
Disulfidptosis: A New Target for Parkinson’s Disease and Cancer


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Identifying the Diagnostic Value of DEDRGs
2.3. In Vivo Experiment Verification
2.3.1. Animal
2.3.2. Western Blot
2.3.3. DEDRG Differential Expression of Pan-Cancer
2.3.4. DEDRG Prognostic Analysis of Pan-Cancer
2.3.5. Survival Curve Analysis of Pan-Cancer
2.3.6. Immune Infiltration and Single-Cell Type Analysis
2.3.7. Mutation and miRNA Analysis of DEDRGs
2.3.8. Molecular Docking and Prediction of Binding Pockets
2.3.9. Statistical Analysis
3. Results
3.1. Biological Functional Enrichment Analysis of DEDRGs
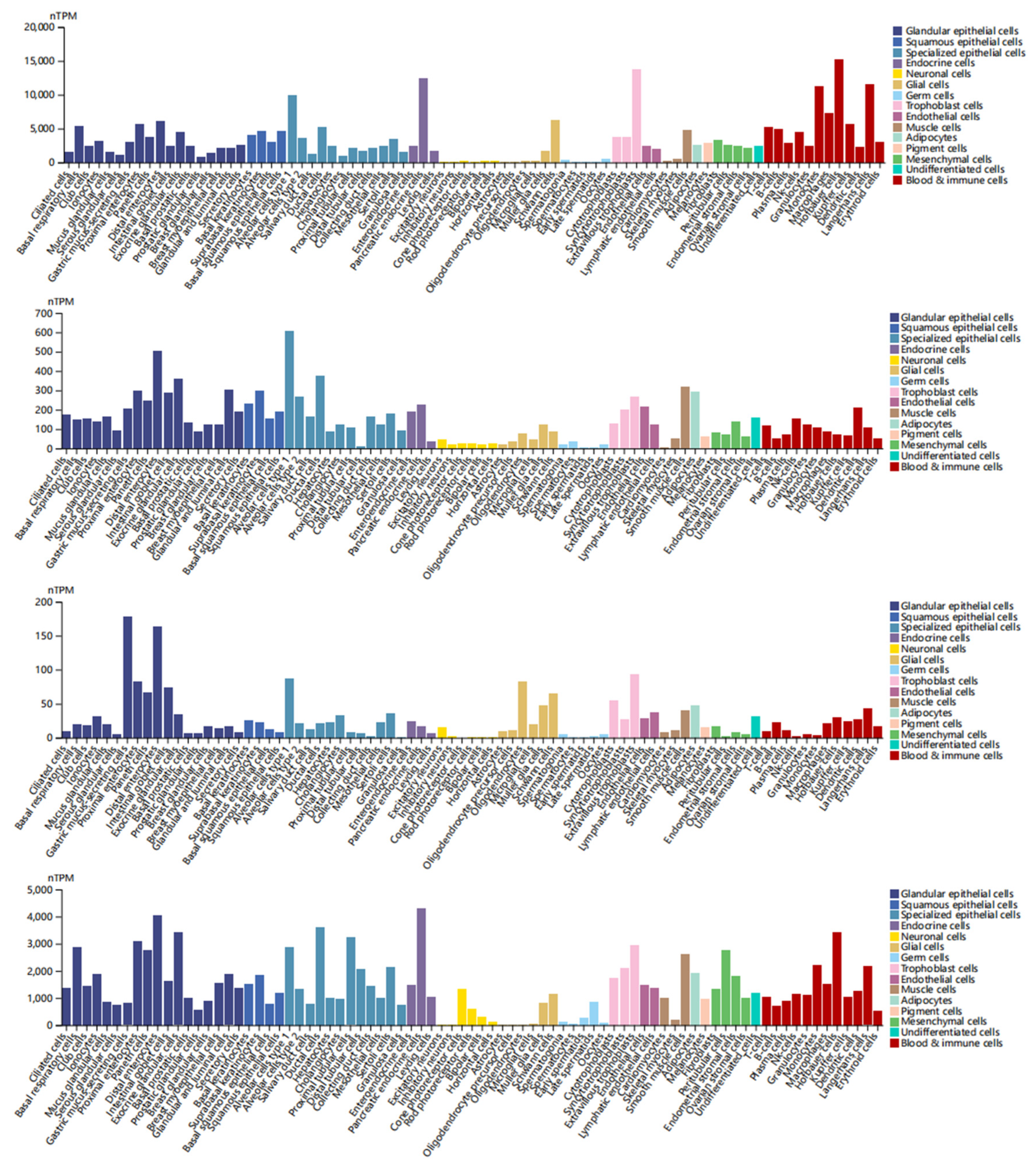
3.2. Analysis of DEDRG Expression in Tissues
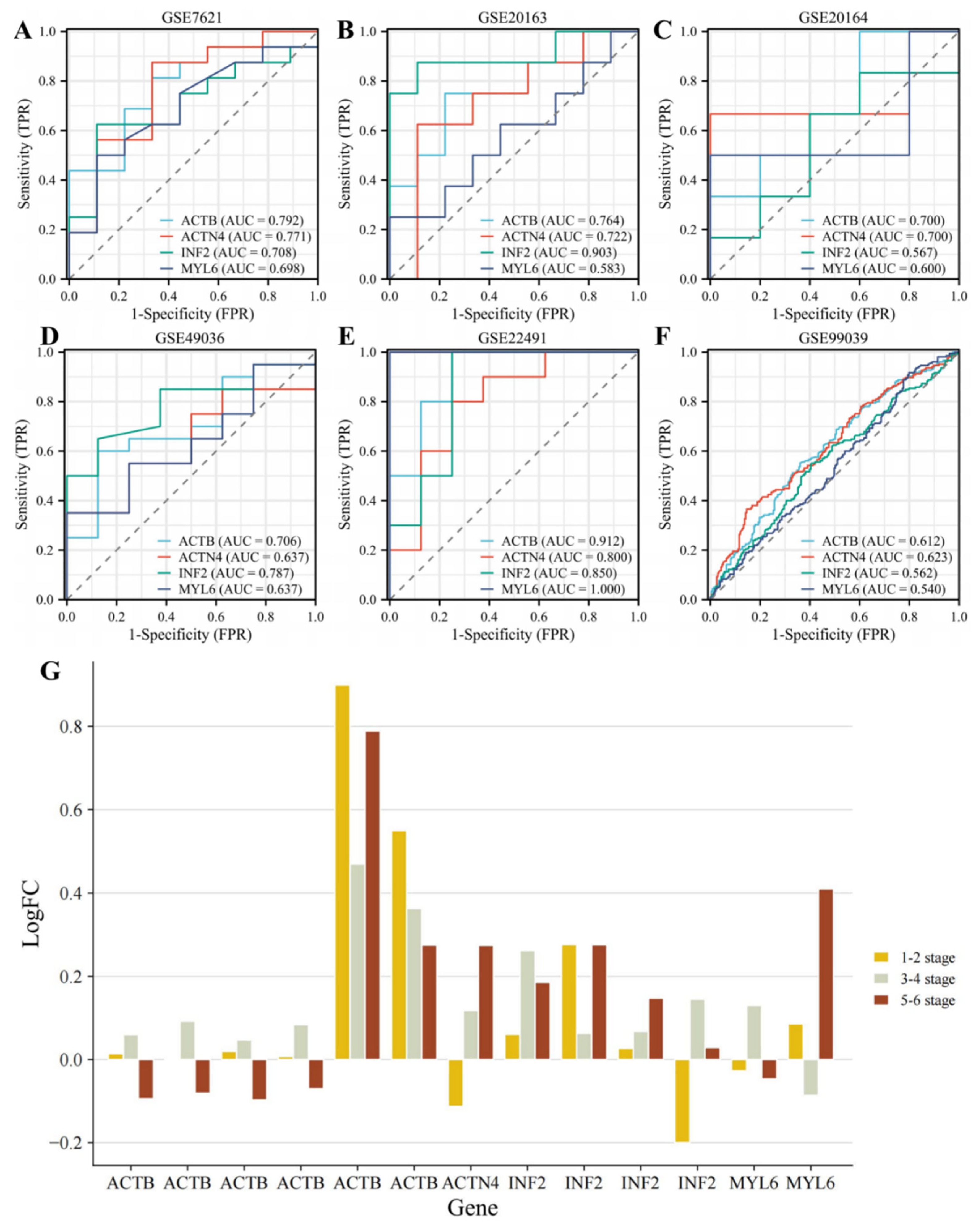
3.3. Identifying the Diagnostic Value of DEDRGs
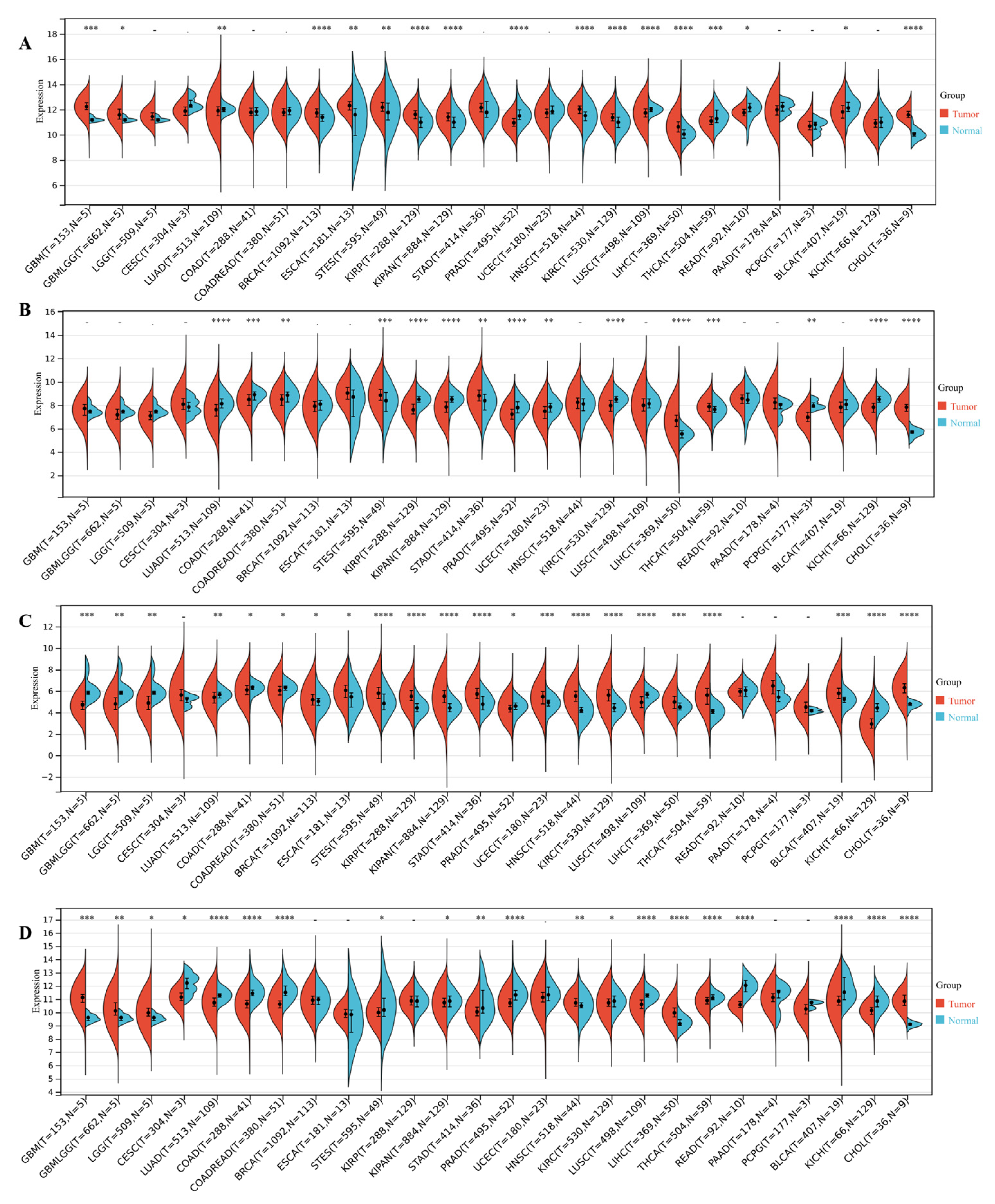
3.4. DEDRG Differential Expression Analysis of Pan-Cancer
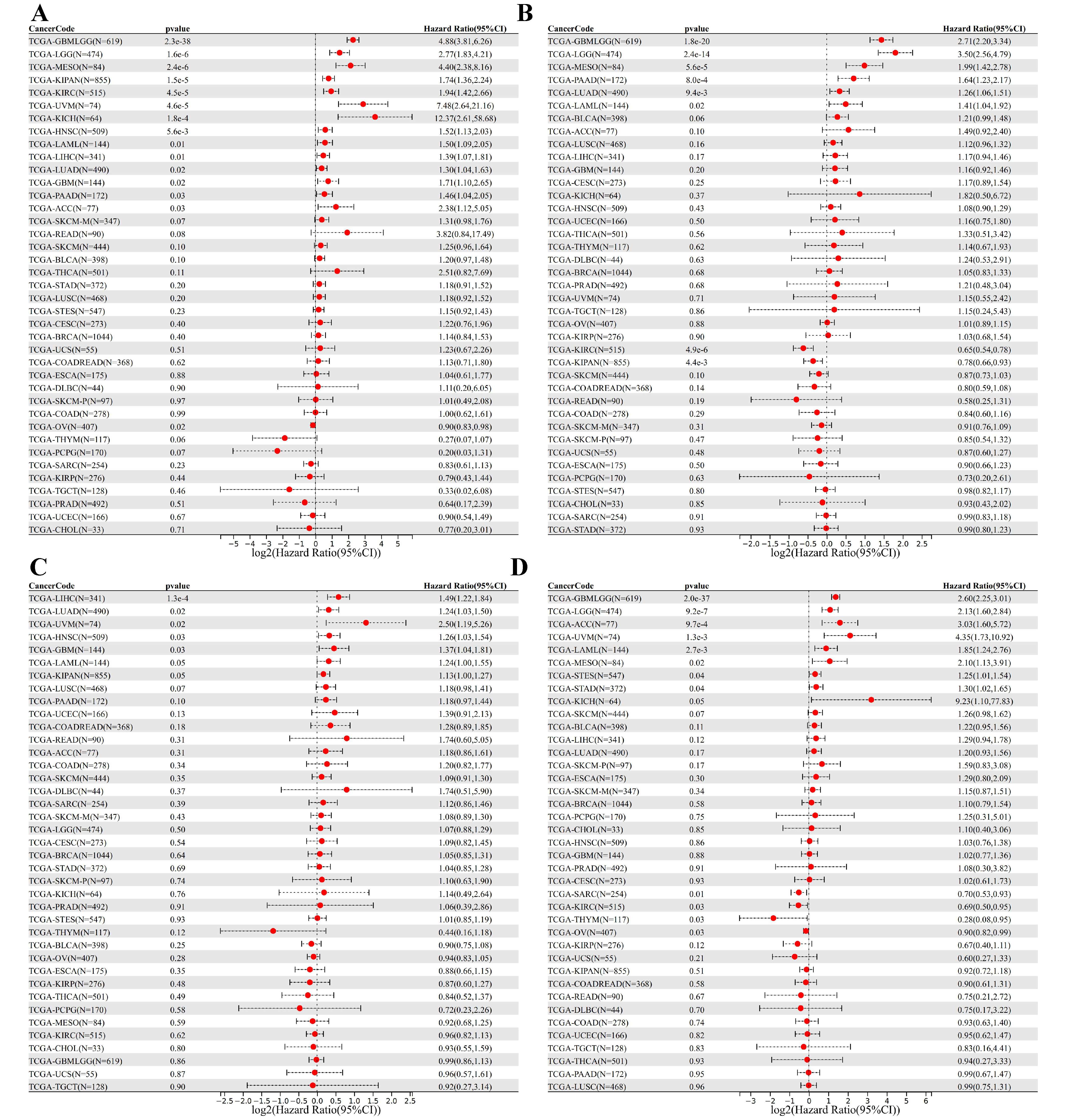
3.5. DEDRG Prognostic Analysis of Pan-Cancer
3.6. Survival Curve Analysis of Pan-Cancer
3.7. Immune Infiltration Analysis
3.8. Single-Cell Type Analysis
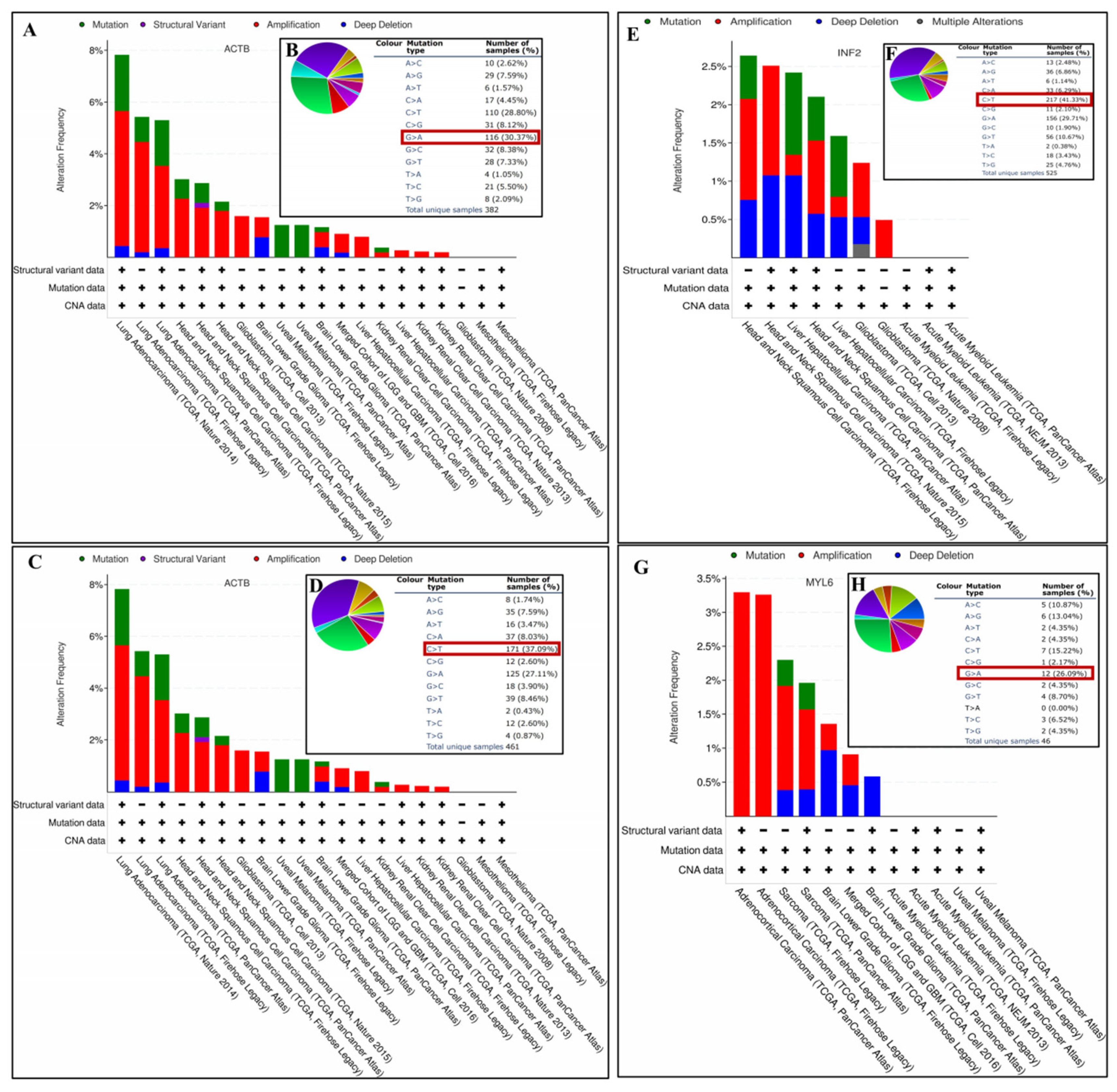
3.9. Gene Mutation Analysis of DEDRGs
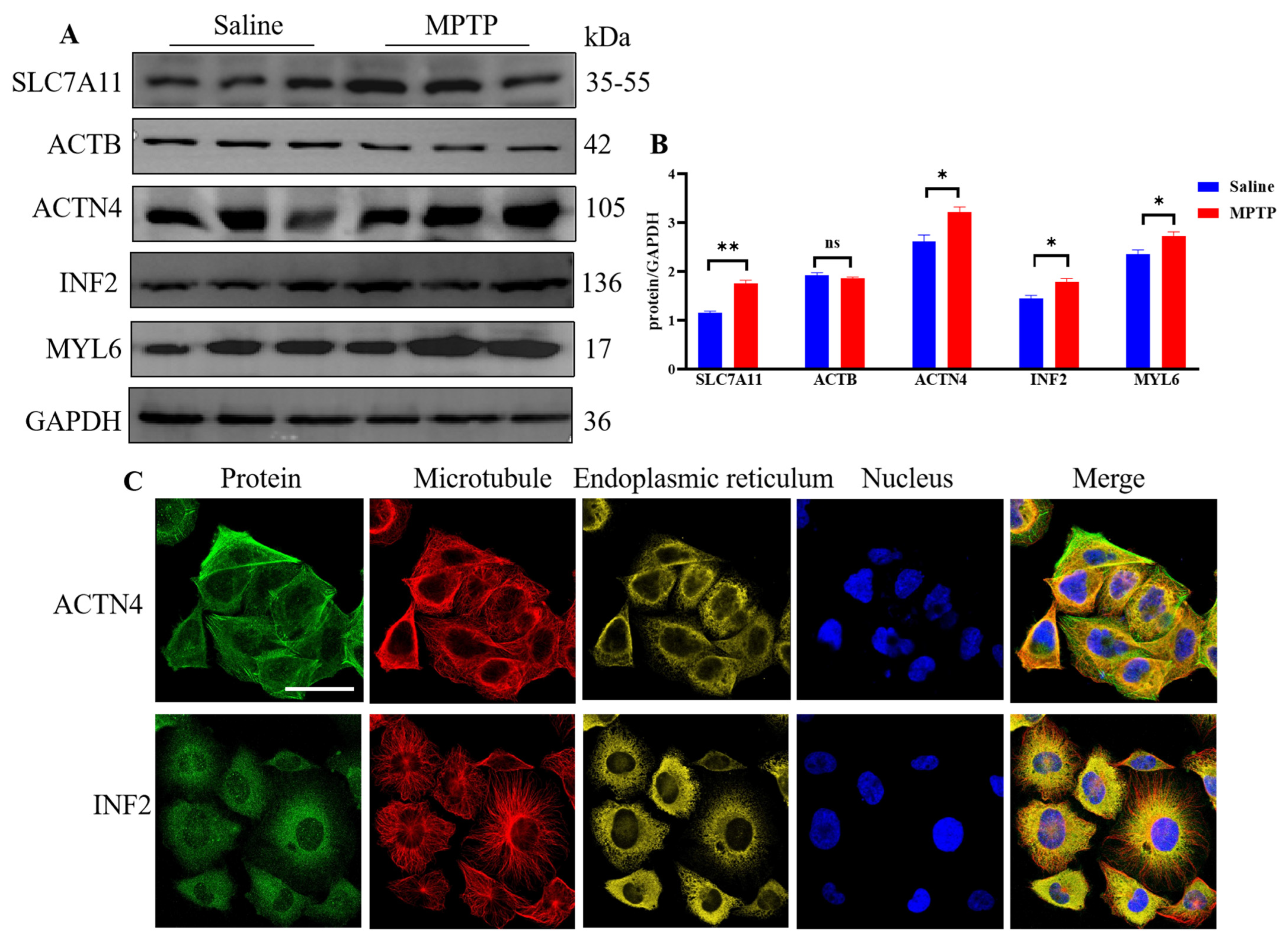
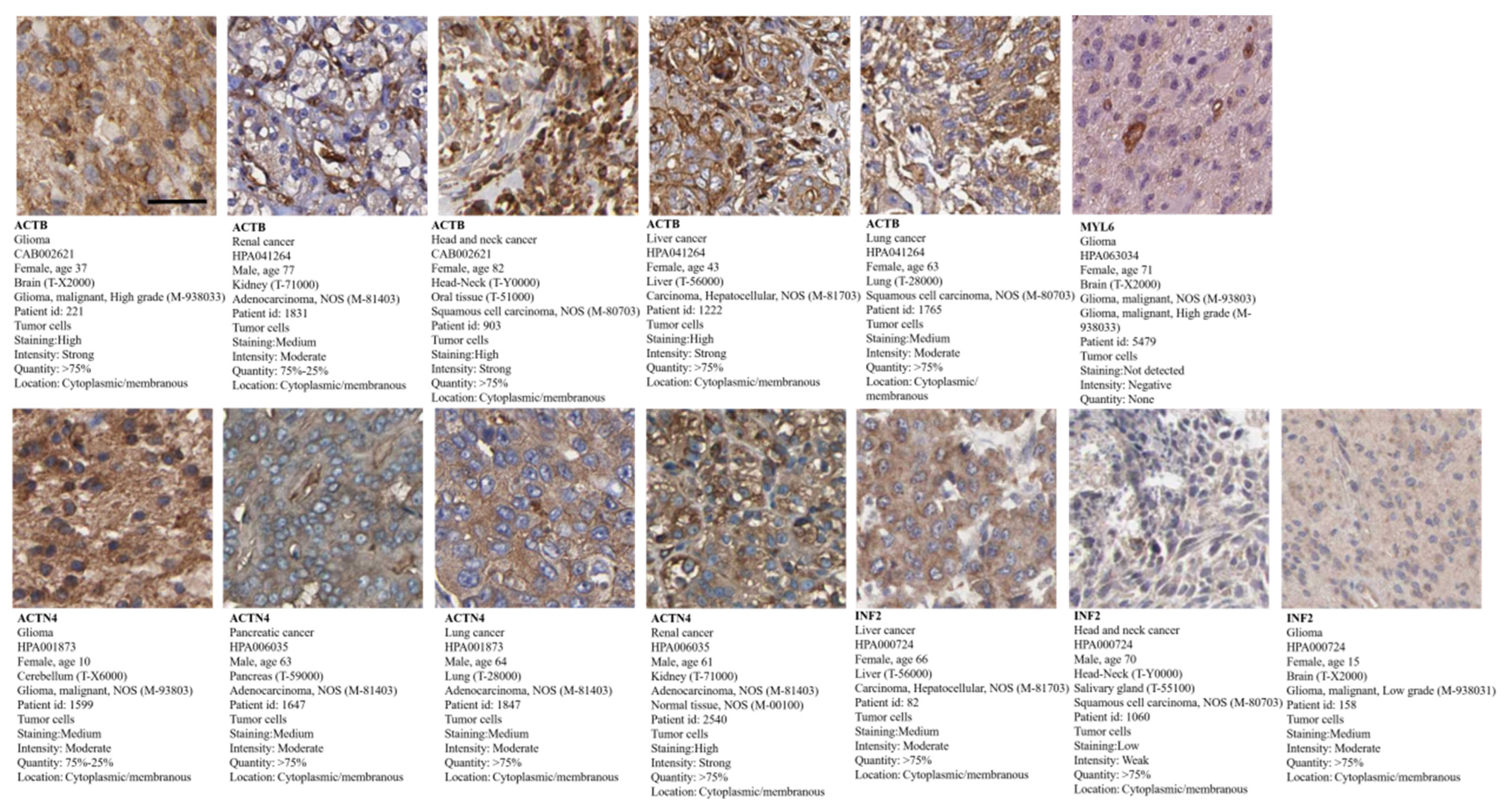
3.10. Tumor Pathological Staining
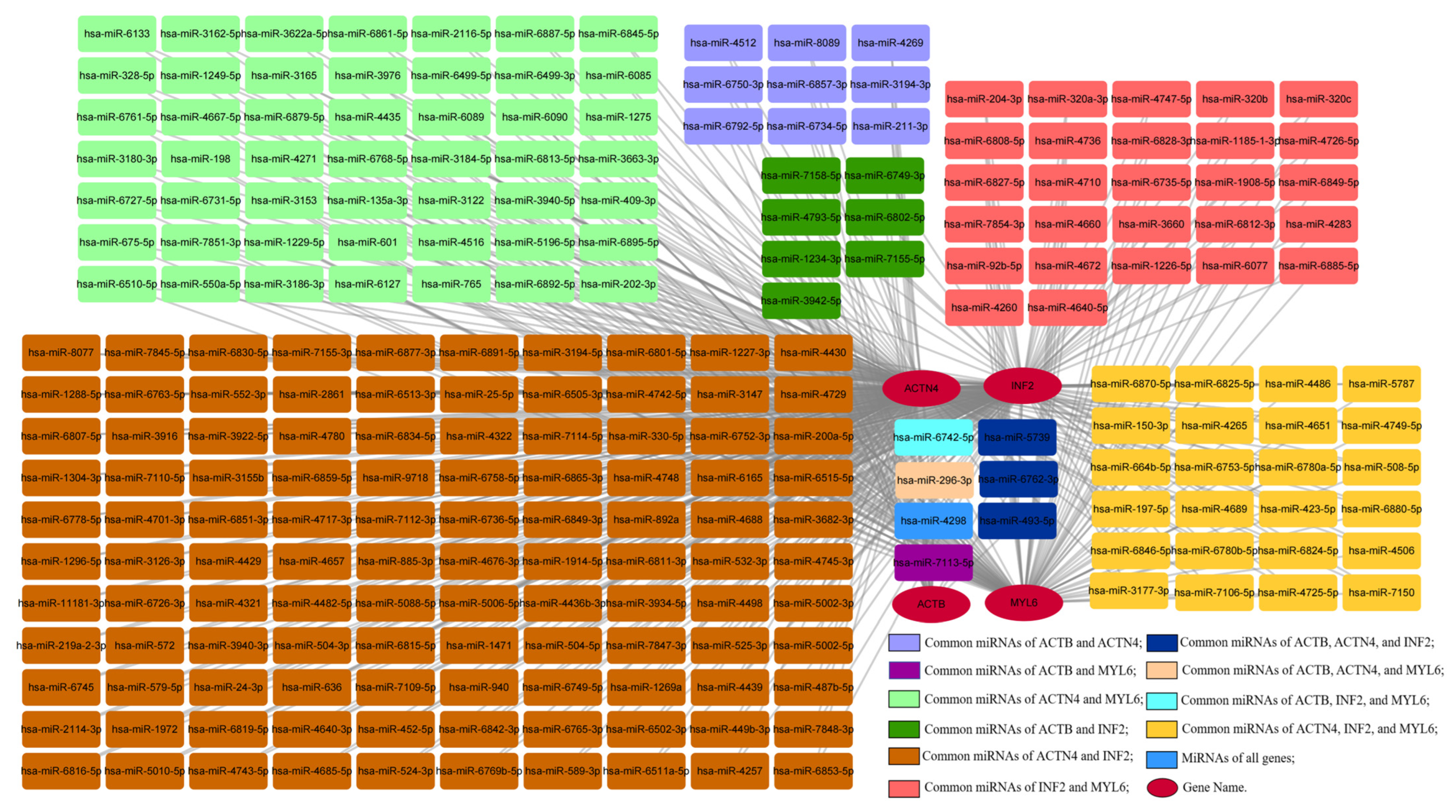
3.11. Construction of the Gene–miRNA Network
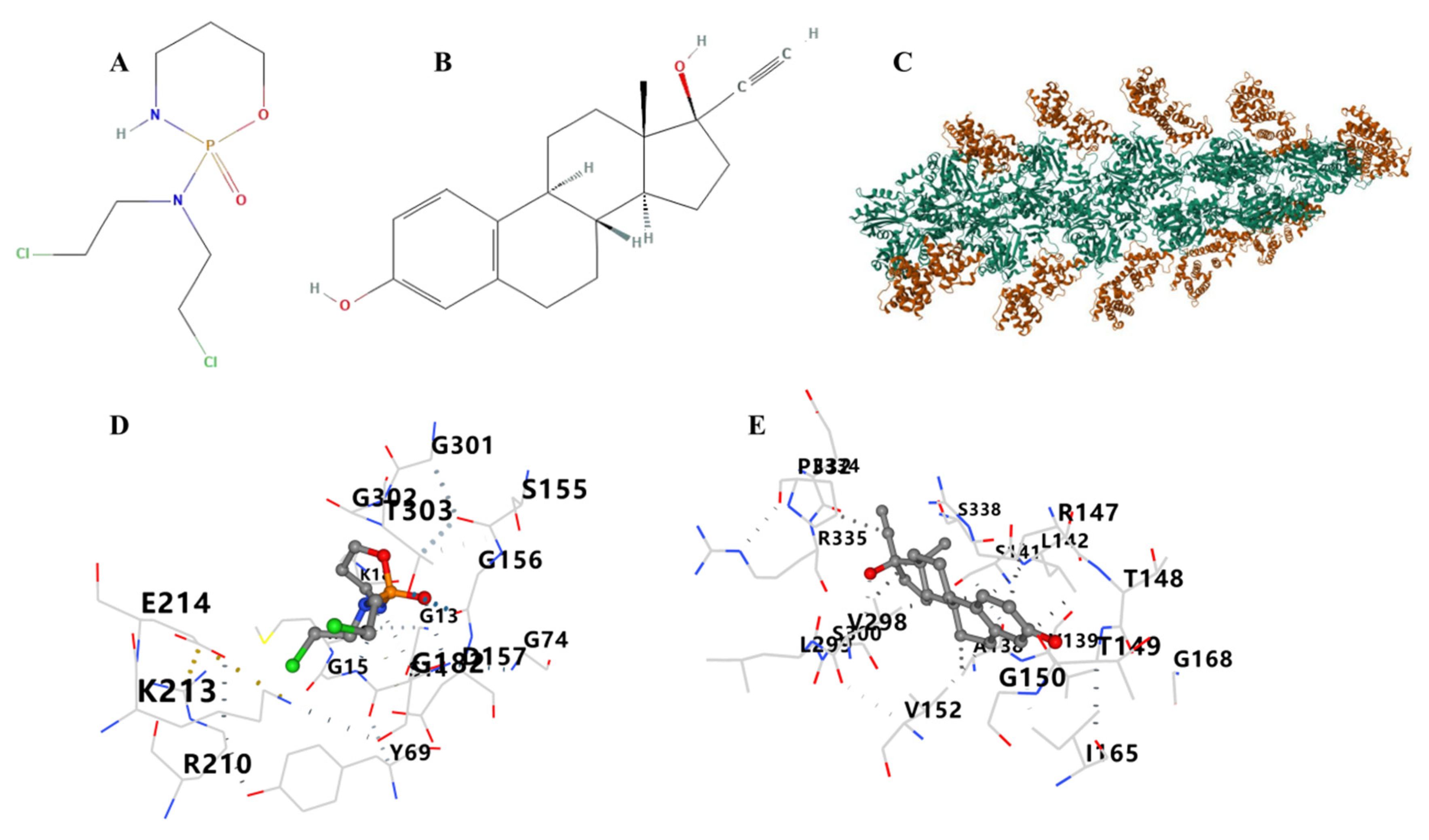
3.12. Therapeutic Drugs and Molecular Docking
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lenart, P.; Novak, J.; Bienertova-Vasku, J. PIWI-piRNA pathway: Setting the pace of aging by reducing DNA damage. Mech. Ageing Dev. 2018, 173, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef]
- Pilato, F.; Profice, P.; Ranieri, F.; Di Iorio, R.; Florio, L.; Di Lazzaro, V. Synaptic plasticity in neurodegenerative diseases evaluated and modulated by in vivo neurophysiological techniques. Mol. Neurobiol. 2012, 46, 563–571. [Google Scholar] [CrossRef]
- Seo, J.; Park, M. Molecular crosstalk between cancer and neurodegenerative diseases. Cell. Mol. Life Sci. 2020, 77, 2659–2680. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Parkinson’s disease-associated protein Parkin: An unusual player in cancer. Cancer Commun. 2018, 38, 40. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hang, Z.; Lei, T.; Zeng, Z.; Cai, S.; Bi, W.; Du, H. Composition of intestinal flora affects the risk relationship between Alzheimer’s disease/Parkinson’s disease and cancer. Biomed. Pharmacother. 2022, 145, 112343. [Google Scholar] [CrossRef]
