
Chimeric Antigen Receptor Cell Therapy: Empowering Treatment Strategies for Solid Tumors



Abstract
1. Introduction
2. What Can Go Wrong with CAR-T Cell Therapy for Solid Tumors?
2.1. Physical Barriers Within the Tumor Microenvironment
2.2. Trafficking and Penetration into Neoplastic Tissue
2.3. Immunosuppressive Tumor Microenvironment
2.4. Tumor-Infiltrating Immune Cells Reversed the Hostile Tumor Immune Environment
2.5. Soluble Inhibitors Impair the Functionality of CAR-T Cells
2.6. Immune Checkpoint Overexpression Hinders the Effector Functions of CAR-T Cells
3. Strategies to Address Challenges in CAR-T Cell Therapy for Solid Tumors
4. Overview of CAR-T Cell Therapy Application in Solid Tumors
4.1. Clinical Insights on CAR-T Cell Applications for Solid and Brain Tumors
4.2. Challenges of CAR-T Cell Immunotherapy for Solid Tumors
5. If CAR-T Therapy Is Unsuccessful, Should We Consider an Alternative Approach?
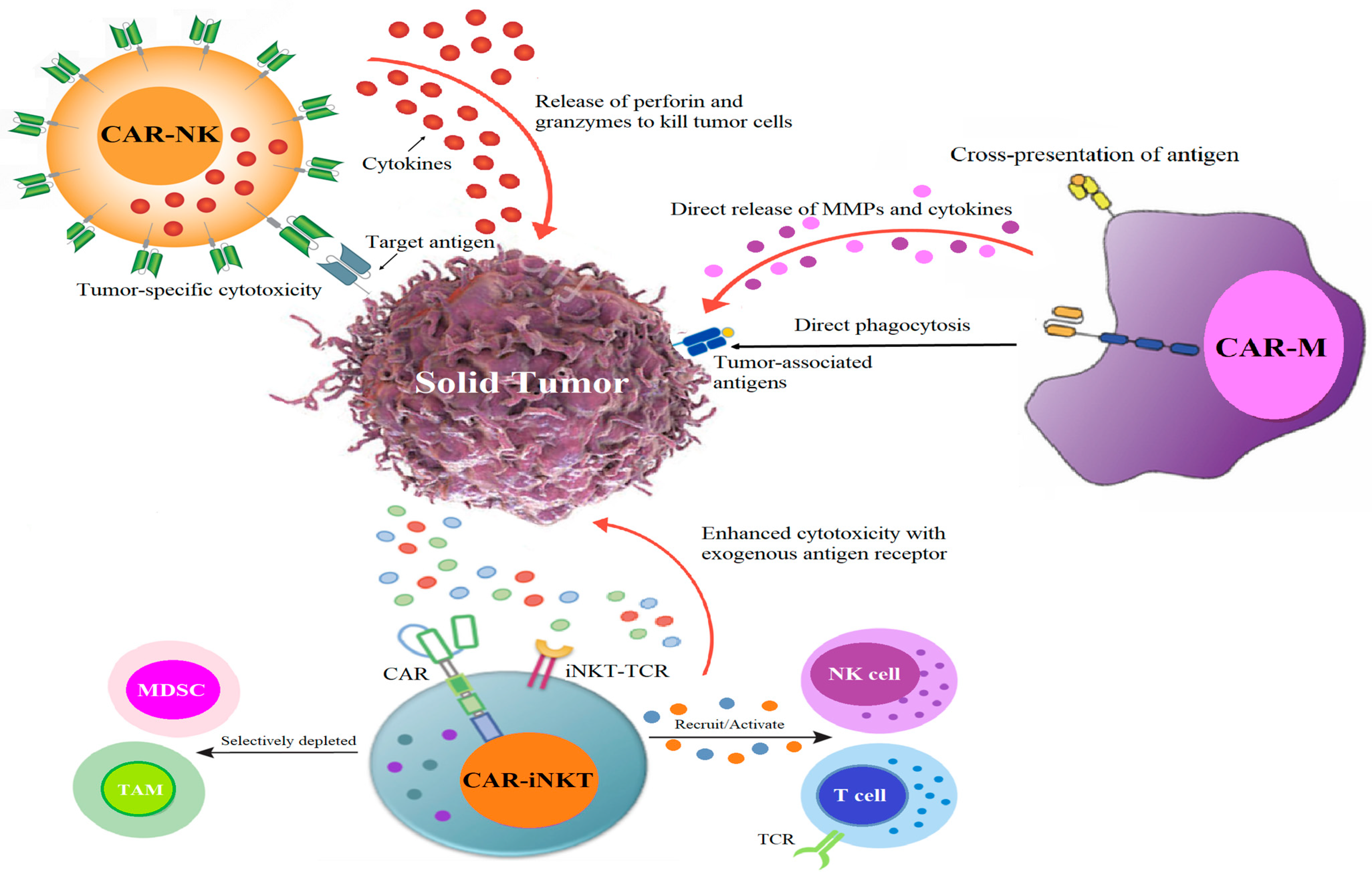
5.1. CAR Race Towards Cancer Immunotherapy: Exploring CAR-NK, CAR-iNKT, and CAR-M Therapies
5.2. CAR-NK: An Encouraging Substitute for CAR-T Therapy
5.3. CAR-iNKT Immunotherapy: A Novel Path for CAR-Based Cancer Immunotherapy
5.3.1. Development of iNKT Cells
5.3.2. Antitumoral Role of iNKT Cells
5.3.3. iNKT Protects from GVHD
5.3.4. Essential Cytokines Enhance CAR-iNKT Activity
5.4. CAR-Macrophage: Pioneering Advancements in Cellular Immunotherapy
5.4.1. Preclinical and Clinical Studies of CAR-Ms
5.4.2. The Advantages, Obstacles, and Prospective Trajectory of CAR-Ms
5.4.3. The Future Direction of CAR-M Therapy
5.4.4. CAR-M Therapy Alongside Additional Immunotherapeutic Approaches
6. Conclusions
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef] [PubMed]
- Chohan, K.L.; Siegler, E.L.; Kenderian, S.S. CAR-T Cell Therapy: The Efficacy and Toxicity Balance. Curr. Hematol. Malig. Rep. 2023, 18, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Choudhery, M.S.; Arif, T.; Mahmood, R.; Harris, D.T. CAR-T-Cell-Based Cancer Immunotherapies: Potentials, Limitations, and Future Prospects. J. Clin. Med. 2024, 13, 3202. [Google Scholar] [CrossRef] [PubMed]
- Kankeu Fonkoua, L.A.; Sirpilla, O.; Sakemura, R.; Siegler, E.L.; Kenderian, S.S. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol. Ther. Oncolytics 2022, 25, 69–77. [Google Scholar] [CrossRef]
- Elahi, R.; Heidary, A.H.; Hadiloo, K.; Esmaeilzadeh, A. Chimeric Antigen Receptor-Engineered Natural Killer (CAR NK) Cells in Cancer Treatment; Recent Advances and Future Prospects. Stem Cell Rev. Rep. 2021, 17, 2081–2106. [Google Scholar] [CrossRef]
- Moscarelli, J.; Zahavi, D.; Maynard, R.; Weiner, L.M. The Next Generation of Cellular Immunotherapy: Chimeric Antigen Receptor-Natural Killer Cells. Transplant. Cell. Ther. 2022, 28, 650–656. [Google Scholar] [CrossRef]
- Marofi, F.; Abdul-Rasheed, O.F.; Rahman, H.S.; Budi, H.S.; Jalil, A.T.; Yumashev, A.V.; Hassanzadeh, A.; Yazdanifar, M.; Motavalli, R.; Chartrand, M.S.; et al. CAR-NK cell in cancer immunotherapy; A promising frontier. Cancer Sci. 2021, 112, 3427–3436. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, W.; Yang, J.; Yang, J.; Wang, W. Chimeric antigen receptor engineered natural killer cells for cancer therapy. Exp. Hematol. Oncol. 2023, 12, 70. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Y.; Dou, Z.; Delfanti, G.; Tsahouridis, O.; Pellegry, C.M.; Zingarelli, M.; Atassi, G.; Woodcock, M.G.; Casorati, G.; et al. CAR-redirected natural killer T cells demonstrate superior antitumor activity to CAR-T cells through multimodal CD1d-dependent mechanisms. Nat. Cancer 2024, 5, 1607–1621. [Google Scholar] [CrossRef]
