_Kim.png)
Advances in Gene Therapy with Oncolytic Viruses and CAR-T Cells and Therapy-Related Groups


Abstract
:1. Introduction
2. Tumor Microenvironment (TME) and Gene Therapy
3. HSV-Based Oncolytic Viruses
4. CAR-T Cell Therapy and Immune Effector Cells
5. The Therapeutic Effect of Ex Vivo or In Vivo Therapy and Issues
6. Aspects of Both CAR-T Cell Therapy and Oncovirotherapy
7. Ex Vivo and In Vivo Gene Editing for Hereditary Diseases
8. Gene Therapy for Hereditary Disease
9. Immune Memory of Gene Therapy with Oncolytic Viruses and CAR-T Cells
10. Issues of Clinical Trials, Setbacks, and Regulatory Hurdles in Gene Therapy with Oncolytic Viruses and CAR-T Cells
10.1. Efficacy and Safety Concerns
10.2. Manufacturing and Delivery Challenges
10.3. Biological Challenges
10.4. Regulatory and Ethical Concerns
11. Mechanistic Pathway of OVs Altering the TME Through Cytokine Signaling
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, L.; Liu, G.; Zheng, L.; Long, H.; Liu, Y. A new era of gene and cell therapy for cancer: A narrative review. Ann. Transl. Med. 2023, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Tjalma, W.A.; Arbyn, M.; Paavonen, J.; van Waes, T.R.; Bogers, J.J. Prophylactic human papillomavirus vaccines: The beginning of the end of cervical cancer. Int. J. Gynecol. Cancer 2004, 14, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Balog, J.E. The moral justification for a compulsory human papillomavirus vaccination program. Am. J. Public Health 2009, 99, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Das, K.R. Why are we still in the 1950s regarding management of cancer of uterine cervix? J. Med. Phys. 2012, 37, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, H.A.; Mauro, G.P. History of radiotherapy in the treatment of uterine cervix cancer: An overview. Rev. Assoc. Med. Bras. 2023, 69, e2023S126. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2022, 10, 1367–1401. [Google Scholar] [CrossRef] [PubMed]
- Rouse, B.T.; Mueller, S.N. Some vexations that challenge viral immunology. F1000Research 2016, 5, F1015. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14 (Suppl. S2), 49. [Google Scholar] [CrossRef] [PubMed]
- Hamad, A.; Yusubalieva, G.M.; Baklaushev, V.P.; Chumakov, P.M.; Lipatova, A.V. Recent Developments in Glioblastoma Therapy: Oncolytic Viruses and Emerging Future Strategies. Viruses 2023, 15, 547. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Chai, H.; Tang, Q.; Shi, Z.; Zhou, L. Clinical advances in oncolytic virus therapy for malignant glioma: A systematic review. Discov. Oncol. 2023, 14, 183. [Google Scholar] [CrossRef] [PubMed]
- Asija, S.; Chatterjee, A.; Goda, J.S.; Yadav, S.; Chekuri, G.; Purwar, R. Oncolytic immunovirotherapy for high-grade gliomas: A novel and an evolving therapeutic option. Front. Immunol. 2023, 14, 1118246. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, F.; Patil, V.; Yefet, L.S.; Singh, O.; Liu, J.; Dang, R.M.A.; Yamaguchi, T.N.; Daras, M.; Cloughesy, T.F.; Colman, H.; et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: A phase 1/2 trial. Nat. Med. 2023, 29, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, M.E. Small DNA tumor viruses and human cancer: Preclinical models of virus infection and disease. Tumour Virus Res. 2022, 14, 200239. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Kaul, R.; Murakami, M.; Robertson, E.S. Tumor viruses and cancer biology: Modulating signaling pathways for therapeutic intervention. Cancer Biol. Ther. 2010, 10, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, P.; Teodoro, J.G. A Renaissance for Oncolytic Adenoviruses? Viruses 2023, 15, 358. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.D.; Nakamura, T.; Russell, S.J.; Peng, K.W. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004, 64, 4919–4926. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Iankov, I.D.; Dispenzieri, A.; Galanis, E. Attenuated oncolytic measles virus strains as cancer therapeutics. Curr. Pharm. Biotechnol. 2012, 13, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Salinas, L.; Naik, S.; Pauszek, S.J.; Peng, K.W.; Russell, S.J.; Rodriguez, L.L. Oncolytic Recombinant Vesicular Stomatitis Virus (VSV) Is Nonpathogenic and Nontransmissible in Pigs, a Natural Host of VSV. Hum. Gene Ther. Clin. Dev. 2017, 28, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, O.V.; Kochneva, G.V.; Netesov, S.V.; Onikienko, S.B.; Chumakov, P.M. Mechanisms of Oncolysis by Paramyxovirus Sendai. Acta Nat. 2015, 7, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Naureen, Z.; Malacarne, D.; Anpilogov, K.; Dautaj, A.; Camilleri, G.; Cecchin, S.; Bressan, S.; Casadei, A.; Albion, E.; Sorrentino, E.; et al. Comparison between American and European legislation in the therapeutical and alimentary bacteriophage usage. Acta Biomed. 