- West, A.B.; Dawson, V.L.; Dawson, T.M. To die or grow: Parkinson’s disease and cancer. Trends Neurosci. 2005, 28, 348–352. [Google Scholar] [CrossRef]
- Westlund, K.; Hougen, A. Cancer as a cause of death among patients with other chronic disease (letter). J. Am. Med. Assoc. 1956, 162, 1003. [Google Scholar] [CrossRef]
- Pritchard, P.B., III; Netsky, M.G. Prevalence of neoplasms and causes of death in paralysis agitans. A necropsy study. Neurology 1973, 23, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.Y.; Tanner, C.M.; Van Den Eeden, S.K.; Albers, K.B.; Leimpeter, A.D.; Nelson, L.M. Comorbid cancer in Parkinson’s disease. Mov. Disord. 2010, 25, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Shi, Y.; Wu, L.; Zhang, H.; Xue, J.; Li, W.; Wang, X.; Zhang, L.; Wang, Q.; Duo, L.; et al. Establishment and verification of a nomogram to predict tumor-specific mortality risk in triple-negative breast cancer: A competing risk model based on the SEER cohort study. Gland Surg. 2022, 11, 1961–1975. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jorgensen, I.; Zhang, Y.; Krantz, B.A.; Miao, E.A. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 2016, 213, 2113–2128. [Google Scholar] [CrossRef]
- Liu, X.; Nie, L.; Zhang, Y.; Yan, Y.; Wang, C.; Colic, M.; Olszewski, K.; Horbath, A.; Chen, X.; Lei, G.; et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 2023, 25, 404–414. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Deng, L.; He, S.; Guo, N.; Tian, W.; Zhang, W.; Luo, L. Molecular mechanisms of ferroptosis and relevance to inflammation. Inflamm. Res. 2023, 72, 281–299. [Google Scholar] [CrossRef]
- Machesky, L.M. Deadly actin collapse by disulfidptosis. Nat. Cell Biol. 2023, 25, 375–376. [Google Scholar] [CrossRef]
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020, 22, 476–486. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Joly, J.H.; Delfarah, A.; Phung, P.S.; Parrish, S.; Graham, N.A. A synthetic lethal drug combination mimics glucose deprivation-induced cancer cell death in the presence of glucose. J. Biol. Chem. 2020, 295, 1350–1365. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Parrini, E.; Conti, V.; Dobyns, W.B.; Guerrini, R. Genetic Basis of Brain Malformations. Mol. Syndromol. 2016, 7, 220–233. [Google Scholar] [CrossRef]
