Pseudoxanthoma Elasticum and Literature Review" Current Issues in Molecular Biology 46, no. 9: 9998-10007. https://doi.org/10.3390/cimb46090597
APA StyleDangreau, L., Hosen, M. J., De Zaeytijd, J., Leroy, B. P., Coucke, P. J., & Vanakker, O. M. (2024). Gonadal Mosaicism as a Rare Inheritance Pattern in Recessive Genodermatoses: Report of Two Cases with Pseudoxanthoma Elasticum and Literature Review. Current Issues in Molecular Biology, 46(9), 9998-10007. https://doi.org/10.3390/cimb46090597