2020, 91, e2020023. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.J.; Khan Mirzaei, M.; Nilsson, A.S. Adapting Drug Approval Pathways for Bacteriophage-Based Therapeutics. Front. Microbiol. 2016, 7, 1209. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Nakagami, H. Development of COVID-19 vaccines utilizing gene therapy technology. Int. Immunol. 2021, 33, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Khuri, F.R.; Nemunaitis, J.; Ganly, I.; Arseneau, J.; Tannock, I.F.; Romel, L.; Gore, M.; Ironside, J.; MacDougall, R.; Heise, C.; et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat. Med. 2000, 6, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Bedford, R.; Abbruzzese, J.L.; Lahoti, S.; Reid, T.R.; Soetikno, R.M.; Kirn, D.H.; Freeman, S.M. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin. Cancer Res. 2003, 9, 555–561. [Google Scholar] [PubMed]
- Nemunaitis, J.; Cunningham, C.; Tong, A.; Post, L.; Netto, G.; Paulson, A.S.; Rich, D.; Blackburn, A.; Sands, B.; Gibson, B.; et al. Pilot trial of intravenous infusion of a replication-selective adenovirus (ONYX-015) in combination with chemotherapy or IL-2 treatment in refractory cancer patients. Cancer Gene Ther. 2003, 10, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Okuno, S.H.; Nascimento, A.G.; Lewis, B.D.; Lee, R.A.; Oliveira, A.M.; Sloan, J.A.; Atherton, P.; Edmonson, J.H.; Erlichman, C.; et al. Phase I-II trial of ONYX-015 in combination with MAP chemotherapy in patients with advanced sarcomas. Gene Ther. 2005, 12, 5. [Google Scholar] [CrossRef]
- Xia, Z.J.; Chang, J.H.; Zhang, L.; Jiang, W.Q.; Guan, Z.Z.; Liu, J.W.; Zhang, Y.; Hu, X.-H.; Wu, G.-H.; Wang, H.-Q.; et al. Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus. Ai Zheng 2004, 23, 1666–1670. [Google Scholar] [PubMed]
- Freytag, S.O.; Stricker, H.; Pegg, J.; Paielli, D.; Pradhan, D.G.; Peabody, J.; DePeralta-Venturina, M.; Xia, X.; Brown, S.; Lu, M.; et al. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high risk prostate cancer. Cancer Res. 2003, 63, 7497–7506. [Google Scholar] [PubMed]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimäki, A.; et al. Phase I study with ONCOS-102 for the treatment of solid tumors—An evaluation of clinical response and exploratory analyses of immune markers. J. Immunother. Cancer. 2016, 4, 17. [Google Scholar] [CrossRef]
- Harrington, K.J.; Hingorani, M.; Tanay, M.A.; Hickey, J.; Bhide, S.A.; Clarke, P.M.; Renouf, L.C.; Thway, K.; Sibtain, A.; McNeish, I.A.; et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin. Cancer Res. 2010, 16, 4005–4015. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef]
- Markert, J.M.; Razdan, S.N.; Kuo, H.-C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A phase I trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Karapangiotou, E.M.; Roulstone, V.; Twigger, K.; Ball, M.; Tanay, M.; Nutting, C.; Newbold, K.; Gore, M.E.; Larkin, J.; Syrigos, K.N.; et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin. Cancer Res. 2012, 18, 2080–2089. [Google Scholar] [CrossRef]
- Rohner, E.; Yang, R.; Foo, K.S.; Goedel, A.; Chien, K.R. Unlocking the promise of mRNA therapeutics. Nat. Biotechnol. 2022, 40, 1586–1600. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Wang, Y.; Liang, S.; Han, R.; Toumi, M. The impact of COVID-19 on the cell and gene therapies industry: Disruptions, opportunities, and future prospects. Drug Discov. Today 2021, 26, 2269–2281. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Toda, M. Historic Overview of Genetic Engineering Technologies for Human Gene Therapy. Neurol. Med. Chir. 2020, 60, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, H. Immune gene therapy of cancer. Turk. J. Med. Sci. 2020, 50, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Khatami, F.; Aghamir, Z.S.; Jahanshahi, F.; Feiz-Abadi, S.A.; Birang, F.; Khoshchehreh, M.; Namazi Shabestari, A.; Aghamir, S.M.K. The Gene Manipulation and Cellular Immunotherapy Combination in the Treatment of Cancer. Iran. J. Biotechnol. 2022, 20, e3094. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, S.; Totmaj, M.A.; Abbasi, M.; Hajazimian, S.; Goleij, P.; Behroozi, J.; Shademan, B.; Isazadeh, A.; Baradaran, B. Chimeric antigen receptor T (CAR-T) cells: Novel cell therapy for hematological malignancies. Cancer Med. 2023, 12, 7844–7858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Huang, X.; Huang, J. CAR-T cell therapy for hematological malignancies: Limitations and optimization strategies. Front. Immunol. 2022, 13, 1019115. [Google Scholar] [CrossRef] [PubMed]