- Quick, Q.; Skalli, O. Alpha-actinin 1 and alpha-actinin 4: Contrasting roles in the survival, motility, and RhoA signaling of astrocytoma cells. Exp. Cell Res. 2010, 316, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, B.; Jones, S.L.; Shiraishi-Yamaguchi, Y.; Lingelbach, M.; Manor, U.; Svitkina, T.M.; Higgs, H.N.; Shih, A.Y.; Halpain, S. INF2-mediated actin filament reorganization confers intrinsic resilience to neuronal ischemic injury. Nat. Commun. 2022, 13, 6037. [Google Scholar] [CrossRef]
- Park, I.; Han, C.; Jin, S.; Lee, B.; Choi, H.; Kwon, J.T.; Kim, D.; Kim, J.; Lifirsu, E.; Park, W.J.; et al. Myosin regulatory light chains are required to maintain the stability of myosin II and cellular integrity. Biochem. J. 2011, 434, 171–180. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Zhang, M.Y.; Deng, M.L.; Weng, N.Q.; Wang, H.Y.; Wu, S.X. Low serum level of miR-485-3p predicts poor survival in patients with glioblastoma. PLoS ONE 2017, 12, e0184969. [Google Scholar] [CrossRef]
- Zhu, L.; Deng, H.; Hu, J.; Huang, S.; Xiong, J.; Deng, J. The promising role of miR-296 in human cancer. Pathol. Res. Pract. 2018, 214, 1915–1922. [Google Scholar] [CrossRef]
- Ameri, A.; Ahmed, H.M.; Pecho, R.D.C.; Arabnozari, H.; Sarabadani, H.; Esbati, R.; Mirabdali, S.; Yazdani, O. Diverse activity of miR-150 in Tumor development: Shedding light on the potential mechanisms. Cancer Cell Int. 2023, 23, 261. [Google Scholar] [CrossRef]
- Bian, W.; Li, Y.; Zhu, H.; Gao, S.; Niu, R.; Wang, C.; Zhang, H.; Qin, X.; Li, S. miR-493 by regulating of c-Jun targets Wnt5a/PD-L1-inducing esophageal cancer cell development. Thorac. Cancer. 2021, 12, 1579–1588. [Google Scholar] [CrossRef]
- Song, M.; Xing, X. miR-6742-5p regulates the invasion and migration of lung adenocarcinoma cells via mediating FGF8/ERK12/MMP9/MMP2 signaling pathway. Aging 2023, 15, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, W.; Li, Y.; Ma, L.; Ji, Z. Leveraging a disulfidptosis-based signature to improve the survival and drug sensitivity of bladder cancer patients. Front. Immunol. 2023, 14, 1198878. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Guo, X.; Guo, Q.; Gupta, N.; Ji, N.; Shen, B.; Xiao, L.; Feng, Y. Metabolic flexibilities and vulnerabilities in the pentose phosphate pathway of the zoonotic pathogen Toxoplasma gondii. PLoS Pathog. 2022, 18, e1010864. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Liu, Q.; Xing, F.; Zeng, C.; Wang, W. Disulfidptosis: A new form of programmed cell death. J. Exp. Clin. Cancer Res. 2023, 42, 137. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gu, Y.; Tang, S.; Wang, Z.; Cai, L.; Lian, H.; Shen, Y.; Zhou, Y. A pan-cancer analysis of the prognostic and immunological role of β-actin (ACTB) in human cancers. Bioengineered 2021, 12, 6166–6185. [Google Scholar] [CrossRef]
- Guo, C.; Liu, S.; Wang, J.; Sun, M.Z.; Greenaway, F.T. ACTB in cancer. Clin. Chim. Acta 2013, 417, 39–44. [Google Scholar] [CrossRef]
- Liu, K.; Gao, R.; Wu, H.; Wang, Z.; Han, G. Single-cell analysis reveals metastatic cell heterogeneity in clear cell renal cell carcinoma. J. Cell. Mol. Med. 2021, 25, 4260–4274. [Google Scholar] [CrossRef]
- Tentler, D.; Lomert, E.; Novitskaya, K.; Barlev, N.A. Role of ACTN4 in Tumorigenesis, Metastasis, and EMT. Cells 2019, 8, 1427. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fung, T.S.; Chakrabarti, R.; Higgs, H.N. The multiple links between actin and mitochondria. Nat. Rev. Mol. Cell Biol. 2023, 24, 651–667. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.Y.; Huang, B.M.; Tang, T.K.; Chao, Y.Y.; Xiao, X.Y.; Lee, P.R.; Yang, L.Y.; Wang, C.Y. Genotoxic stress-activated DNA-PK-p53 cascade and autophagy cooperatively induce ciliogenesis to maintain the DNA damage response. Cell Death Differ. 2021, 28, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Kriger, D.; Novitskaya, K.; Vasileva, G.; Lomert, E.; Aksenov, N.D.; Barlev, N.A.; Tentler, D. Alpha-actnin-4 (ACTN4) selectively affects the DNA double-strand breaks repair in non-small lung carcinoma cells. Biol. Direct. 2022, 17, 40. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Casadonte, L.; Verhoeff, B.J.; Piek, J.J.; VanBavel, E.; Spaan, J.A.E.; Siebes, M. Influence of increased heart rate and aortic pressure on resting indices of functional coronary stenosis severity. Basic Res. Cardiol. 2017, 112, 61. [Google Scholar] [CrossRef]
