
Molecular Docking and Pharmacological In Silico Evaluation of Camptothecin and Related Ligands as Promising HER2-Targeted Therapies for Breast Cancer

 ,
,  ,
,  , ,
, ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Molecular Docking Procedure
2.2. Molecular Dynamic
2.3. Prediction of Pharmacological Properties
3. Results
3.1. Molecular Docking
3.1.1. Coupling Energies (ΔG) and Dissociation Constants of Ligands Against the HER2 and EGFR Receptors
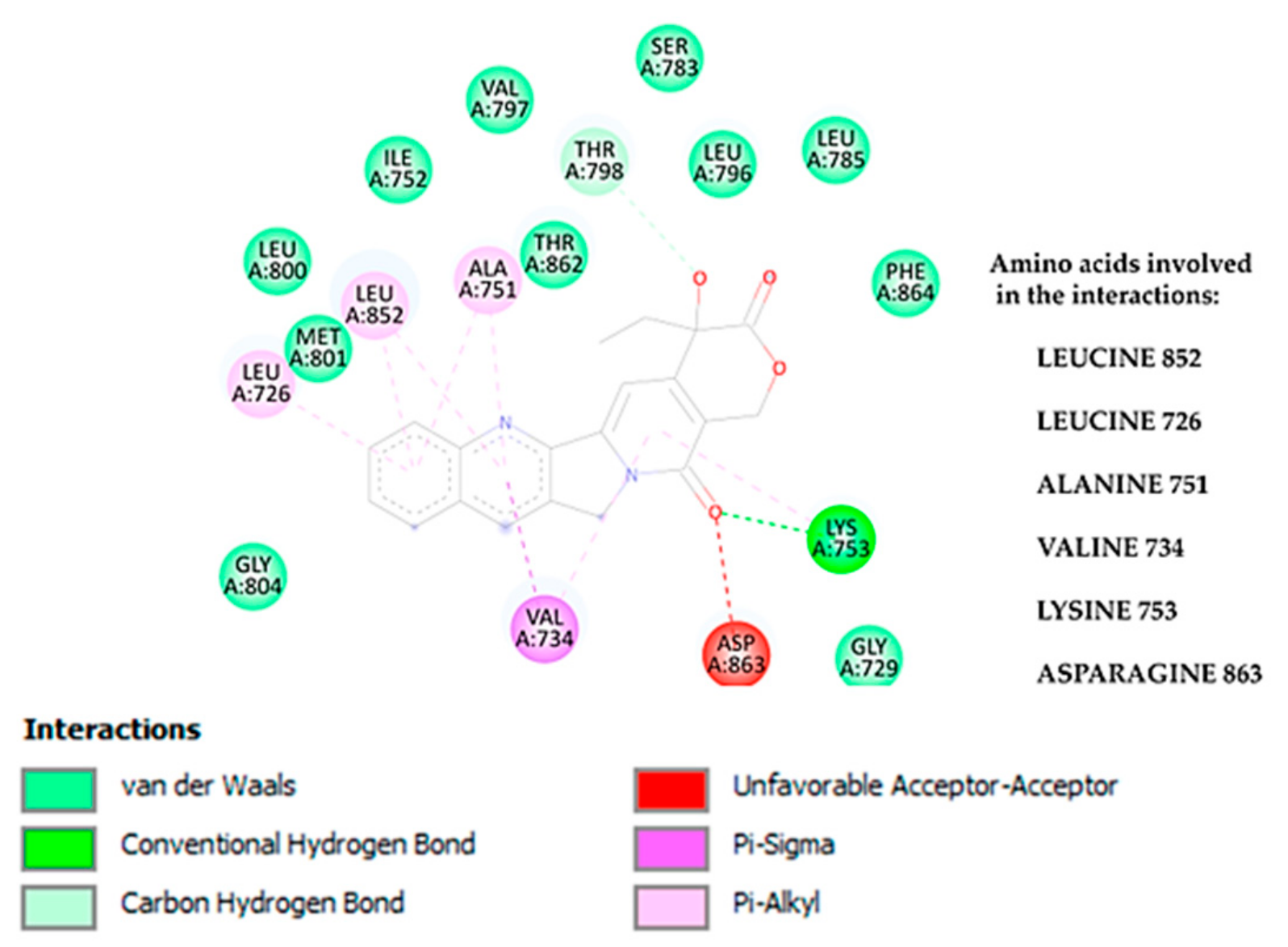
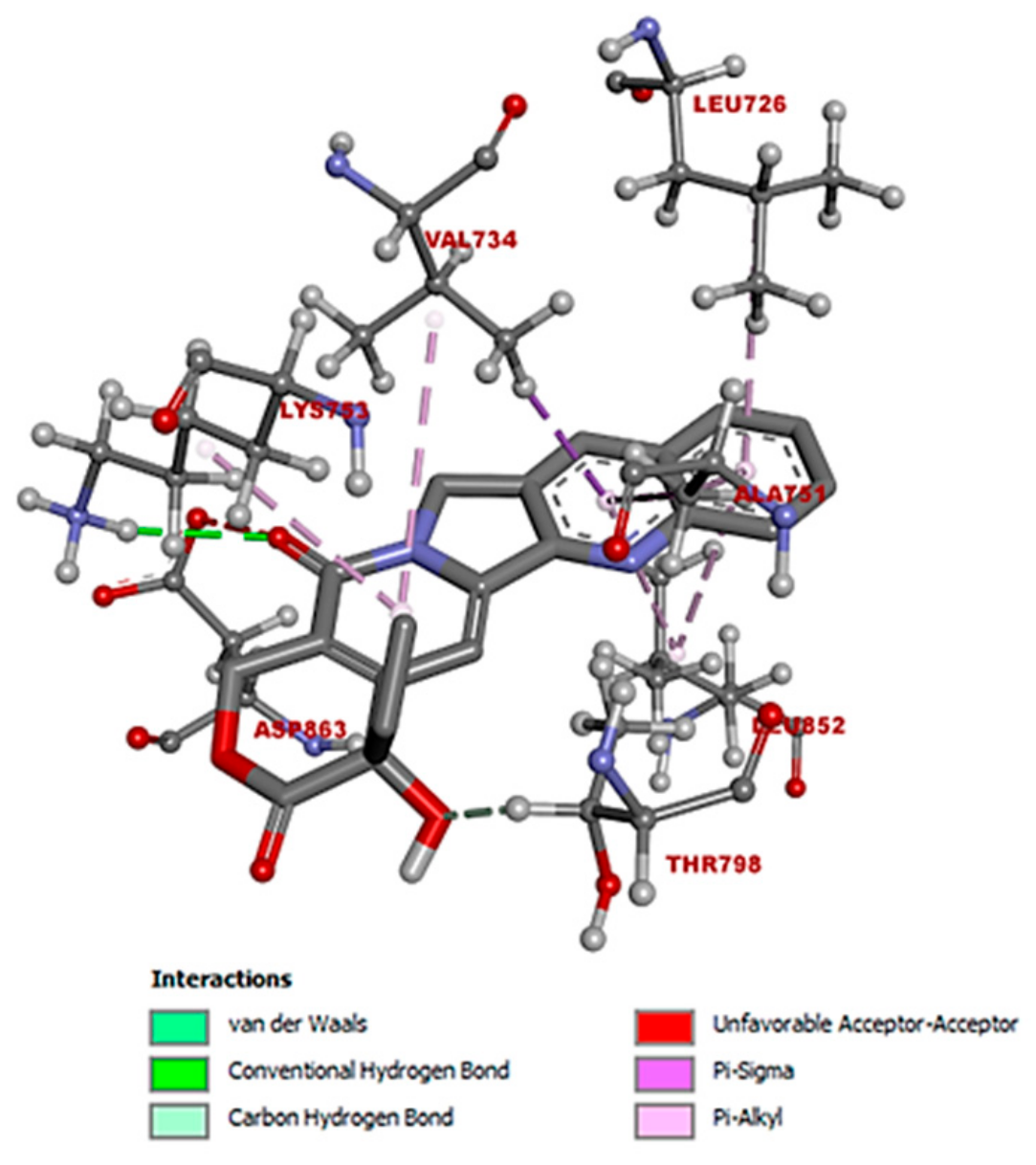
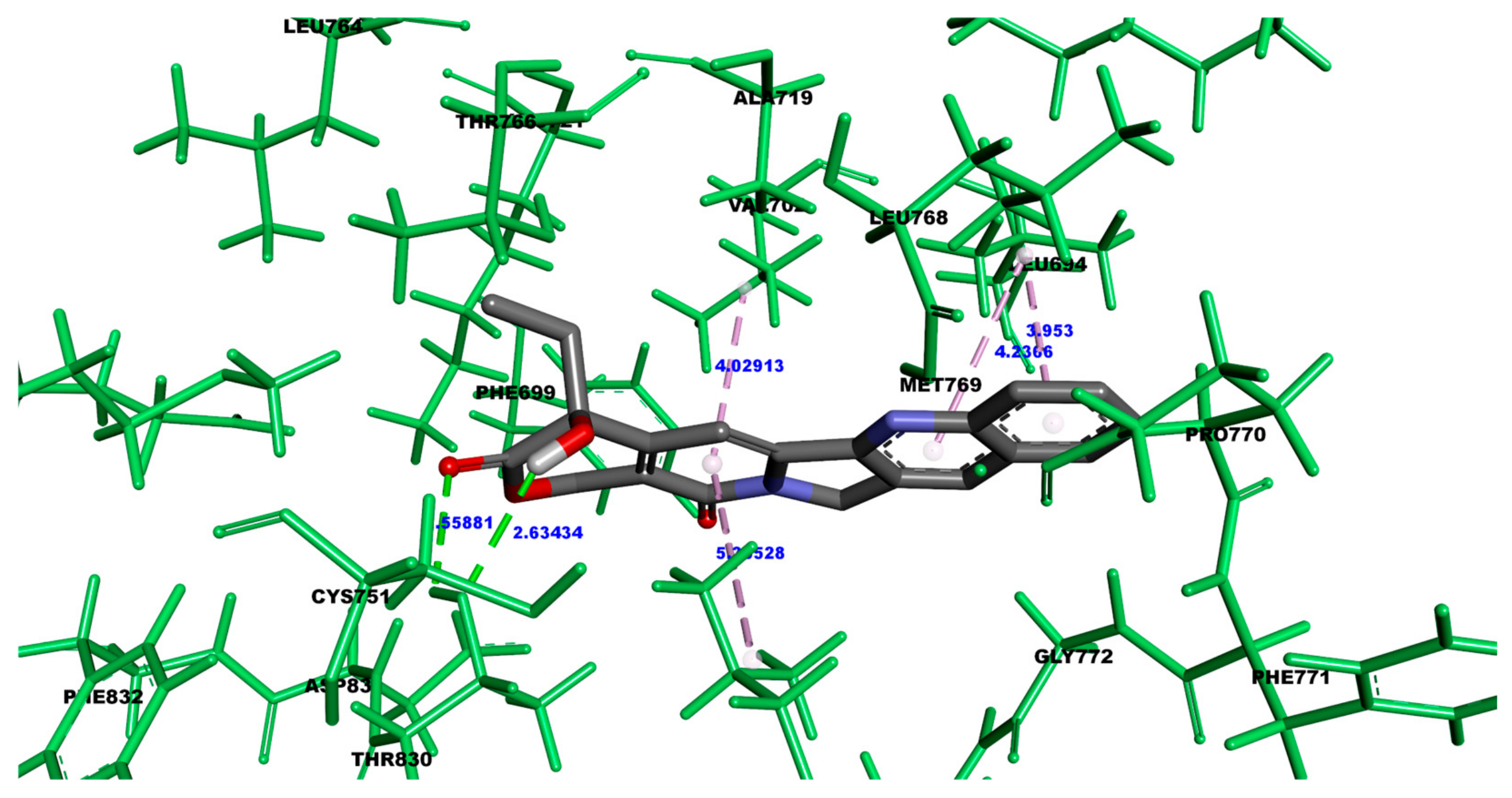
3.1.2. Molecular Interactions Between Camptothecin and HER2
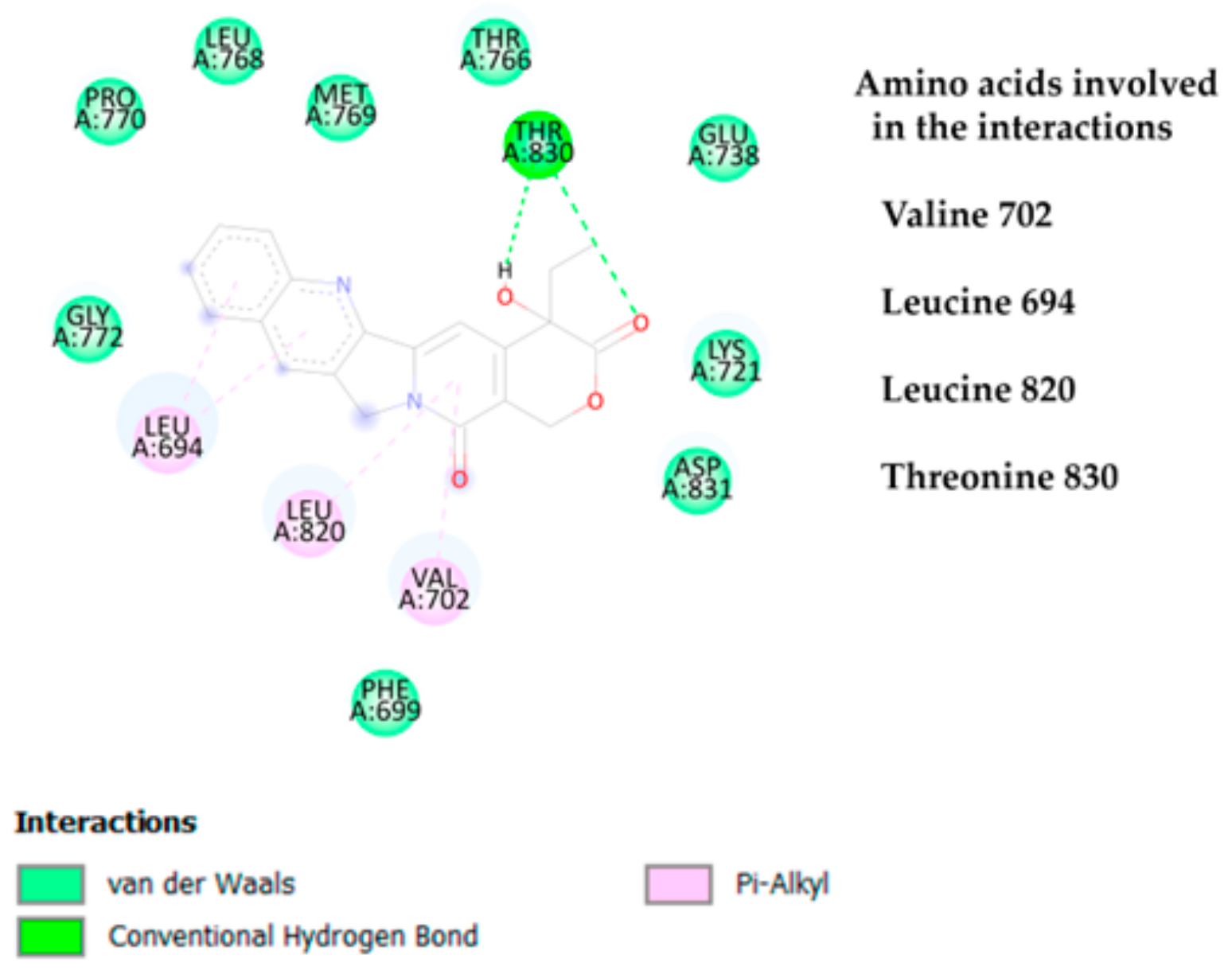
3.1.3. Molecular Interactions Between Camptothecin and EGFR
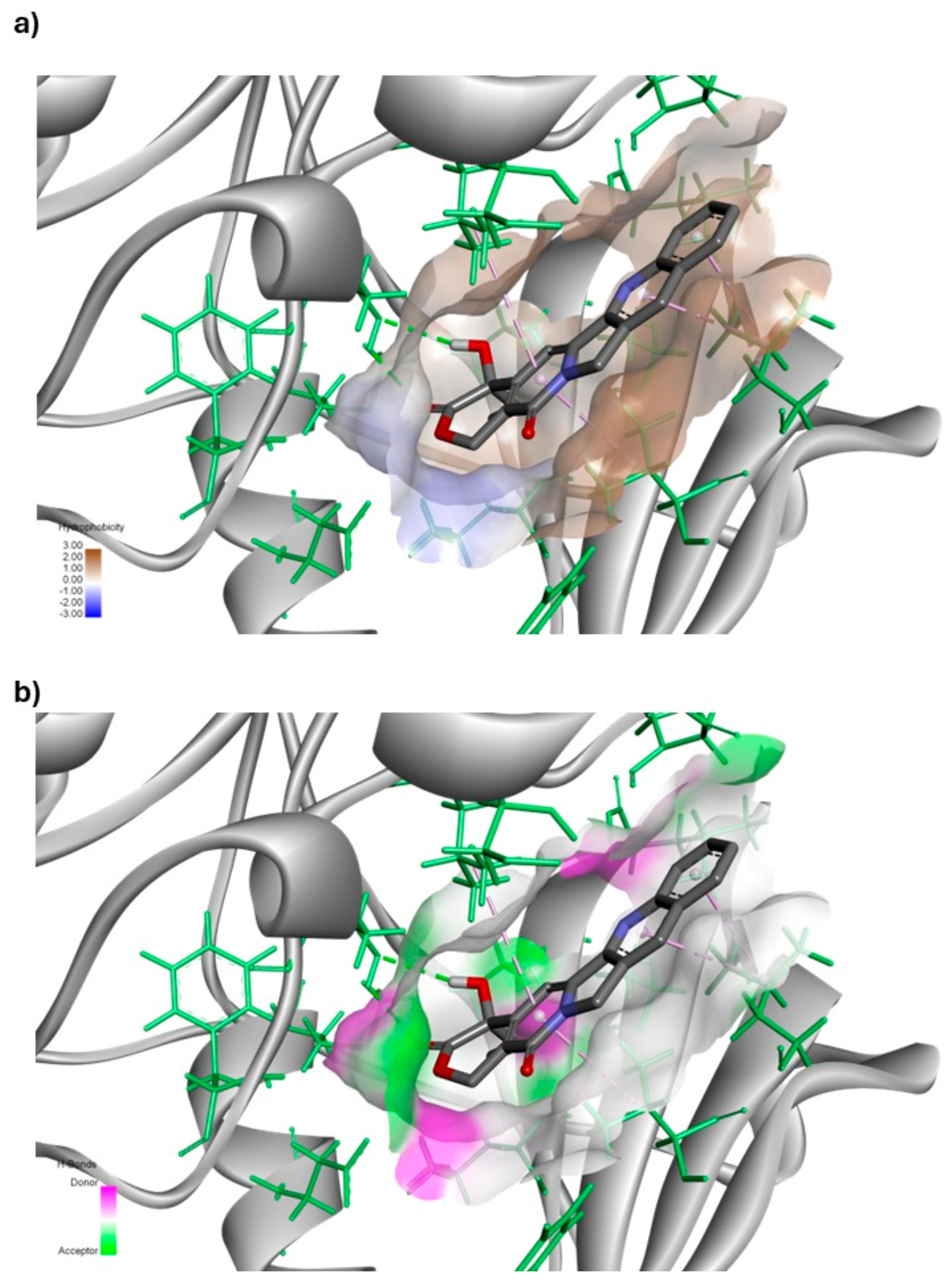
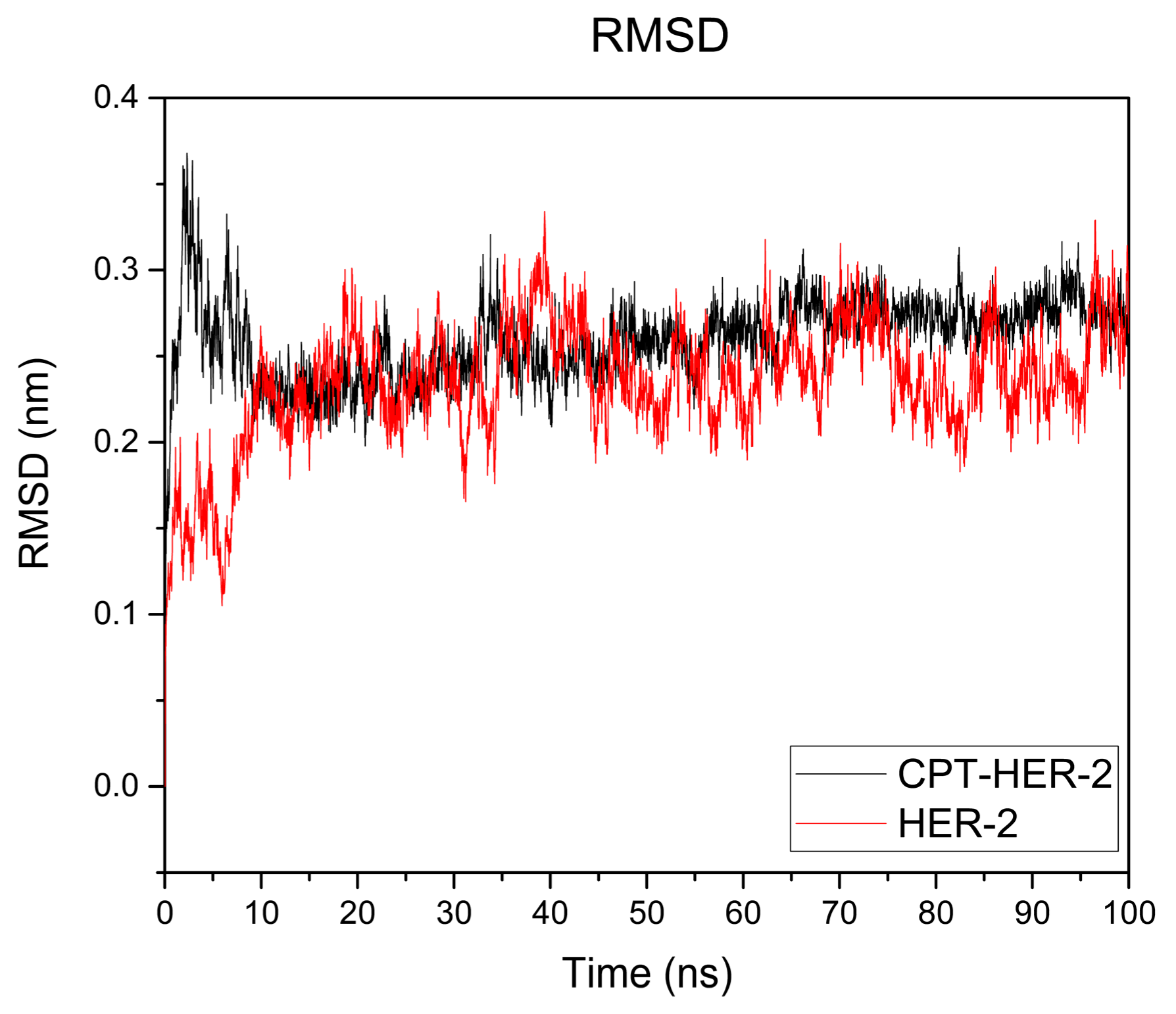
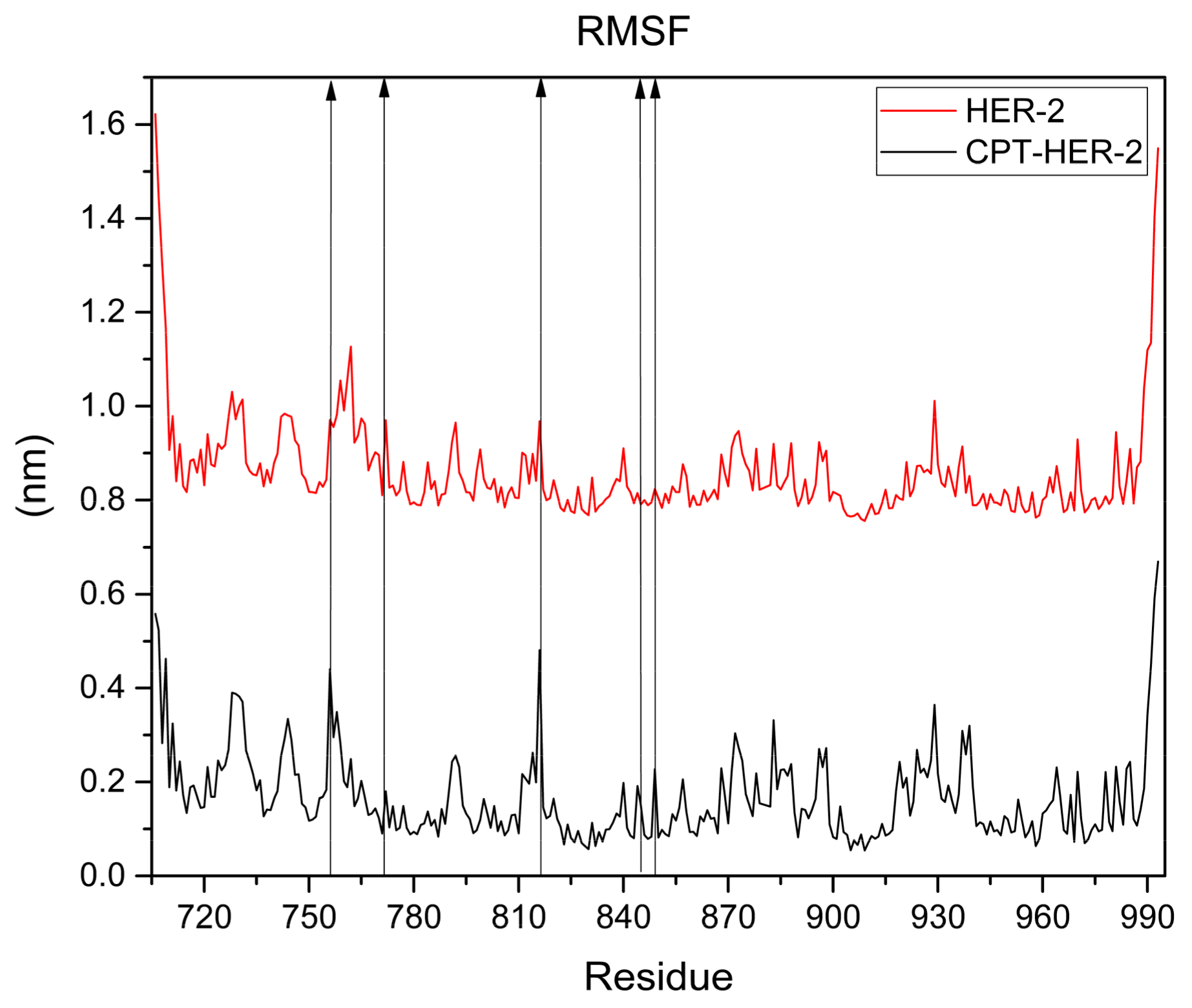
3.1.4. Molecular Dynamic (MD) Simulation
3.1.5. Pharmacological Properties
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Cancer Statistics. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 26 December 2024).
- Gluz, O.; Liedtke, C.; Gottschalk, N.; Pusztai, L.; Nitz, U.; Harbeck, N. Triple-Negative Breast Cancer—Current Status and Future Directions. Ann. Oncol. 2009, 20, 1913–1927. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Gnant, M. Breast Cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, S.G. Breast Cancer: An Overview of Current Therapeutic Strategies, Challenge, and Perspectives. Breast Cancer Targets Ther. 2023, 15, 721–730. [Google Scholar] [CrossRef]
- Obidiro, O.; Battogtokh, G.; Akala, E.O. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics 2023, 15, 1796. [Google Scholar] [CrossRef]
- Jiang, N.; Lin, J.J.; Wang, J.; Zhang, B.N.; Li, A.; Chen, Z.Y.; Guo, S.; Li, B.B.; Duan, Y.Z.; Yan, R.Y.; et al. Novel Treatment Strategies for Patients with HER2-Positive Breast Cancer Who Do Not Benefit from Current Targeted Therapy Drugs (Review). Exp. Ther. Med. 2018, 16, 2183–2192. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2024, 344, 783–792. [Google Scholar] [CrossRef]
- Andre, F.; Ismaila, N.; Henry, N.L.; Somerfield, M.R.; Bast, R.C.; Barlow, W.; Collyar, D.E.; Hammond, M.E.; Kuderer, N.M.; Liu, M.C.; et al. Use of Biomarkers to Guide Decisions on Adjuvant Systemic Therapy for Women With Early-Stage Invasive Breast Cancer: ASCO Clinical Practice Guideline Update—Integration of Results From TAILORx. J. Clin. Oncol. 2019, 37, 1956–1964. [Google Scholar] [CrossRef]
- Harris, L.N.; Ismaila, N.; McShane, L.M.; Andre, F.; Collyar, D.E.; Gonzalez-Angulo, A.M.; Hammond, E.H.; Kuderer, N.M.; Liu, M.C.; Mennel, R.G.; et al. Use of Biomarkers to Guide Decisions on Adjuvant Systemic Therapy for Women With Early-Stage Invasive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2016, 34, 1134–1150. [Google Scholar] [CrossRef]
- Jayasekera, J.; Mandelblatt, J.S. Systematic Review of the Cost Effectiveness of Breast Cancer Prevention, Screening, and Treatment Interventions. J. Clin. Oncol. 2019, 38, 332–350. [Google Scholar] [CrossRef]
- Njerua, S.N.; Matasyoh, J.; Mwaniki, C.G.; Mwendia, C.M.; Kobia, G.K. A Review of Some Phytochemicals Commonly Found in Medicinal. Int. J. Med. Plants Cit. 2013, 105, 135–140. Available online: https://www.researchgate.net/publication/236258875 (accessed on 4 January 2025).
- Haines, D.D.; Cowan, F.M.; Tosaki, A. Evolving Strategies for Use of Phytochemicals in Prevention and Long-Term Management of Cardiovascular Diseases (CVD). Int. J. Mol. Sci. 2024, 25, 6176. [Google Scholar] [CrossRef] [PubMed]
- Howes, M.J.R.; Simmonds, M.S.J. The Role of Phytochemicals as Micronutrients in Health and Disease. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Nirmal, P.; Kumar, M.; Jose, A.; Tomer, V.; Oz, E.; Proestos, C.; Zeng, M.; Elobeid, T.; Sneha, V.; et al. Major Phytochemicals: Recent Advances in Health Benefits and Extraction Method. Molecules 2023, 28, 887. [Google Scholar] [CrossRef]
- Muscolo, A.; Mariateresa, O.; Giulio, T.; Mariateresa, R. Oxidative Stress: The Role of Antioxidant Phytochemicals in the Prevention and Treatment of Diseases. Int. J. Mol. Sci. 2024, 25, 3264. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Gan, R.-Y.; Li, S.; Zhou, Y.; Li, A.-N.; Xu, D.-P.; Li, H.-B. Antioxidant Phytochemicals for the Prevention and Treatment of Chronic Diseases. Molecules 2015, 20, 21138–21156. [Google Scholar] [CrossRef]
- Venditto, V.J.; Simanek, E.E. Cancer Therapies Utilizing the Camptothecins: A Review of the in Vivo Literature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef]
- Khaiwa, N.; Maarouf, N.R.; Darwish, M.H.; Alhamad, D.W.M.; Sebastian, A.; Hamad, M.; Omar, H.A.; Orive, G.; Al-Tel, T.H. Camptothecin’s Journey from Discovery to WHO Essential Medicine: Fifty Years of Promise. Eur. J. Med. Chem. 2021, 223, 113639. [Google Scholar] [CrossRef]
- Saleem, A.; Edwards, T.K.; Rasheed, Z.; Rubin, E.H. Mechanisms of Resistance to Camptothecins. Ann. N. Y. Acad. Sci. 2000, 922, 46–55. [Google Scholar] [CrossRef]
- Gupta, E.; Vyas, V.; Ahmed, F.; Sinko, P.; Cook, T.; Rubin, E. Pharmacokinetics of Orally Administered Camptothecins. Ann. N. Y. Acad. Sci. 2000, 922, 195–204. [Google Scholar] [CrossRef]
- Takimoto, C.H.; Wright, J.; Arbuck, S.G. Clinical Applications of the Camptothecins. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 1998, 1400, 107–119. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB Receptors and Cancer: The Complexity of Targeted Inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M. The Oncogene HER2: Its Signaling and Transforming Functions and Its Role in Human Cancer Pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef]
- Baselga, J.; Swain, S.M. Novel Anticancer Targets: Revisiting ERBB2 and Discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development q Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and Drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef]
- Pollastri, M.P. Overview on the Rule of Five. Curr. Protoc. Pharmacol. 2010, 49, 9.12.1–9.12.8. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Saura, C.; Oliveira, M.; Feng, Y.-H.; Dai, M.-S.; Chen, S.-W.; Hurvitz, S.A.; Kim, S.-B.; Moy, B.; Delaloge, S.; Gradishar, W.; et al. Neratinib Plus Capecitabine Versus Lapatinib Plus Capecitabine in HER2-Positive Metastatic Breast Cancer Previously Treated with ≥2 HER2-Directed Regimens: Phase III NALA Trial. J. Clin. Oncol. 2020, 38, 3138–3148. [Google Scholar] [CrossRef]
- Natesan, S.; Sugumaran, A.; Ponnusamy, C.; Thiagarajan, V.; Palanichamy, R.; Kandasamy, R. Chitosan Stabilized Camptothecin Nanoemulsions: Development, Evaluation and Biodistribution in Preclinical Breast Cancer Animal Mode. Int. J. Biol. Macromol. 2017, 104, 1846–1852. [Google Scholar] [CrossRef] [PubMed]
- Galatage, S.T.; Trivedi, R.; Bhagwat, D.A. Oral Self-Emulsifying Nanoemulsion Systems for Enhancing Dissolution, Bioavailability and Anticancer Effects of Camptothecin. J. Drug Deliv. Sci. Technol. 2022, 78, 103929. [Google Scholar] [CrossRef]
- Yang, S.C.; Zhu, J.B. Preparation and Characterization of Camptothecin Solid Lipid Nanoparticles. Drug Dev. Ind. Pharm. 2002, 28, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes. (s.f.). BIOVIA Draw for Academics. Available online: https://discover.3ds.com/biovia-draw-academic (accessed on 1 January 2025).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 ; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2022; Available online: https://gaussian.com/ (accessed on 14 February 2025).
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods I: Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Aertgeerts, K.; Skene, R.; Yano, J.; Sang, B.-C.; Zou, H.; Snell, G.; Jennings, A.; Iwamoto, K.; Habuka, N.; Hirokawa, A.; et al. Structural Analysis of the Mechanism of Inhibition and Allosteric Activation of the Kinase Domain of HER2 Protein. J. Biol. Chem. 2011, 286, 18756–18765. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Dassault Systèmes. (s.f.). Discovery Studio Visualization. Available online: https://www.3ds.com/products/biovia/discovery-studio/visualization (accessed on 3 January 2025).