- Haslauer, T.; Greil, R.; Zaborsky, N.; Geisberger, R. CAR T-Cell Therapy in Hematological Malignancies. Int. J. Mol. Sci. 2021, 22, 8996. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jiang, G. The journey of CAR-T therapy in hematological malignancies. Mol. Cancer 2022, 21, 194. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xia, Y.; Chen, R.; Gao, P.; Duan, S. Unveiling a novel fusion gene enhances CAR T cell therapy for solid tumors. Mol. Cancer 2024, 23, 98. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, C.D.; Corrigan, D.; Zheng, X.Y.; Galbo, P.M., Jr.; Wang, S.; Liu, Y.; Wei, Y.; Suo, L.; Cui, W.; Mercado, N.; et al. TOP CAR with TMIGD2 as a safe and effective costimulatory domain in CAR cells treating human solid tumors. Sci. Adv. 2024, 10, eadk1857. [Google Scholar] [CrossRef] [PubMed]
- Adhikarla, V.; Awuah, D.; Caserta, E.; Minnix, M.; Kuznetsov, M.; Krishnan, A.; Wong, J.Y.C.; Shively, J.E.; Wang, X.; Pichiorri, F.; et al. Designing combination therapies for cancer treatment: Application of a mathematical framework combining CAR T-cell immunotherapy and targeted radionuclide therapy. Front. Immunol. 2024, 15, 1358478. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Zhang, J.; Xu, W.; Qian, Q.; Zhang, G.; Yuan, Q.; Lv, Y.; Zhang, H.; Shen, C.; Wang, W. Global research trends in CAR-T cell therapy for solid tumors: A comprehensive visualization and bibliometric study (2012–2023). Hum. Vaccin. Immunother. 2024, 20, 2338984. [Google Scholar] [CrossRef] [PubMed]
- Khaniya, A.; Rad, S.M.A.H.; Halpin, J.; Tawinwung, S.; McLellan, A.; Suppipat, K.; Hirankarn, N. Development of a compact bidirectional promoter-driven dual chimeric antigen receptor (CAR) construct targeting CD19 and CD20 in the Sleeping Beauty (SB) transposon system. J. Immunother. Cancer 2024, 12, e008555. [Google Scholar] [CrossRef] [PubMed]
- Abudureheman, T.; Zhou, H.; Yang, L.T.; Huang, X.S.; Jing, J.J.; Duan, C.W.; Chen, K.M. Construction of Switch Modules for CAR-T Cell Treatment Using a Site-Specific Conjugation System. Bioconjug. Chem. 2024, 35, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, K.; Yue, L.; Zuo, D.; Sheng, J.; Lan, S.; Zhao, Z.; Dong, S.; Hu, S.; Chen, X.; et al. Mesothelin CAR-T cells expressing tumor-targeted immunocytokine IL-12 yield durable efficacy and fewer side effects. Pharmacol. Res. 2024, 203, 107186. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.; Agarwalla, P.; Song, F.; Jansson, A.; Dotti, G.; Brudno, Y. Implantable CAR T cell factories enhance solid tumor treatment. Biomaterials 2024, 308, 122580. [Google Scholar] [CrossRef] [PubMed]
- Sakunrangsit, N.; Khuisangeam, N.; Inthanachai, T.; Yodsurang, V.; Taechawattananant, P.; Suppipat, K.; Tawinwung, S. Incorporating IL7 receptor alpha signaling in the endodomain of B7H3-targeting chimeric antigen receptor T cells mediatesantitumor activity in glioblastoma. Cancer Immunol. Immunother. 2024, 73, 98. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, M.; Chu, Y.; Zhou, L.; You, Y.; Pang, X.; Yang, S.; Zhang, L.; Chen, L.; Zhu, L.; et al. Dawn of CAR-T cell therapy in autoimmune diseases. Chin. Med. J. 2024, 137, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.M.; Chand Thakuri, B.K.; Nurmukhambetova, S.; Lee, J.J.; Hu, P.; Tran, N.Q.; Steimle, B.; Dash, P.; Schneider, D. Armored TGFbetaRIIDN ROR1-CAR T cells reject solid tumors and resist suppression by constitutively-expressed and treatment-induced TGFbeta1. J. Immunother. Cancer 2024, 12, e008261. [Google Scholar] [CrossRef] [PubMed]
- García-García, L.G.; Sánchez, E.; Ivanova, M.; Pastora, K.; Alcántara-Sánchez, C.; García-Martínez, J.; Martín-Antonio, B.; Ramírez, M.; González-Murillo, Á. Choosing T-cell sources determines CAR-T cell activity in neuroblastoma. Front. Immunol. 2024, 15, 1375833. [Google Scholar] [CrossRef] [PubMed]
- Rotolo, A.; