


Tauroursodeoxycholic Acid Confers Protection Against Oxidative Stress via Autophagy Induction in Retinal Pigment Epithelial Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Quantitative PCR of Bile Acid Receptors in iPSC-RPE and ARPE-19 Cells
2.3. MTT Assay
2.4. Western Blots
2.5. Immunocytochemistry in iPSC-Derived RPE and ARPE-19 Cells
2.6. Statistical Analysis
3. Results
3.1. Detection and Baseline Expression of Bile Acid Receptors in RPE Cells
3.2. TUDCA Protects Against H2O2-Induced Cell Death
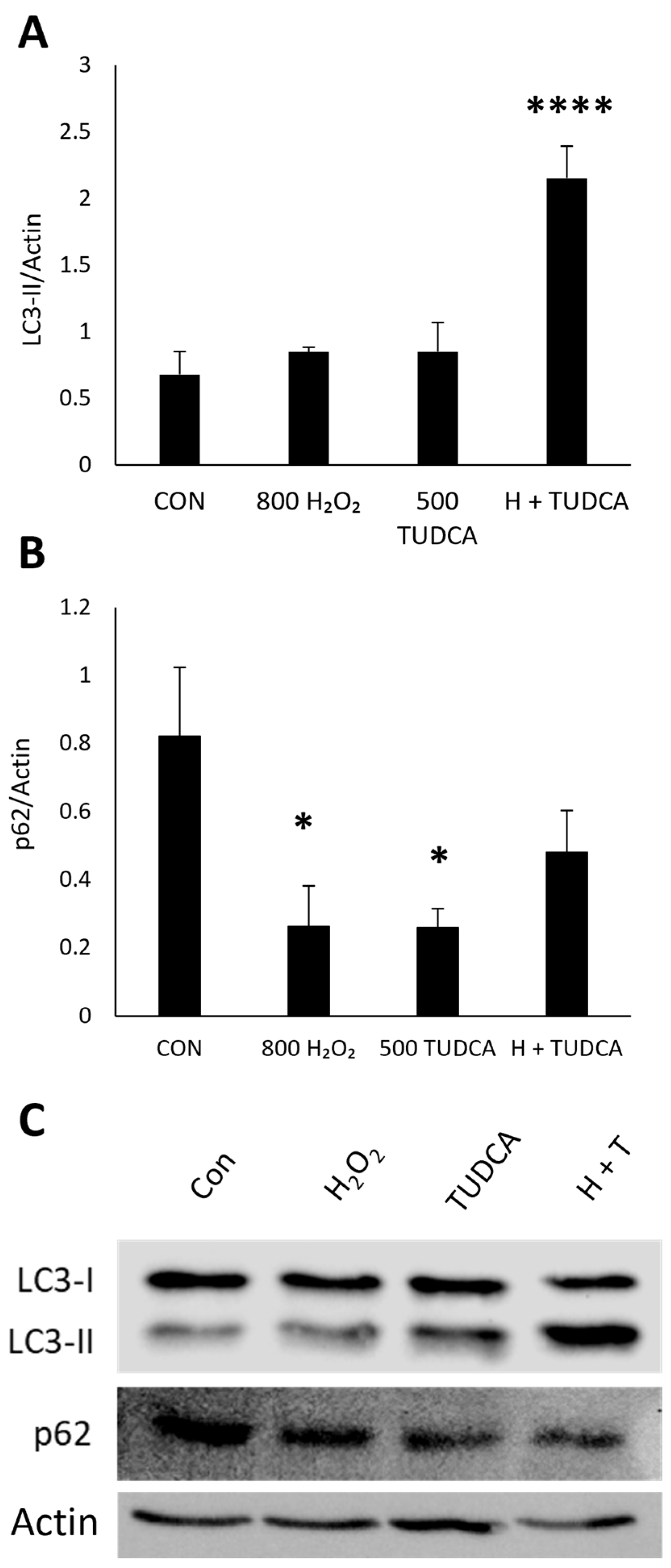
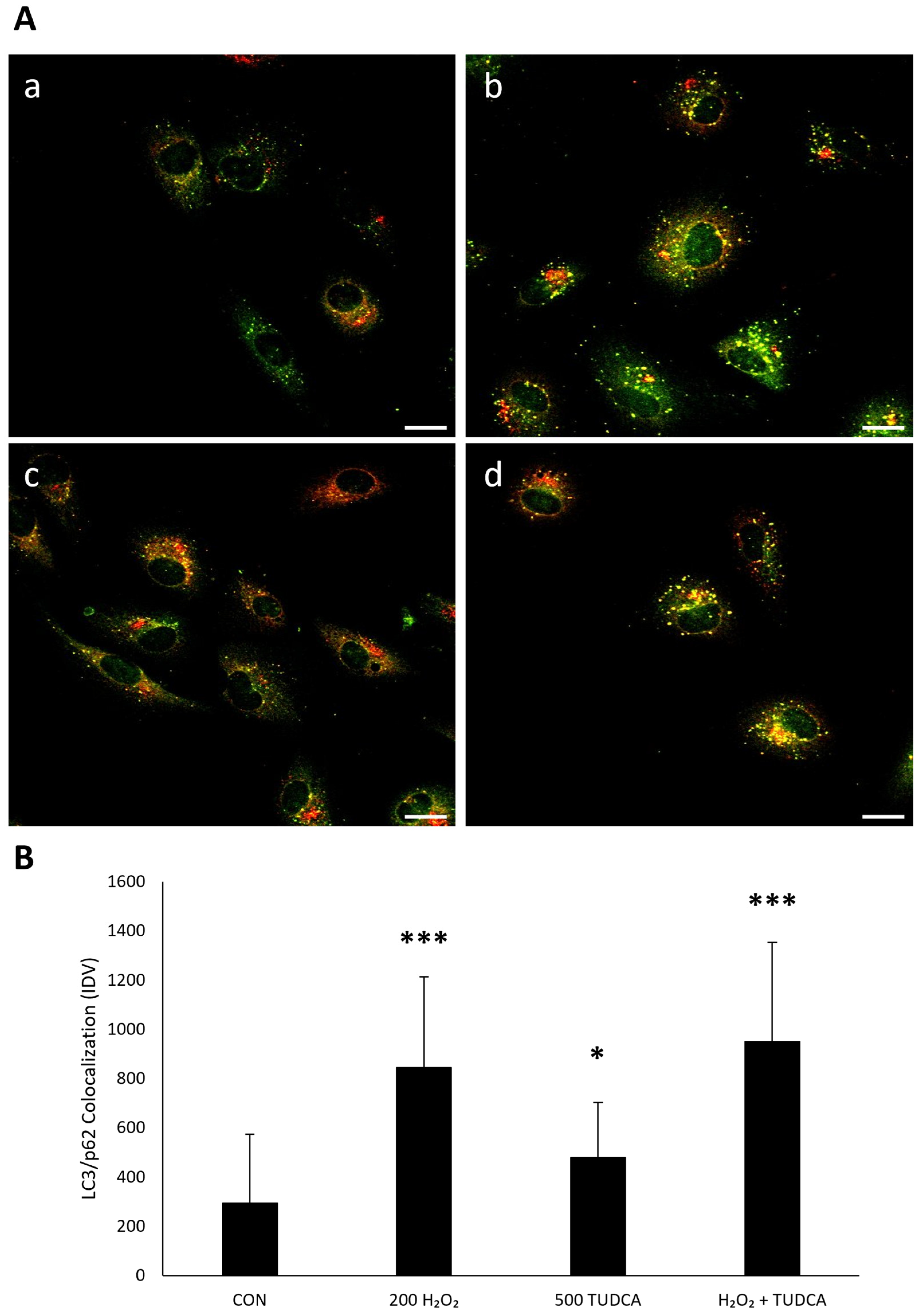
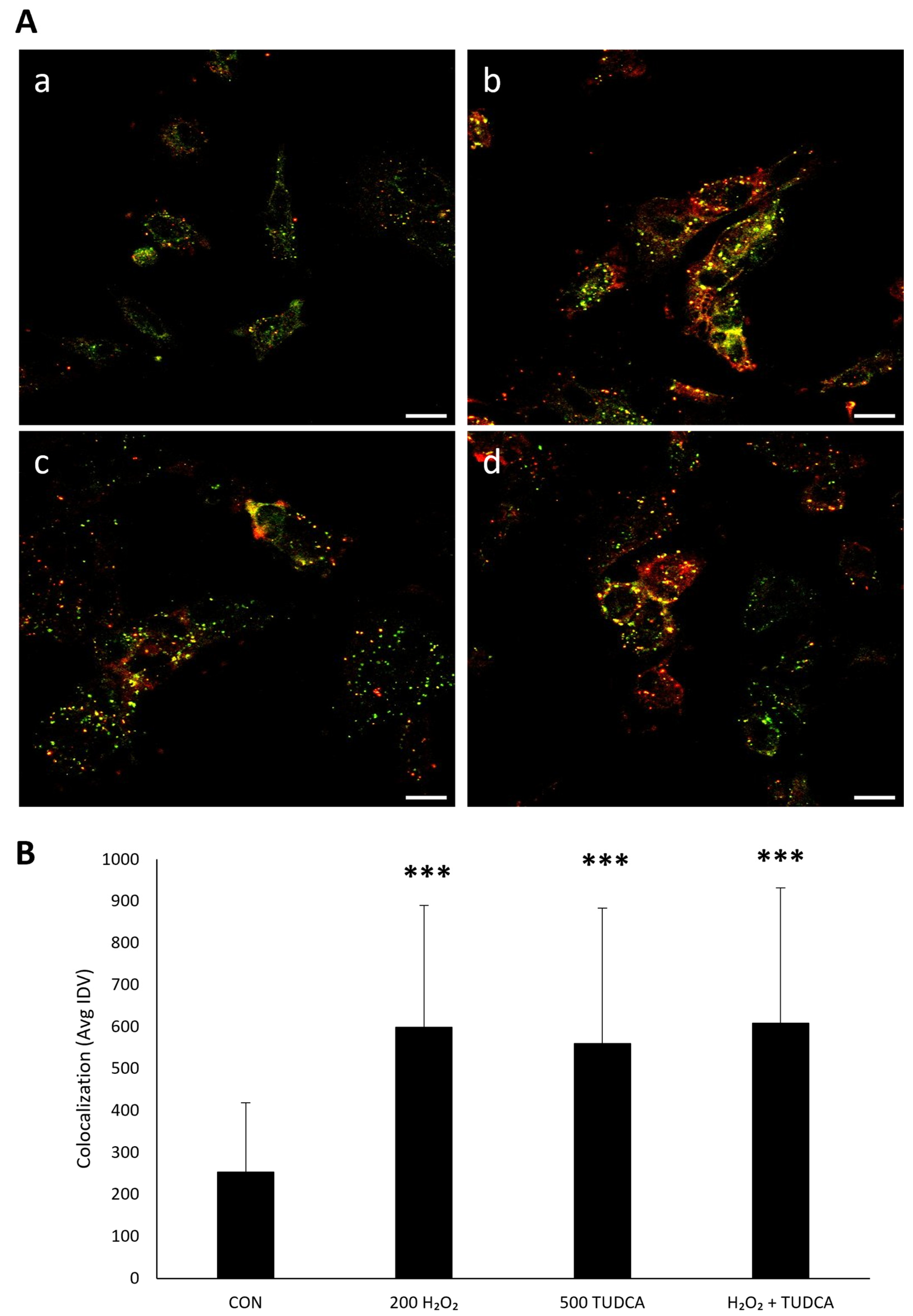
3.3. TUDCA Initiates RPE Autophagy by Increasing LC3-II Expression and LC3/p62 Colocalization
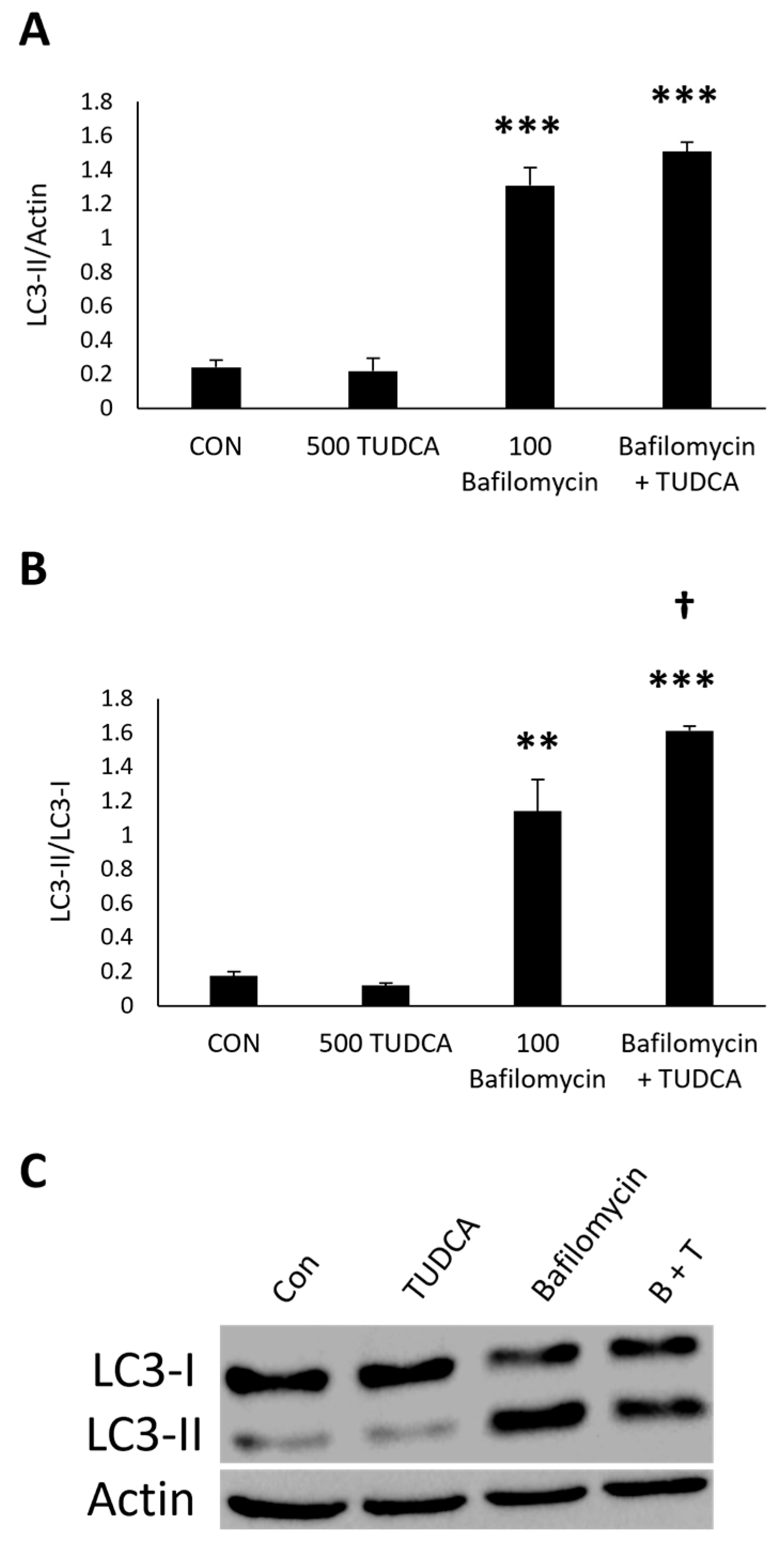
3.4. TUDCA Increases Autophagy Flux in the Presence of Bafilomycin
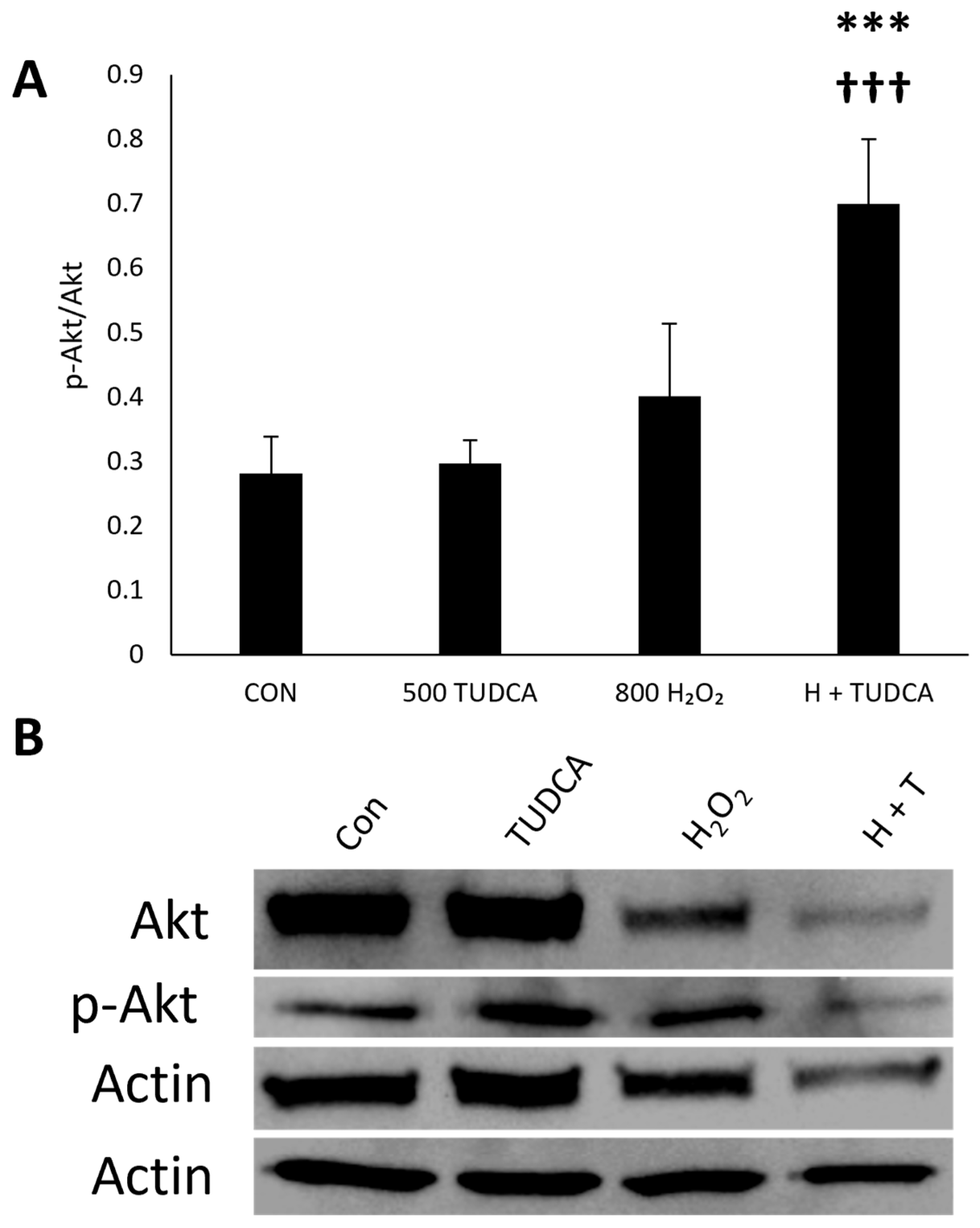
3.5. TUDCA Initiates RPE Autophagy Through Atg Proteins
3.6. TUDCA Activates Autophagy in iPSC-Derived RPE Cells
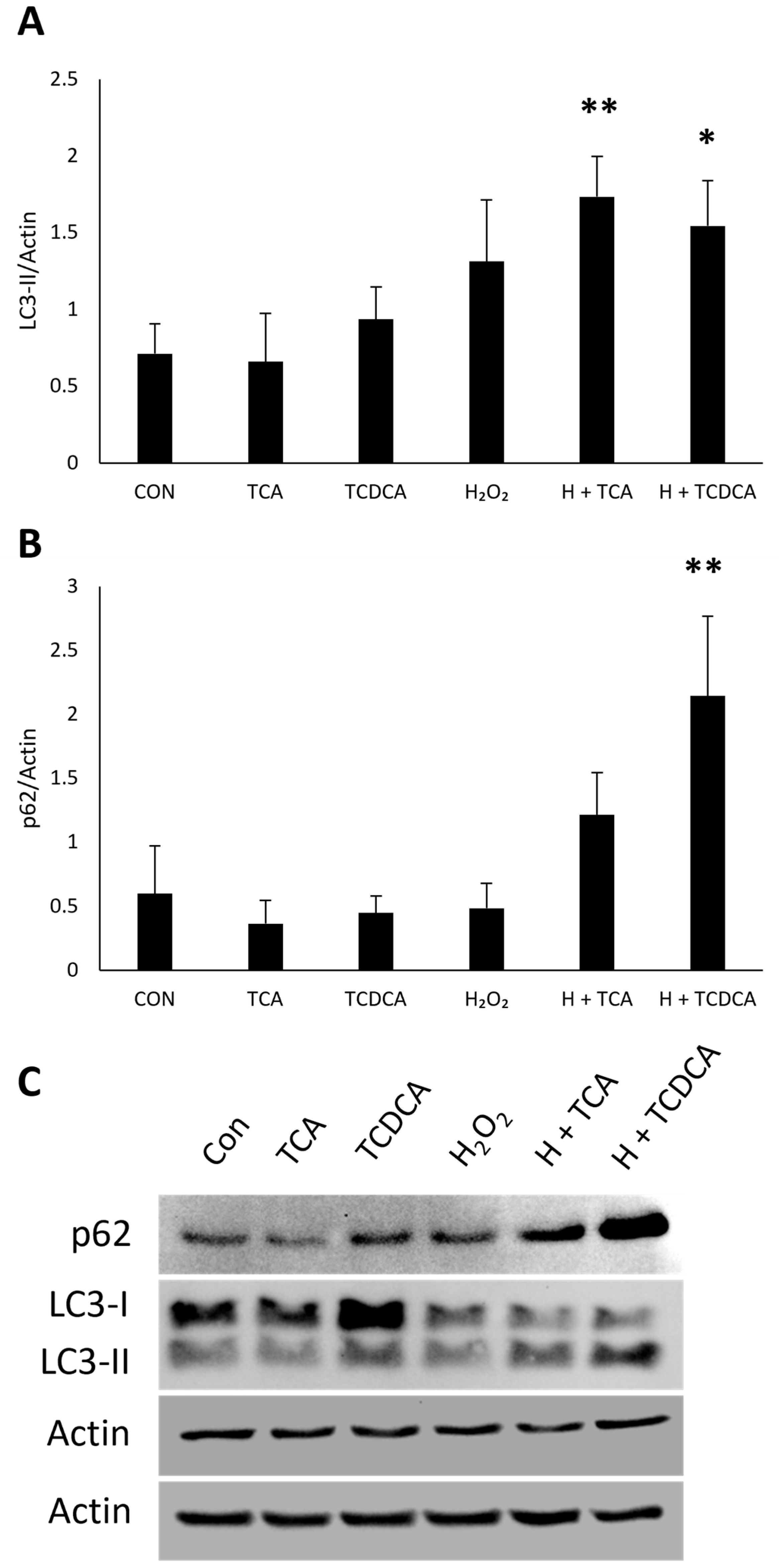
3.7. RPE Autophagy Induction by Other Bile Acids
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruan, Y.; Jiang, S.; Gericke, A. Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels. Int. J. Mol. Sci. 2021, 22, 1296. [Google Scholar] [CrossRef]
- de Jong, E.K.; Geerlings, M.J.; den Hollander, A.I. Chapter 10—Age-Related Macular Degeneration. In Genetics and Genomics of Eye Disease; Gao, X.R., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 155–180. ISBN 978-0-12-816222-4. [Google Scholar]
- Gallenga, C.E.; Lonardi, M.; Pacetti, S.; Violanti, S.S.; Tassinari, P.; Di Virgilio, F.; Tognon, M.; Perri, P. Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa. Antioxidants 2021, 10, 848. [Google Scholar] [CrossRef]