- Li, J.; Chen, L.; Xiong, Y.; Zheng, X.; Xie, Q.; Zhou, Q.; Shi, L.; Wu, C.; Jiang, J.; Wang, H. Knockdown of PD-L1 in Human Gastric Cancer Cells Inhibits Tumor Progression and Improves the Cytotoxic Sensitivity to CIK Therapy. Cell. Physiol. Biochem. 2017, 41, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, H.R.; Gao, D.; Pararasa, C. Redox regulation in metabolic programming and inflammation. Redox Biol. 2017, 12, 50–57. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, P.; Guo, J.; Hu, N.; Wang, S.; Li, D.; Hu, S.; Ren, J.; Cao, F.; Chen, Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017, 13, 498–507. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, C.; Yu, N.; Si, L.; Zhu, L.; Zeng, A.; Liu, Z.; Wang, X. INF2 regulates oxidative stress-induced apoptosis in epidermal HaCaT cells by modulating the HIF1 signaling pathway. Biomed. Pharmacother. 2019, 111, 151–161. [Google Scholar] [CrossRef]
- Sellers, J.R.; Heissler, S.M. Nonmuscle myosin-2 isoforms. Curr. Biol. 2019, 29, R275–R278. [Google Scholar] [CrossRef]
- Vierthaler, M.; Sun, Q.; Wang, Y.; Steinfass, T.; Poelchen, J.; Hielscher, T.; Novak, D.; Umansky, V.; Utikal, J. ADCK2 Knockdown Affects the Migration of Melanoma Cells via MYL6. Cancers 2022, 14, 1071. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, J.; Chen, A.; Liao, R.; Duan, Y.; Xu, Y.; Tao, L. Combined transplantation of neural stem cells and bone marrow mesenchymal stem cells promotes neuronal cell survival to alleviate brain damage after cardiac arrest via microRNA-133b incorporated in extracellular vesicles. Aging 2021, 13, 262–278. [Google Scholar] [CrossRef] [PubMed]
- Mead, B.; Tomarev, S. Bone Marrow-Derived Mesenchymal Stem Cells-Derived Exosomes Promote Survival of Retinal Ganglion Cells Through miRNA-Dependent Mechanisms. Stem Cells Transl. Med. 2017, 6, 1273–1285. [Google Scholar] [CrossRef]
- Vishnoi, A.; Rani, S. miRNA Biogenesis and Regulation of Diseases: An Updated Overview. Methods Mol. Biol. 2023, 2595, 1–12. [Google Scholar] [CrossRef]
- Li, S.; Lei, Z.; Sun, T. The role of microRNAs in neurodegenerative diseases: A review. Cell Biol. Toxicol. 2023, 39, 53–83. [Google Scholar] [CrossRef]
- Hill, M.; Tran, N. miRNA interplay: Mechanisms and consequences in cancer. Dis. Model Mech. 2021, 14, dmm047662. [Google Scholar] [CrossRef]