- Moraes Ribeiro, E.; Secker, K.A.; Nitulescu, A.M.; Schairer, R.; Keppeler, H.; Wesle, A.; Schmid, H.; Schmitt, A.; Neuber, B.; Chmiest, D.; et al. PD-1 checkpoint inhibition enhances the antilymphoma activity of CD19-CAR-iNKT cells that retain their ability to prevent alloreactivity. J. Immunother. Cancer 2024, 12, e007829. [Google Scholar] [CrossRef]
- Lu, J.; Ma, Y.; Li, Q.; Xu, Y.; Xue, Y.; Xu, S. CAR Macrophages: A promising novel immunotherapy for solid tumors and beyond. Biomark. Res. 2024, 12, 86. [Google Scholar] [CrossRef]
- Su, S.; Lei, A.; Wang, X.; Lu, H.; Wang, S.; Yang, Y.; Li, N.; Zhang, Y.; Zhang, J. Induced CAR-Macrophages as a Novel Therapeutic Cell Type for Cancer Immune Cell Therapies. Cells 2022, 11, 1652. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, Y.; Jia, J.; Fang, Y.; Yang, Y.; Yuan, W.; Hu, J. Advances in Engineered Macrophages: A New Frontier in Cancer Immunotherapy. Cell Death Dis. 2024, 15, 238. [Google Scholar] [CrossRef] [PubMed]
- Donnadieu, E.; Dupré, L.; Pinho, L.G.; Cotta-de-Almeida, V. Surmounting the obstacles that impede effective CAR T cell trafficking to solid tumors. J. Leukoc. Biol. 2020, 108, 1067–1079. [Google Scholar] [CrossRef]
- Yuan, Z.; Li, Y.; Zhang, S.; Wang, X.; Dou, H.; Yu, X.; Zhang, Z.; Yang, S.; Xiao, M. Extracellular matrix remodeling in tumor progression and immune escape: From mechanisms to treatments. Mol. Cancer 2023, 22, 48. [Google Scholar] [CrossRef]
- Rojas-Quintero, J.; Díaz, M.P.; Palmar, J.; Galan-Freyle, N.J.; Morillo, V.; Escalona, D.; González-Torres, H.J.; Torres, W.; Navarro-Quiroz, E.; Rivera-Porras, D.; et al. Car T Cells in Solid Tumors: Overcoming Obstacles. Int. J. Mol. Sci. 2024, 25, 4170. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Liu, Z.L.; Chen, H.H.; Zheng, L.L.; Sun, L.P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for Cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, H.; Li, F.; Huang, S.; Chen, F.; Li, Y. Bright future or blind alley? CAR-T cell therapy for solid tumors. Front. Immunol. 2023, 14, 1045024. [Google Scholar] [CrossRef]
- Daei Sorkhabi, A.; Mohamed Khosroshahi, L.; Sarkesh, A.; Mardi, A.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Baradaran, B. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front. Immunol. 2023, 14, 1113882. [Google Scholar] [CrossRef]
- Guzman, G.; Pellot, K.; Reed, M.R.; Rodriguez, A. CAR T-cells to treat brain tumors. Brain Res. Bull. 2023, 196, 76–98. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Riddell, S.R. Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J. Immunol. 2018, 200, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Foeng, J.; Comerford, I.; McColl, S.R. Harnessing the chemokine system to home CAR-T cells into solid tumors. Cell Rep. Med. 2022, 3, 100543. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, Y.; Shao, Y.; Zhang, Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J. Hematol. Oncol. 2020, 13, 54. [Google Scholar] [CrossRef]
- Charrot, S.; Hallam, S. CAR-T Cells: Future Perspectives. Hemasphere 2019, 3, e188. [Google Scholar] [CrossRef]
- Liu, J.; Bai, Y.; Li, Y.; Li, X.; Luo, K. Reprogramming the immunosuppressive tumor microenvironment through nanomedicine: An immunometabolism perspective. EBioMedicine 2024, 107, 105301. [Google Scholar] [CrossRef]
- Xia, X.; Yang, Z.; Lu, Q.; Liu, Z.; Wang, L.; Du, J.; Li, Y.; Yang, D.H.; Wu, S. Reshaping the tumor immune microenvironment to improve CAR-T cell-based cancer immunotherapy. Mol. Cancer 2024, 23, 175. [Google Scholar] [CrossRef]