- Collins, S.L.; Stine, J.G.; Bisanz, J.E.; Okafor, C.D.; Patterson, A.D. Bile Acids and the Gut Microbiota: Metabolic Interactions and Impacts on Disease. Nat. Rev. Microbiol. 2023, 21, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile Acids as Regulatory Molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Win, A.; Delgado, A.; Jadeja, R.N.; Martin, P.M.; Bartoli, M.; Thounaojam, M.C. Pharmacological and Metabolic Significance of Bile Acids in Retinal Diseases. Biomolecules 2021, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Boatright, J.H.; Moring, A.G.; McElroy, C.; Phillips, M.J.; Do, V.T.; Chang, B.; Hawes, N.L.; Boyd, A.P.; Sidney, S.S.; Stewart, R.E.; et al. Tool from Ancient Pharmacopoeia Prevents Vision Loss. Mol. Vis. 2006, 12, 1706–1714. [Google Scholar]
- Drack, A.V.; Dumitrescu, A.V.; Bhattarai, S.; Gratie, D.; Stone, E.M.; Mullins, R.; Sheffield, V.C. TUDCA Slows Retinal Degeneration in Two Different Mouse Models of Retinitis Pigmentosa and Prevents Obesity in Bardet-Biedl Syndrome Type 1 Mice. Investig. Ophthalmol. Vis. Sci. 2012, 53, 100–106. [Google Scholar] [CrossRef]
- Fernández-Sánchez, L.; Lax, P.; Pinilla, I.; Martín-Nieto, J.; Cuenca, N. Tauroursodeoxycholic Acid Prevents Retinal Degeneration in Transgenic P23H Rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4998–5008. [Google Scholar] [CrossRef]
- Lawson, E.C.; Bhatia, S.K.; Han, M.K.; Aung, M.H.; Ciavatta, V.; Boatright, J.H.; Pardue, M.T. Tauroursodeoxycholic Acid Protects Retinal Function and Structure in Rd1 Mice. In Proceedings of the Retinal Degenerative Diseases; Bowes Rickman, C., LaVail, M.M., Anderson, R.E., Grimm, C., Hollyfield, J., Ash, J., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 431–436. [Google Scholar]
- Phillips, M.J.; Walker, T.A.; Choi, H.-Y.; Faulkner, A.E.; Kim, M.K.; Sidney, S.S.; Boyd, A.P.; Nickerson, J.M.; Boatright, J.H.; Pardue, M.T. Tauroursodeoxycholic Acid Preservation of Photoreceptor Structure and Function in the Rd10 Mouse through Postnatal Day 30. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2148–2155. [Google Scholar] [CrossRef]
- Shiraya, T.; Araki, F.; Ueta, T.; Fukunaga, H.; Totsuka, K.; Arai, T.; Uemura, A.; Moriya, K.; Kato, S. Ursodeoxycholic Acid Attenuates the Retinal Vascular Abnormalities in Anti-PDGFR-β Antibody-Induced Pericyte Depletion Mouse Models. Sci. Rep. 2020, 10, 977. [Google Scholar] [CrossRef]
- Beli, E.; Yan, Y.; Moldovan, L.; Vieira, C.P.; Gao, R.; Duan, Y.; Prasad, R.; Bhatwadekar, A.; White, F.A.; Townsend, S.D.; et al. Restructuring of the Gut Microbiome by Intermittent Fasting Prevents Retinopathy and Prolongs Survival in Db/Db Mice. Diabetes 2018, 67, 1867–1879. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, W.; Xie, T.-H.; Zou, J.; Nie, X.; Wang, X.; Zhang, M.-Y.; Wang, Z.-Y.; Gu, S.; Zhuang, M.; et al. TGR5 Receptor Activation Attenuates Diabetic Retinopathy through Suppression of RhoA/ROCK Signaling. FASEB J. 2020, 34, 4189–4203. [Google Scholar] [CrossRef]
- Alhasani, R.H.; Almarhoun, M.; Zhou, X.; Reilly, J.; Patterson, S.; Zeng, Z.; Shu, X. Tauroursodeoxycholic Acid Protects Retinal Pigment Epithelial Cells from Oxidative Injury and Endoplasmic Reticulum Stress In Vitro. Biomedicines 2020, 8, 367. [Google Scholar] [CrossRef]
- Wang, C.; Yuan, J.; Qin, D.; Gu, J.; Zhao, B.; Zhang, L.; Zhao, D.; Chen, J.; Hou, X.; Yang, N.; et al. Protection of Tauroursodeoxycholic Acid on High Glucose-Induced Human Retinal Microvascular Endothelial Cells Dysfunction and Streptozotocin-Induced Diabetic Retinopathy Rats. J. Ethnopharmacol. 2016, 185, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.J.; Kim, J.H.; Yu, H.G. Ursodeoxycholic Acid and Tauroursodeoxycholic Acid Suppress Choroidal Neovascularization in a Laser-Treated Rat Model. J. Ocul. Pharmacol. Ther. 2010, 26, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.P.; Park, Y.; Parks, M.B.; Burgess, L.G.; Uppal, K.; Lee, K.; Jones, D.P.; Brantley, M.A., Jr. Metabolome-Wide Association Study of Neovascular Age-Related Macular Degeneration. PLoS ONE 2013, 8, e72737. [Google Scholar] [CrossRef]
- Mitchell, S.L.; Uppal, K.; Williamson, S.M.; Liu, K.; Burgess, L.G.; Tran, V.; Umfress, A.C.; Jarrell, K.L.; Cooke Bailey, J.N.; Agarwal, A.; et al. The Carnitine Shuttle Pathway Is Altered in Patients With Neovascular Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4978–4985. [Google Scholar] [CrossRef]
- Warden, C.; Barnett, J.M.; Brantley, M.A. Taurocholic Acid Inhibits Features of Age-Related Macular Degeneration in Vitro. Exp. Eye Res. 2020, 193, 107974. [Google Scholar] [CrossRef]
- Warden, C.; Brantley, M.A. Glycine-Conjugated Bile Acids Protect RPE Tight Junctions against Oxidative Stress and Inhibit Choroidal Endothelial Cell Angiogenesis In Vitro. Biomolecules 2021, 11, 626. [Google Scholar] [CrossRef]
- Golestaneh, N.; Chu, Y.; Xiao, Y.-Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional Autophagy in RPE, a Contributing Factor in Age-Related Macular Degeneration. Cell Death Dis. 2017, 8, e2537. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W.; Ding, J.; et al. Dysregulated Autophagy in the RPE Is Associated with Increased Susceptibility to Oxidative Stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Lv, G.; Li, R.; Liu, W.-T.; Zong, C.; Ye, F.; Li, X.-Y.; Yang, X.; Jiang, J.-H.; Hou, X.-J.; et al. Glycochenodeoxycholate Promotes Hepatocellular Carcinoma Invasion and Migration by AMPK/mTOR Dependent Autophagy Activation. Cancer Lett. 2019, 454, 215–223. [Google Scholar] [CrossRef]
- Wu, Z.; Yao, H.; Xu, H.; Wang, Y.; Hu, W.; Lou, G.; Zhang, L.; Huang, C.; Jiang, C.; Zhou, S.; et al. Inhibition of eNOS by L-NAME Resulting in Rat Hind Limb Developmental Defects through PFKFB3 Mediated Angiogenetic Pathway. Sci. Rep. 2020, 10, 16754. [Google Scholar] [CrossRef]
- Gibson, S.B. Chapter Thirteen—Investigating the Role of Reactive Oxygen Species in Regulating Autophagy. In Methods in Enzymology; Cadenas, E., Packer, L., Eds.; Hydrogen Peroxide and Cell Signaling, Part C; Academic Press: Cambridge, MA, USA, 2013; Volume 528, pp. 217–235. [Google Scholar]
- Rusten, T.E.; Stenmark, H. P62, an Autophagy Hero or Culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to Interpret LC3 Immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 Prevents Maturation of Autophagic Vacuoles by Inhibiting Fusion between Autophagosomes and Lysosomes in Rat Hepatoma Cell Line, H-4-II-E Cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef]
- Redmann, M.; Benavides, G.A.; Berryhill, T.F.; Wani, W.Y.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Barnes, S.; Darley-Usmar, V.M.; Zhang, J. Inhibition of Autophagy with Bafilomycin and Chloroquine Decreases Mitochondrial Quality and Bioenergetic Function in Primary Neurons. Redox Biol. 2017, 11, 73–81. [Google Scholar] [CrossRef]
- Ye, X.; Zhou, X.-J.; Zhang, H. Exploring the Role of Autophagy-Related Gene 5 (ATG5) Yields Important Insights Into Autophagy in Autoimmune/Autoinflammatory Diseases. Front. Immunol. 2018, 9, 2334. [Google Scholar] [CrossRef]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, H.; Qu, T.; Luo, J.; An, P.; Ren, F.; Luo, Y.; Li, Y. mTORC2: A Multifaceted Regulator of Autophagy. Cell Commun. Signal. 2023, 21, 4. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.-M.; Feng, Y.; Wang, J.; Shi, R.; Jiang, X. Regulation of Autophagy by Coordinated Action of mTORC1 and Protein Phosphatase 2A. Nat. Commun. 2015, 6, 8048. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Pester, J.M.; McDowell, M.; Soza, S.; Montecucco, A.; Lee-Fruman, K.K. Identification of S6K2 as a Centrosome-Located Kinase. FEBS Lett. 2007, 581, 4058–4064. [Google Scholar] [CrossRef]
- Memmott, R.M.; Dennis, P.A. Akt-Dependent and -Independent Mechanisms of mTOR Regulation in Cancer. Cell. Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.C.; Aotaki-keen, A.E.; Putkey, F.R.; Hjelmeland, L.M. ARPE-19, A Human Retinal Pigment Epithelial Cell Line with Differentiated Properties. Exp. Eye Res. 1996, 62, 155–170. [Google Scholar] [CrossRef]
- Westenskow, P.D.; Moreno, S.K.; Krohne, T.U.; Kurihara, T.; Zhu, S.; Zhang, Z.; Zhao, T.; Xu, Y.; Ding, S.; Friedlander, M. Using Flow Cytometry to Compare the Dynamics of Photoreceptor Outer Segment Phagocytosis in iPS-Derived RPE Cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6282–6290. [Google Scholar] [CrossRef]
- Hanafi, N.I.; Mohamed, A.S.; Sheikh Abdul Kadir, S.H.; Othman, M.H.D. Overview of Bile Acids Signaling and Perspective on the Signal of Ursodeoxycholic Acid, the Most Hydrophilic Bile Acid, in the Heart. Biomolecules 2018, 8, 159. [Google Scholar] [CrossRef]
- Hazim, R.A.; Volland, S.; Yen, A.; Burgess, B.L.; Williams, D.S. Rapid Differentiation of the Human RPE Cell Line, ARPE-19, Induced by Nicotinamide. Exp. Eye Res. 2018, 179, 18. [Google Scholar] [CrossRef]
- Smith, E.N.; D’Antonio-Chronowska, A.; Greenwald, W.W.; Borja, V.; Aguiar, L.R.; Pogue, R.; Matsui, H.; Benaglio, P.; Borooah, S.; D’Antonio, M.; et al. Human iPSC-Derived Retinal Pigment Epithelium: A Model System for Prioritizing and Functionally Characterizing Causal Variants at AMD Risk Loci. Stem Cell Rep. 2019, 12, 1342–1353. [Google Scholar] [CrossRef]
- Zhang, M.-Y.; Zhu, L.; Zheng, X.; Xie, T.-H.; Wang, W.; Zou, J.; Li, Y.; Li, H.-Y.; Cai, J.; Gu, S.; et al. TGR5 Activation Ameliorates Mitochondrial Homeostasis via Regulating the PKCδ/Drp1-HK2 Signaling in Diabetic Retinopathy. Front. Cell Dev. Biol. 2022, 9, 759421. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, P.; Ma, X.; Qiao, F.; Ma, Y.; Qing, S.; Zhang, Y.; Wang, Y.; Cui, W. Tauroursodeoxycholic Acid (TUDCA) Alleviates Endoplasmic Reticulum Stress of Nuclear Donor Cells under Serum Starvation. PLoS ONE 2018, 13, e0196785. [Google Scholar] [CrossRef]
- Chen, F.; Ge, Z.; Li, N.; Yu, Z.; Wu, R.; Zhao, Y.; He, X.; Cai, G. TUDCA Protects against Tunicamycin-Induced Apoptosis of Dorsal Root Ganglion Neurons by Suppressing Activation of ER Stress. Exp. Ther. Med. 2022, 24, 509. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-J.; Abudureyimu, M.; Zhang, Z.-H.; Tao, J.; Ceylan, A.F.; Lin, J.; Yu, W.; Reiter, R.J.; Ashrafizadeh, M.; Guo, J.; et al. Inhibition of ER Stress Using Tauroursodeoxycholic Acid Rescues Obesity-Evoked Cardiac Remodeling and Contractile Anomalies through Regulation of Ferroptosis. Chem. Biol. Interact. 2024, 398, 111104. [Google Scholar] [CrossRef]
- Mantopoulos, D.; Murakami, Y.; Comander, J.; Thanos, A.; Roh, M.; Miller, J.W.; Vavvas, D.G. Tauroursodeoxycholic Acid (TUDCA) Protects Photoreceptors from Cell Death after Experimental Retinal Detachment. PLoS ONE 2011, 6, e24245. [Google Scholar] [CrossRef]
- Li, J.; Huang, Z.; Jin, Y.; Liang, L.; Li, Y.; Xu, K.; Zhou, W.; Li, X. Neuroprotective Effect of Tauroursodeoxycholic Acid (TUDCA) on In Vitro and In Vivo Models of Retinal Disorders: A Systematic Review. Curr. Neuropharmacol. 2024, 22, 1374–1390. [Google Scholar] [CrossRef] [PubMed]
- Oveson, B.C.; Iwase, T.; Hackett, S.F.; Lee, S.Y.; Usui, S.; Sedlak, T.W.; Snyder, S.H.; Campochiaro, P.A.; Sung, J.U. Constituents of Bile, Bilirubin and TUDCA, Protect against Oxidative Stress-Induced Retinal Degeneration. J. Neurochem. 2011, 116, 144–153. [Google Scholar] [CrossRef]
- Daruich, A.; Picard, E.; Boatright, J.H.; Behar-Cohen, F. Review: The Bile Acids Urso- and Tauroursodeoxycholic Acid as Neuroprotective Therapies in Retinal Disease. Mol. Vis. 2019, 25, 610–624. [Google Scholar]
- Chang, Y.; Yang, T.; Ding, H.; Wang, Z.; Liang, Q. Tauroursodeoxycholic Acid Protects Rat Spinal Cord Neurons after Mechanical Injury through Regulating Neuronal Autophagy. Neurosci. Lett. 2022, 776, 136578. [Google Scholar] [CrossRef]
- Huang, X.; Wu, L.; Kuang, Y.; Li, X.; Deng, X.; Liang, X.; Li, L.; Yang, H.; Huang, Z.; Lu, D.; et al. Tauroursodeoxycholic Acid Mediates Endoplasmic Reticulum Stress and Autophagy in Adrenocortical Carcinoma Cells. Oncol. Lett. 2019, 18, 6475–6482. [Google Scholar] [CrossRef]
- Ryter, S.W.; Cloonan, S.M.; Choi, A.M.K. Autophagy: A Critical Regulator of Cellular Metabolism and Homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Murase, H.; Tsuruma, K.; Shimazawa, M.; Hara, H. TUDCA Promotes Phagocytosis by Retinal Pigment Epithelium via MerTK Activation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2511–2518. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.C.; Zhou, Z.; Green, D.R.; et al. Non-Canonical Autophagy Promotes the Visual Cycle. Cell 2013, 154, 365. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Green, D.R. LC3-Associated Phagocytosis at a Glance. J. Cell Sci. 2019, 132, jcs222984. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Reveles, J.; Dhingra, A.; Alexander, D.; Bragin, A.; Philp, N.J.; Boesze-Battaglia, K. Phagocytosis-Dependent Ketogenesis in Retinal Pigment Epithelium. J. Biol. Chem. 2017, 292, 8038–8047. [Google Scholar] [CrossRef]
- Mannack, G.; Graf, D.; Donner, M.M.; Richter, L.; Görg, B.; vom Dahl, S.; Häussinger, D.; Schliess, F. Taurolithocholic Acid-3 Sulfate Impairs Insulin Signaling in Cultured Rat Hepatocytes and Perfused Rat Liver. Cell. Physiol. Biochem. 2008, 21, 137–150. [Google Scholar] [CrossRef]
- Turdi, S.; Hu, N.; Ren, J. Tauroursodeoxycholic Acid Mitigates High Fat Diet-Induced Cardiomyocyte Contractile and Intracellular Ca2+ Anomalies. PLoS ONE 2013, 8, e63615. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubieta, D.; Warden, C.; Bhattacharya, S.; Brantley, M.A., Jr. Tauroursodeoxycholic Acid Confers Protection Against Oxidative Stress via Autophagy Induction in Retinal Pigment Epithelial Cells. Curr. Issues Mol. Biol. 2025, 47, 224. https://doi.org/10.3390/cimb47040224
Zubieta D, Warden C, Bhattacharya S, Brantley MA Jr. Tauroursodeoxycholic Acid Confers Protection Against Oxidative Stress via Autophagy Induction in Retinal Pigment Epithelial Cells. Current Issues in Molecular Biology. 2025; 47(4):224. https://doi.org/10.3390/cimb47040224
Chicago/Turabian StyleZubieta, Daniella, Cassandra Warden, Sujoy Bhattacharya, and Milam A. Brantley, Jr. 2025. "Tauroursodeoxycholic Acid Confers Protection Against Oxidative Stress via Autophagy Induction in Retinal Pigment Epithelial Cells" Current Issues in Molecular Biology 47, no. 4: 224. https://doi.org/10.3390/cimb47040224
APA StyleZubieta, D., Warden, C., Bhattacharya, S., & Brantley, M. A., Jr. (2025). Tauroursodeoxycholic Acid Confers Protection Against Oxidative Stress via Autophagy Induction in Retinal Pigment Epithelial Cells. Current Issues in Molecular Biology, 47(4), 224. https://doi.org/10.3390/cimb47040224