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software News and Updates AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Islam, S.M.; Huang, L.; Rui, H.; Zhu, A.; Lee, H.S.; Qi, Y.; Han, W.; Vanommeslaeghe, K.; et al. Chapter Eight—CHARMM-GUI PDB Manipulator for Advanced Modeling and Simulations of Proteins Containing Nonstandard Residues. In Advances in Protein Chemistry and Structural Biology; Karabencheva-Christova, T., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 96, pp. 235–265. ISBN 1876-1623. [Google Scholar]
- Park, S.-J.; Kern, N.; Brown, T.; Lee, J.; Im, W. CHARMM-GUI PDB Manipulator: Various PDB Structural Modifications for Biomolecular Modeling and Simulation. J. Mol. Biol. 2023, 435, 167995. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.E. Genetic Algorithms in Search, Optimization and Machine Learning; Addison-Wesley: Boston, MA, USA, 1989; Volume 7, 401p. [Google Scholar]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. ILOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Kacprzak, K.M.; Sierakowska, A. Chemistry and Biology of Camptothecin and Their Derivatives. Nat. Prod. 2013, 69, 643–682. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H. Nanoparticle-Based Drug Delivery Systems for Enhanced Tumor-Targeting Treatment. Molecules 2019, 15, 1–27. [Google Scholar] [CrossRef]
- Jerez, Y.; Herrero, B.; Arregui, M.; Morón, B. Neratinib for the Treatment of Early-Stage, Hormone Receptor-Positive, HER2-Overexpressed Breast Cancer. Future Oncol. 2020, 16, 1165–1177. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | HER2 | EGFR | ||
|---|---|---|---|---|
| Energy (kcal/mol) | Kd | Energy (kcal/mol) | Kd | |
| Neratinib | −7.92 | 1.57 μM | −5.53 | 88.20 μM |
| Oleanolic Acid | −8.17 | 1.02 μM | −5.98 | 41.67 μM |
| Ursolic Acid | −7.91 | 1.60 μM | −5.81 | 54.76 μM |
| Acronine/Acronycine | −7.96 | 1.47 μM | −5.68 | 68.37 μM |
| α-Peltatin | −8.09 | 1.17 μM | −4.38 | 619.16 μM |
| Camptothecin | −8.03 | 1.30 μM | −6.00 | 40.01 μM |
| Panaxadiol | −7.99 | 1.39 μM | −5.18 | 158.94 μM |
| Ligand | Log p | MW | nOHNH | nON | No. Violations |
|---|---|---|---|---|---|
| Camptothecin | 2.03 | 348.36 | 1 | 6 | 0 |
| Neratinib | 5.33 | 557.05 | 2 | 9 | 2 |
| Oleanolic Acid | 6.72 | 456.71 | 2 | 3 | 1 |
| Ursolic Acid | 6.79 | 456.71 | 2 | 3 | 1 |
| Acronycine | 3.85 | 321.38 | 0 | 4 | 0 |
| α-Peltatin | 1.74 | 400.38 | 2 | 8 | 0 |
| Panaxadiol | 6.98 | 460.74 | 2 | 3 | 1 |
| Ligand | Water Solubility | GI Absorption | Lipophilicity | CYP Inhibitor | BBB Permeant |
|---|---|---|---|---|---|
| Camptothecin | Moderately soluble | High | 2.20 | No | No |
| Neratinib | Poorly soluble | Low | 4.24 | Yes | No |
| Oleanolic Acid | Poorly soluble | Low | 6.07 | No | No |
| Ursolic Acid | Poorly soluble | Low | 5.93 | No | No |
| Acronycine | Moderately soluble | High | 3.43 | Yes | Yes |
| α-Peltatin | Moderately soluble | High | 2.72 | Yes | No |
| Panaxadiol | Poorly soluble | High | 5.90 | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millan-Casarrubias, E.J.; García-Tejeda, Y.V.; González-De la Rosa, C.H.; Ruiz-Mazón, L.; Hernández-Rodríguez, Y.M.; Cigarroa-Mayorga, O.E. Molecular Docking and Pharmacological In Silico Evaluation of Camptothecin and Related Ligands as Promising HER2-Targeted Therapies for Breast Cancer. Curr. Issues Mol. Biol. 2025, 47, 193. https://doi.org/10.3390/cimb47030193
Millan-Casarrubias EJ, García-Tejeda YV, González-De la Rosa CH, Ruiz-Mazón L, Hernández-Rodríguez YM, Cigarroa-Mayorga OE. Molecular Docking and Pharmacological In Silico Evaluation of Camptothecin and Related Ligands as Promising HER2-Targeted Therapies for Breast Cancer. Current Issues in Molecular Biology. 2025; 47(3):193. https://doi.org/10.3390/cimb47030193
Chicago/Turabian StyleMillan-Casarrubias, Elmer Joel, Yunia Verónica García-Tejeda, Claudia Haydée González-De la Rosa, Lucero Ruiz-Mazón, Yazmín Mariela Hernández-Rodríguez, and Oscar Eduardo Cigarroa-Mayorga. 2025. "Molecular Docking and Pharmacological In Silico Evaluation of Camptothecin and Related Ligands as Promising HER2-Targeted Therapies for Breast Cancer" Current Issues in Molecular Biology 47, no. 3: 193. https://doi.org/10.3390/cimb47030193
APA StyleMillan-Casarrubias, E. J., García-Tejeda, Y. V., González-De la Rosa, C. H., Ruiz-Mazón, L., Hernández-Rodríguez, Y. M., & Cigarroa-Mayorga, O. E. (2025). Molecular Docking and Pharmacological In Silico Evaluation of Camptothecin and Related Ligands as Promising HER2-Targeted Therapies for Breast Cancer. Current Issues in Molecular Biology, 47(3), 193. https://doi.org/10.3390/cimb47030193