- Cortellino, S.; Longo, V.D. Metabolites and Immune Response in Tumor Microenvironments. Cancers 2023, 15, 3898. [Google Scholar] [CrossRef]
- Shi, R.; Tang, Y.Q.; Miao, H. Metabolism in tumor microenvironment: Implications for cancer immunotherapy. MedComm 2020, 1, 47–68. [Google Scholar] [CrossRef]
- Zheng, Y.; Xu, R.; Chen, X.; Lu, Y.; Zheng, J.; Lin, Y.; Lin, P.; Zhao, X.; Cui, L. Metabolic gatekeepers: Harnessing tumor-derived metabolites to optimize T cell-based immunotherapy efficacy in the tumor microenvironment. Cell Death Dis. 2024, 15, 775. [Google Scholar] [CrossRef] [PubMed]
- Poorebrahim, M.; Melief, J.; Pico de Coaña, Y.; Wickström, S.L.; Cid-Arregui, A.; Kiessling, R. Counteracting CAR T cell dysfunction. Oncogene 2021, 40, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, S.; Wang, D.; Liu, S.; Xiao, T.; Gu, W.; Yang, H.; Wang, H.; Yang, M.; Chen, P. Metabolic reprogramming and immune evasion: The interplay in the tumor microenvironment. Biomark. Res. 2024, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Li, K.; Ni, Y.; Liang, X.; Zhao, X. Myeloid-Derived Suppressor Cells: Implications in the Resistance of Malignant Tumors to T Cell-Based Immunotherapy. Front. Cell Dev. Biol. 2021, 9, 707198. [Google Scholar] [CrossRef]
- Lu, J.; Luo, Y.; Rao, D.; Wang, T.; Lei, Z.; Chen, X.; Zhang, B.; Li, Y.; Liu, B.; Xia, L.; et al. Myeloid-derived suppressor cells in cancer: Therapeutic targets to overcome tumor immune evasion. Exp. Hematol. Oncol. 2024, 13, 39. [Google Scholar] [CrossRef]
- Dysthe, M.; Parihar, R. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 117–140. [Google Scholar]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in Cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef]
- Huang, R.; Kang, T.; Chen, S. The role of tumor-associated macrophages in tumor immune evasion. J. Cancer Res. Clin. Oncol. 2024, 150, 238. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Maalej, K.M.; Merhi, M.; Inchakalody, V.P.; Mestiri, S.; Alam, M.; Maccalli, C.; Cherif, H.; Uddin, S.; Steinhoff, M.; Marincola, F.M.; et al. CAR-cell therapy in the era of solid tumor treatment: Current challenges and emerging therapeutic advances. Mol. Cancer 2023, 22, 20. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Anderson, K.C. B cell maturation antigen (BCMA)-based immunotherapy for multiple myeloma. Expert Opin. Biol. Ther. 2019, 19, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Akbari, P.; Katsarou, A.; Daghighian, R.; van Mil, L.W.H.G.; Huijbers, E.J.M.; Griffioen, A.W.; van Beijnum, J.R. Directing CAR T cells towards the tumor vasculature for the treatment of solid tumors. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188701. [Google Scholar] [CrossRef]
- Boccalatte, F.; Mina, R.; Aroldi, A.; Leone, S.; Suryadevara, C.M.; Placantonakis, D.G.; Bruno, B. Advances and Hurdles in CAR T Cell Immune Therapy for Solid Tumors. Cancers 2022, 14, 5108. [Google Scholar] [CrossRef]
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.E.; Badie, B. Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front. Immunol. 2017, 8, 1850. [Google Scholar] [CrossRef]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef]
- Lin, X.; Kang, K.; Chen, P.; Zeng, Z.; Li, G.; Xiong, W.; Yi, M.; Xiang, B. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol. Cancer 2024, 23, 108. [Google Scholar] [CrossRef]
- Lv, Y.; Luo, X.; Xie, Z.; Qiu, J.; Yang, J.; Deng, Y.; Long, R.; Tang, G.; Zhang, C.; Zuo, J. Prospects and challenges of CAR-T cell therapy combined with ICIs. Front. Oncol. 2024, 14, 1368732. [Google Scholar] [CrossRef]
- Najafi, S.; Mortezaee, K. Modifying CAR-T cells with anti-checkpoints in cancer immunotherapy: A focus on anti PD-1/PD-L1 antibodies. Life Sci. 2024, 338, 122387. [Google Scholar] [CrossRef]
- Ai, K.; Liu, B.; Chen, X.; Huang, C.; Yang, L.; Zhang, W.; Weng, J.; Du, X.; Wu, K.; Lai, P. Optimizing CAR-T cell therapy for solid tumors: Current challenges and potential strategies. J. Hematol. Oncol. 2024, 17, 105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Z.; Wang, T.; Wang, X.F.; Zhang, Y.Q.; Song, S.X.; Ma, C.Q. Improving the ability of CAR-T cells to hit solid tumors: Challenges and strategies. Pharmacol. Res. 2022, 175, 106036. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Li, J.; Zhao, X.; Wu, Y.; Chen, L. CAR-T cell therapy: Developments, challenges and expanded applications from cancer to autoimmunity. Front. Immunol. 2025, 15, 1519671. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Pan, S.; Wei, X.; Xu, X.; Wei, Q. Arming CAR-T cells with cytokines and more: Innovations in the fourth-generation CAR-T development. Mol. Ther. 2023, 31, 3146–3162. [Google Scholar] [CrossRef]
- Whilding, L.M.; Maher, J. CAR T-cell immunotherapy: The path from the by-road to the freeway? Mol. Oncol. 2015, 9, 1994–2018. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Wagner, J.; Wickman, E.; DeRenzo, C.; Gottschalk, S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol. Ther. 2020, 28, 2320–2339. [Google Scholar] [CrossRef]
- Hou, B.; Tang, Y.; Li, W.; Zeng, Q.; Chang, D. Efficiency of CAR-T Therapy for Treatment of Solid Tumor in Clinical Trials: A Meta-Analysis. Dis. Markers 2019, 2019, 3425291. [Google Scholar] [CrossRef]
- Dagar, G.; Gupta, A.; Masoodi, T.; Nisar, S.; Merhi, M.; Hashem, S.; Chauhan, R.; Dagar, M.; Mirza, S.; Bagga, P.; et al. Harnessing the potential of CAR-T cell therapy: Progress, challenges, and future directions in hematological and solid tumor treatments. J. Transl. Med. 2023, 21, 449. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- De Marco, R.C.; Monzo, H.J.; Ojala, P.M. CAR T Cell Therapy: A Versatile Living Drug. Int. J. Mol. Sci. 2023, 24, 6300. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Rui, W.; Zhao, X.; Lin, X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell. Mol. Immunol. 2021, 18, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- O’Cathail, S.M.; Pokrovska, T.D.; Maughan, T.S.; Fisher, K.D.; Seymour, L.W.; Hawkins, M.A. Combining Oncolytic Adenovirus with Radiation-A Paradigm for the Future of Radiosensitization. Front. Oncol. 2017, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, X.; Zhang, W.; Gao, R.; Wei, H.; Yu, C.Y. Current advances in modulating tumor hypoxia for enhanced therapeutic efficacy. Acta Biomater. 2024, 176, 1–27. [Google Scholar] [CrossRef]
- McDonald, P.C.; Chafe, S.C.; Dedhar, S. Overcoming Hypoxia-Mediated Tumor Progression: Combinatorial Approaches Targeting pH Regulation, Angiogenesis and Immune Dysfunction. Front. Cell Dev. Biol. 2016, 4, 27. [Google Scholar] [CrossRef]
- Tang, J.; Zou, Y.; Li, L.; Lu, F.; Xu, H.; Ren, P.; Bai, F.; Niedermann, G.; Zhu, X. BAY 60-6583 Enhances the Antitumor Function of Chimeric Antigen Receptor-Modified T Cells Independent of the Adenosine A2b Receptor. Front. Pharmacol. 2021, 12, 619800. [Google Scholar] [CrossRef]
- Mane, M.M.; Cohen, I.J.; Ackerstaff, E.; Shalaby, K.; Ijoma, J.N.; Ko, M.; Maeda, M.; Albeg, A.S.; Vemuri, K.; Satagopan., J.; et al. Lactate Dehydrogenase A Depletion Alters MyC-CaP Tumor Metabolism, Microenvironment, and CAR T Cell Therapy. Mol. Ther.-Oncolytics 2020, 18, 382–395. [Google Scholar] [CrossRef]
- Huang, M.; Deng, J.; Gao, L.; Zhou, J. Innovative strategies to advance CAR T cell therapy for solid tumors. Am. J. Cancer Res. 2020, 10, 1979–1992. [Google Scholar]
- Kouro, T.; Himuro, H.; Sasada, T. Exhaustion of CAR T cells: Potential causes and solutions. J. Transl. Med. 2022, 20, 239. [Google Scholar] [CrossRef]
- Yi, M.; Zheng, X.; Niu, M.; Zhu, S.; Ge, H.; Wu, K. Combination strategies with PD-1/PD-L1 blockade: Current advances and future directions. Mol. Cancer 2022, 21, 28. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- de Campos, N.S.P.; de Oliveira Beserra, A.; Pereira, P.H.B.; Chaves, A.S.; Fonseca, F.L.A.; da Silva Medina, T.; Dos Santos, T.G.; Wang, Y.; Marasco, W.A.; Suarez, E.R. Immune Checkpoint Blockade via PD-L1 Potentiates More CD28-Based than 4-1BB-Based Anti-Carbonic Anhydrase IX Chimeric Antigen Receptor T Cells. Int. J. Mol. Sci. 2022, 23, 5448. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.; Townsend, M.; O’Neill, K. Tumor Microenvironment Immunosuppression: A Roadblock to CAR T-Cell Advancement in Solid Tumors. Cells 2022, 11, 3626. [Google Scholar] [CrossRef] [PubMed]
- Gatto, L.; Ricciotti, I.; Tosoni, A.; Di Nunno, V.; Bartolini, S.; Ranieri, L.; Franceschi, E. CAR-T cells neurotoxicity from consolidated practice in hematological malignancies to fledgling experience in CNS tumors: Fill the gap. Front. Oncol. 2023, 13, 1206983. [Google Scholar] [CrossRef]
- Li, W.; Wu, L.; Huang, C.; Liu, R.; Li, Z.; Liu, L.; Shan, B. Challenges and strategies of clinical application of CAR-T therapy in the treatment of tumors-a narrative review. Ann. Transl. Med. 2020, 8, 1093. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Li, J.; Chen, P.; Ma, W. The next frontier in immunotherapy: Potential and challenges of CAR-macrophages. Exp. Hematol. Oncol. 2024, 13, 76. [Google Scholar] [CrossRef]
- Shi, Y.; Tomczak, K.; Li, J.; Ochieng, J.K.; Lee, Y.; Haymaker, C. Next-Generation Immunotherapies to Improve Anticancer Immunity. Front. Pharmacol. 2021, 11, 566401. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, G. CAR-iNKT cell therapy: Mechanisms, advantages, and challenges. Curr. Res. Transl. Med. 2024, 73, 103488. [Google Scholar] [CrossRef]
- Sadri, M.; Heidari, S.; Faridzadeh, A.; Roozbehani, M.; Toosi, S.; Mahmoudian, R.A.; Hoseinzadeh, A.; Salmani Fard, M.T.; Arab, F.L.; Fard, S.R.; et al. Potential applications of macrophages in cancer immunotherapy. Biomed. Pharmacother. 2024, 178, 117161. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, G.; Chai, D.; Dang, Y.; Zheng, J.; Li, H. iNKT: A new avenue for CAR-based cancer immunotherapy. Transl. Oncol. 2022, 17, 101342. [Google Scholar] [CrossRef] [PubMed]
- Lim, O.; Jung, M.Y.; Hwang, Y.K.; Shin, E.C. Present and Future of Allogeneic Natural Killer Cell Therapy. Front. Immunol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for Cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, X.; Zhang, X.; Aziz, A.U.R.; Wang, D. CAR-NK Cell Therapy: A Transformative Approach to Overcoming Oncological Challenges. Biomolecules 2024, 14, 1035. [Google Scholar] [CrossRef]
- Sun, X.; Wu, Y.; Li, H.; Zhao, A.; Niu, T. Harmonizing efficacy and safety: The potentials of CAR-NK in effectively addressing severe toxicities of CAR-T therapy in mantle cell lymphoma. Int. J. Surg. 2024, 110, 5871–5872. [Google Scholar] [CrossRef]
- Strizova, Z.; Benesova, I.; Bartolini, R.; Novysedlak, R.; Cecrdlova, E.; Foley, L.K.; Striz, I. M1/M2 macrophages and their overlaps—Myth or reality? Clin. Sci. 2023, 137, 1067–1093. [Google Scholar] [CrossRef]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on “Off-The-Shelf” Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef]
- Berrien-Elliott, M.M.; Jacobs, M.T.; Fehniger, T.A. Allogeneic natural killer cell therapy. Blood 2023, 141, 856–868. [Google Scholar] [CrossRef]
- Maia, A.; Tarannum, M.; Romee, R. Genetic Manipulation Approaches to Enhance the Clinical Application of NK Cell-Based Immunotherapy. Stem Cells Transl. Med. 2024, 13, 230–242. [Google Scholar] [CrossRef]
- Robbins, G.M.; Wang, M.; Pomeroy, E.J.; Moriarity, B.S. Nonviral genome engineering of natural killer cells. Stem Cell Res. Ther. 2021, 12, 350. [Google Scholar] [CrossRef]
- Dong, W.; Kantor, B. Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives. Viruses 2021, 13, 1288. [Google Scholar] [CrossRef]
- Chong, Z.X.; Yeap, S.K.; Ho, W.Y. Transfection types, methods and strategies: A technical review. PeerJ 2021, 9, e11165. [Google Scholar] [CrossRef]
- Ucha, M.; Štach, M.; Kaštánková, I.; Rychlá, J.; Vydra, J.; Lesný, P.; Otáhal, P. Good manufacturing practice-grade generation of CD19 and CD123-specific CAR-T cells using piggyBac transposon and allogeneic feeder cells in patients diagnosed with B-cell non-Hodgkin lymphoma and acute myeloid leukemia. Front. Immunol. 2024, 15, 1415328. [Google Scholar]
- Wrona, E.; Borowiec, M.; Potemski, P. CAR-NK Cells in the Treatment of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5899. [Google Scholar] [CrossRef]
- Yu, Y. The Function of NK Cells in Tumor Metastasis and NK Cell-Based Immunotherapy. Cancers 2023, 15, 2323. [Google Scholar] [CrossRef]
- Khawar, M.B.; Sun, H. CAR-NK Cells: From Natural Basis to Design for Kill. Front. Immunol. 2021, 12, 707542. [Google Scholar] [CrossRef]
- Teng, R.; Wang, Y.; Lv, N.; Zhang, D.; Williamson, R.A.; Lei, L.; Chen, P.; Lei, L.; Wang, B.; Fu, J.; et al. Hypoxia Impairs NK Cell Cytotoxicity through SHP-1-Mediated Attenuation of STAT3 and ERK Signaling Pathways. J. Immunol. Res. 2020, 2020, 4598476. [Google Scholar] [CrossRef]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 70. [Google Scholar] [CrossRef]
- Riggan, L.; Shah, S.; O’Sullivan, T.E. Arrested development: Suppression of NK cell function in the tumor microenvironment. Clin. Transl. Immunol. 2021, 10, e1238. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, F.; Wang, Y. Hypoxic tumor microenvironment: Destroyer of natural killer cell function. Chin. J. Cancer Res. 2024, 36, 138–150. [Google Scholar] [CrossRef]
- Hadiloo, K.; Tahmasebi, S.; Esmaeilzadeh, A. CAR-NKT cell therapy: A new promising paradigm of cancer immunotherapy. Cancer Cell Int. 2023, 23, 86. [Google Scholar] [CrossRef]
- Carreño, L.J.; Saavedra-Ávila, N.A.; Porcelli, S.A. Synthetic glycolipid activators of natural killer T cells as immunotherapeutic agents. Clin. Transl. Immunol. 2016, 5, e69. [Google Scholar] [CrossRef]
- Kitayama, S.; Zhang, R.; Liu, T.Y.; Ueda, N.; Iriguchi, S.; Yasui, Y.; Kawai, Y.; Tatsumi, M.; Hirai, N.; Mizoro, Y.; et al. Cellular Adjuvant Properties, Direct Cytotoxicity of Re-differentiated Vα24 Invariant NKT-like Cells from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2016, 6, 213–227. [Google Scholar] [CrossRef]
- Kim, S.; Lalani, S.; Parekh, V.V.; Wu, L.; Van Kaer, L. Glycolipid ligands of invariant natural killer T cells as vaccine adjuvants. Expert Rev. Vaccines 2008, 7, 1519–1532. [Google Scholar] [CrossRef]
- Hung, J.T.; Huang, J.R.; Yu, A.L. Tailored design of NKT-stimulatory glycolipids for polarization of immune responses. J. Biomed. Sci. 2017, 24, 22. [Google Scholar] [CrossRef]
- Liu, Y.; Dang, Y.; Zhang, C.; Liu, L.; Cai, W.; Li, L.; Fang, L.; Wang, M.; Xu, S.; Wang, G.; et al. IL-21-armored B7H3 CAR-iNKT cells exert potent antitumor effects. iScience 2023, 27, 108597. [Google Scholar] [CrossRef]
- Maas-Bauer, K.; Lohmeyer, J.K.; Hirai, T.; Ramos, T.L.; Fazal, F.M.; Litzenburger, U.M.; Yost, K.E.; Ribado, J.V.; Kambham, N.; Wenokur, A.S.; et al. Invariant natural killer T-cell subsets have diverse graft-versus-host-disease-preventing and antitumor effects. Blood 2021, 138, 858–870. [Google Scholar] [CrossRef]
- Matsuda, J.L.; Mallevaey, T.; Scott-Browne, J.; Gapin, L. CD1d-restricted iNKT cells, the ’Swiss-Army knife’ of the immune system. Curr. Opin. Immunol. 2008, 20, 358–368. [Google Scholar] [CrossRef]
- Li, Y.R.; Zeng, S.; Dunn, Z.S.; Zhou, Y.; Li, Z.; Yu, J.; Wang, Y.C.; Ku, J.; Cook, N.; Kramer, A.; et al. Off-the-shelf third-party HSC-engineered iNKT cells for ameliorating GvHD while preserving GvL effect in the treatment of blood cancers. iScience 2022, 25, 104859. [Google Scholar] [CrossRef]
- Sim, G.C.; Radvanyi, L. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 377–390. [Google Scholar] [CrossRef]
- Yang, Y.; Lundqvist, A. Immunomodulatory Effects of IL-2 and IL-15; Implications for Cancer Immunotherapy. Cancers 2020, 12, 3586. [Google Scholar] [CrossRef]
- Huang, T.; Bei, C.; Hu, Z.; Li, Y. CAR-macrophage: Breaking new ground in cellular immunotherapy. Front. Cell Dev. Biol. 2024, 12, 1464218. [Google Scholar] [CrossRef]
- Meng, S.; Hara, T.; Miura, Y.; Ishii, H. Fibroblast activation protein constitutes a novel target of chimeric antigen receptor T-cell therapy in solid tumors. Cancer Sci. 2024, 115, 3532–3542. [Google Scholar] [CrossRef]
- Feng, F.; Shen, J.; Qi, Q.; Zhang, Y.; Ni, S. Empowering brain tumor management: Chimeric antigen receptor macrophage therapy. Theranostics 2024, 14, 5725–5742. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.U.H.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Herb, M.; Schatz, V.; Hadrian, K.; Hos, D.; Holoborodko, B.; Jantsch, J.; Brigo, N. Macrophage variants in laboratory research: Most are well done, but some are RAW. Front. Cell. Infect. Microbiol. 2024, 14, 1457323. [Google Scholar] [CrossRef]
- Moroni, F.; Dwyer, B.J.; Graham, C.; Pass, C.; Bailey, L.; Ritchie, L.; Mitchell, D.; Glover, A.; Laurie, A.; Doig, S.; et al. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat. Med. 2019, 25, 1560–1565. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.; Dunmall, L.C.; Wang, Y.Y.; Fan, Z.; Cheng, Z.; Wang, Y. The dilemmas and possible solutions for CAR-T cell therapy application in solid tumors. Cancer Lett. 2024, 591, 216871. [Google Scholar] [CrossRef]
- Hadiloo, K.; Taremi, S.; Heidari, M.; Esmaeilzadeh, A. The CAR macrophage cells, a novel generation of chimeric antigen-based approach against solid tumors. Biomark. Res. 2023, 11, 103. [Google Scholar] [CrossRef]
- Giorgioni, L.; Ambrosone, A.; Cometa, M.F.; Salvati, A.L.; Nisticò, R.; Magrelli, A. Revolutionizing CAR T-Cell Therapies: Innovations in Genetic Engineering and Manufacturing to Enhance Efficacy and Accessibility. Int. J. Mol. Sci. 2024, 25, 10365. [Google Scholar] [CrossRef]
- Amiri, M.; Moaveni, A.K.; Majidi Zolbin, M.; Shademan, B.; Nourazarian, A. Optimizing cancer treatment: The synergistic potential of CAR-T cell therapy and CRISPR/Cas9. Front. Immunol. 2024, 15, 1462697. [Google Scholar] [CrossRef]
- Lin, H.; Yang, X.; Ye, S.; Huang, L.; Mu, W. Antigen escape in CAR-T cell therapy: Mechanisms and overcoming strategies. Biomed. Pharmacother. 2024, 178, 117252. [Google Scholar] [CrossRef]
- Gumber, D.; Wang, L.D. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef]
- Mishra, H.K.; Kalyuzhny, A. Revolutionizing Cancer Treatments through Stem Cell-Derived CAR T Cells for Immunotherapy: Opening New Horizons for the Future of Oncology. Cells 2024, 13, 1516. [Google Scholar] [CrossRef]
- Chen, T.; Wang, M.; Chen, Y.; Liu, Y. Current challenges and therapeutic advances of CAR-T cell therapy for solid tumors. Cancer Cell Int. 2024, 24, 133. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Current Challenges | Strategies and Outcomes |
|---|---|
| Enhancing CAR-T cell efficacy by targeting the TME:
|
| Overcoming hypoxia in the TME is crucial for improving cancer treatment outcomes: |
| Enhanced anti-tumor effects of hypoxic tumor mesenchymal stem cells:
|
| CAR-T cell exhaustion impairs the efficacy of immunotherapies, leading to reduced persistence and killing activity of these engineered T cells [69]:
|
| Innovative engineering strategies to enhance CAR-T cell effectiveness in solid tumors:
|
| CAR-T cell therapy resistance to TME-induced immunosuppression [73]:
|
| Type of Therapy | Mechanisms | Advantages | Limitations | Study Number (ClincalTrials.gov) |
|---|---|---|---|---|
| CAR-T cells | CARs bind to tumor cell surface antigens, eliciting anti-tumoral effects through inflammatory cytokines, cytolytic effector function, and the activation of the graft-versus-tumor effect through TNF-related apoptosis-inducing ligand binding to death receptors. |
|
| As of 25 January 2023, there are 1087 CAR-T cell clinical trials listed on ClinicalTrials.gov |
| CAR-NK cells | NK cells have spontaneous cytotoxic activity and can generate target cell death independent of tumor antigen. |
|
| NCT03056339 NCT03415100 NCT02944162 |
| CAR-iNKT cells | iNKT cells exhibit NK-like cytotoxicity, capable of directly eliminating tumor cells and infected cells through the secretion of granzyme and perforin, as well as via Fas/FasL-mediated pathways. |
|
| NCT03774654 |
| CAR-M cells | CAR-Ms effectively destroy cancer cells through tumor-associated antigen-induced phagocytosis, secreting pro-inflammatory cytokines that activate T cells and remodel the TME, bolstering the immune response against tumors. | Potential advantages in homing in on and infiltrating solid tumors. | Unlike T cells, macrophages do not proliferate effectively either in vitro or in vivo after injection, which restricts their therapeutic potential. | NCT04660929 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaing, T.-H.; Hsiao, Y.-W.; Wang, Y.-L. Chimeric Antigen Receptor Cell Therapy: Empowering Treatment Strategies for Solid Tumors. Curr. Issues Mol. Biol. 2025, 47, 90. https://doi.org/10.3390/cimb47020090
Jaing T-H, Hsiao Y-W, Wang Y-L. Chimeric Antigen Receptor Cell Therapy: Empowering Treatment Strategies for Solid Tumors. Current Issues in Molecular Biology. 2025; 47(2):90. https://doi.org/10.3390/cimb47020090
Chicago/Turabian StyleJaing, Tang-Her, Yi-Wen Hsiao, and Yi-Lun Wang. 2025. "Chimeric Antigen Receptor Cell Therapy: Empowering Treatment Strategies for Solid Tumors" Current Issues in Molecular Biology 47, no. 2: 90. https://doi.org/10.3390/cimb47020090
APA StyleJaing, T.-H., Hsiao, Y.-W., & Wang, Y.-L. (2025). Chimeric Antigen Receptor Cell Therapy: Empowering Treatment Strategies for Solid Tumors. Current Issues in Molecular Biology, 47(2), 90. https://doi.org/10.3390/cimb47020090