- Hussen, B.M.; Hidayat, H.J.; Salihi, A.; Sabir, D.K.; Taheri, M.; Ghafouri-Fard, S. MicroRNA: A signature for cancer progression. Biomed. Pharmacother. 2021, 138, 111528. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Zhao, J.X.; Jiang, M.Y.; Yang, L.P.; Sun, M.L.; Wang, H.W. MiR-193 promotes cell proliferation and invasion by ING5/PI3K/AKT pathway of triple-negative breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3122–3129. [Google Scholar] [CrossRef]
- Majumder, A.; Sen, D. Artificial intelligence in cancer diagnostics and therapy: Current perspectives. Indian J. Cancer 2021, 58, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, M.; Wojtusciszyn, A.; Favre, L.; Boughorbel, S.; Shan, J.; Letaief, K.B.; Pitteloud, N.; Chouchane, L. Precision medicine in the era of artificial intelligence: Implications in chronic disease management. J. Transl. Med. 2020, 18, 472. [Google Scholar] [CrossRef]
- Gupta, R.; Kumari, S.; Senapati, A.; Ambasta, R.K.; Kumar, P. New era of artificial intelligence and machine learning-based detection, diagnosis, and therapeutics in Parkinson’s disease. Ageing Res. Rev. 2023, 90, 102013. [Google Scholar] [CrossRef] [PubMed]
- Bhinder, B.; Gilvary, C.; Madhukar, N.S.; Elemento, O. Artificial Intelligence in Cancer Research and Precision Medicine. Cancer Discov. 2021, 11, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Prelaj, A.; Miskovic, V.; Zanitti, M.; Trovo, F.; Genova, C.; Viscardi, G.; Rebuzzi, S.E.; Mazzeo, L.; Provenzano, L.; Kosta, S.; et al. Artificial intelligence for predictive biomarker discovery in immuno-oncology: A systematic review. Ann. Oncol. 2024, 35, 29–65. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer’s disease: A meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Dhapola, R.; Reddy, D.H. Apoptosis in Alzheimer’s disease: Insight into the signaling pathways and therapeutic avenues. Apoptosis 2023, 28, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Vidović, M.; Rikalovic, M.G. Alpha-Synuclein Aggregation Pathway in Parkinson’s Disease: Current Status and Novel Therapeutic Approaches. Cells 2022, 11, 1732. [Google Scholar] [CrossRef]
- Mätlik, K.; Baffuto, M.; Kus, L.; Deshmukh, A.L.; Davis, D.A.; Paul, M.R.; Carroll, T.S.; Caron, M.C.; Masson, J.Y.; Pearson, C.E.; et al. Cell-type-specific CAG repeat expansions and toxicity of mutant Huntingtin in human striatum and cerebellum. Nat. Genet. 2024, 56, 383–394. [Google Scholar] [CrossRef]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Yoshida, G.J. Regulation of heterogeneous cancer-associated fibroblasts: The molecular pathology of activated signaling pathways. J. Exp. Clin. Cancer Res. 2020, 39, 112. [Google Scholar] [CrossRef]
- Wang, X.; Lin, J.; Li, Z.; Wang, M. In what area of biology has a “new” type of cell death been discovered? Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188955. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Abbreviation |
|---|---|
| Adrenocortical Cancer | ACC |
| Bladder Cancer | BLCA |
| Breast Cancer | BRCA |
| Cervical Cancer | CESC |
| Bile Duct Cancer | CHOL |
| Colon Cancer | COAD |
| Colon and Rectal Cancer | COADREAD |
| Large B-cell Lymphoma | DLBC |
| Esophageal Cancer | ESCA |
| FFPE Pilot Phase II | FPPP |
| Glioblastoma | GBM |
| Lower Grade Glioma and Glioblastoma | GBMLGG |
| Head and Neck Cancer | HNSC |
| Kidney Chromophobe | KICH |
| Kidney Clear Cell Carcinoma | KIRC |
| Kidney Papillary Cell Carcinoma | KIRP |
| Acute Myeloid Leukemia | LAML |
| Lower Grade Glioma | LGG |
| Liver Cancer | LIHC |
| Lung Adenocarcinoma | LUAD |
| Lung Cancer | LUNG |
| Lung Squamous Cell Carcinoma | LUSC |
| Mesothelioma | MESO |
| Ovarian Cancer | OV |
| Pancreatic Cancer | PAAD |
| Pan-Cancer | PANCAN |
| Pheochromocytoma and Paraganglioma | PCPG |
| Prostate Cancer | PRAD |
| Rectal Cancer | READ |
| Sarcoma | SARC |
| Melanoma | SKCM |
| Stomach Cancer | STAD |
| Testicular Cancer | TGCT |
| Thyroid Cancer | THCA |
| Thymoma | THYM |
| Endometrioid Cancer | UCEC |
| Uterine Carcinosarcoma | UCS |
| Ocular melanomas | UVM |
| CurPocket ID | Vina Score | Contact Residues | |
|---|---|---|---|
| ACTB–cyclophosphamide | C3 | −5.5 | Chain A: ASP11 GLY13 SER14 GLY15 MET16 LYS18 TYR69 GLY74 GLN137 SER155 GLY156 ASP157 GLY182 ARG183 ARG210 LYS213 GLU214 GLY301 GLY302 THR303 MET305 TYR306 LYS336 |
| C2 | −5.4 | Chain A: ASP11 ASN12 GLY13 SER14 GLY15 MET16 LYS18 TYR69 GLY74 GLU107 ALA108 PRO109 LEU110 ASN111 PRO112 LYS113 ARG116 ILE136 GLN137 ALA138 VAL139 SER141 LEU142 SER145 GLY146 ARG147 THR149 GLY150 VAL152 ASP154 SER155 GLY156 ASP157 HIS161 ILE165 PRO172 ILE175 ARG177 GLY182 ARG183 VAL298 LEU299 SER300 GLY301 GLY302 THR303 ILE330 PRO332 ARG335 LYS336 SER338 VAL339 HIS371 CYS374 PHE375 | |
| C1 | −5.0 | Chain A: ASP3 ASP4 PHE21 ARG28 ALA29 TYR91 ASN92 GLU93 LEU94 ARG95 VAL96 ALA97 PRO98 GLU99 GLU100 Chain B: ASP394 CYS397 TYR398 LEU466 LYS467 PHE468 LYS483 LYS484 LEU487 GLY488 LEU489 TRP491 GLN492 ARG495 PHE496 LEU499 GLN500 LEU502 LYS503 ARG506 MET514 THR515 ASP516 ALA517 VAL604 ASN605 GLN606 LYS607 | |
| C5 | −4.0 | Chain B: ASP408 SER409 TYR410 ASN412 GLU416 ASP417 VAL418 ARG419 ASN420 TRP422 ILE423 GLU426 PRO444 PRO445 ILE446 LYS447 LYS452 VAL479 | |
| C4 | −3.6 | Chain B: ASP408 SER409 TYR410 ASN412 GLU416 ASP417 VAL418 ARG419 ASN420 TRP422 ILE423 GLU426 PRO444 PRO445 ILE446 LYS447 LYS452 VAL479 | |
| ACTB–ethinyl estradiol | C2 | −10.0 | Chain A: ASP11 ASN12 GLY13 SER14 GLY15 MET16 LYS18 TYR69 HIS73 GLU107 ALA108 PRO109 LEU110 ASN111 PRO112 LYS113 ARG116 ILE136 GLN137 ALA138 VAL139 SER141 LEU142 SER145 GLY146 ARG147 THR148 THR149 GLY150 ILE151 VAL152 SER155 GLY156 ASP157 HIS161 ILE165 GLY168 PRO172 ILE175 ARG177 GLY182 ARG183 THR186 LYS213 GLU214 VAL298 LEU299 SER300 GLY301 GLY302 THR303 PRO332 PRO333 GLU334 ARG335 LYS336 TYR337 SER338 VAL339 HIS371 ARG372 CYS374 PHE375 |
| C3 | −8.3 | Chain A: ASP11 ASN12 GLY13 SER14 GLY15 MET16 LYS18 TYR69 GLN137 SER155 GLY156 ASP157 GLY182 ARG183 LEU185 THR186 ARG210 LYS213 GLU214 LYS215 CYS217 GLY301 GLY302 THR303 MET305 TYR306 PRO307 LYS336 TYR337 SER338 VAL339 | |
| C5 | −6.0 | Chain B: ASP408 SER409 TYR410 VAL411 ASN412 GLU416 ASP417 VAL418 ARG419 ASN420 TRP422 ILE423 GLU426 LYS443 PRO444 PRO445 ILE446 LYS452 VAL479 | |
| C4 | −5.6 | Chain A: GLN59 SER60 LYS61 ARG62 GLY63 ILE64 GLY197 TYR198 SER199 PHE200 THR202 THR203 ALA204 GLU205 ILE208 ASP211 LYS215 TYR240 LEU242 PRO243 ASP244 GLN246 VAL247 ILE248 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Kong, X.; Wei, J. Disulfidptosis: A New Target for Parkinson’s Disease and Cancer. Curr. Issues Mol. Biol. 2024, 46, 10038-10064. https://doi.org/10.3390/cimb46090600
Liu T, Kong X, Wei J. Disulfidptosis: A New Target for Parkinson’s Disease and Cancer. Current Issues in Molecular Biology. 2024; 46(9):10038-10064. https://doi.org/10.3390/cimb46090600
Chicago/Turabian StyleLiu, Tingting, Xiangrui Kong, and Jianshe Wei. 2024. "Disulfidptosis: A New Target for Parkinson’s Disease and Cancer" Current Issues in Molecular Biology 46, no. 9: 10038-10064. https://doi.org/10.3390/cimb46090600
APA StyleLiu, T., Kong, X., & Wei, J. (2024). Disulfidptosis: A New Target for Parkinson’s Disease and Cancer. Current Issues in Molecular Biology, 46(9), 10038-10064. https://doi.org/10.3390/cimb46090600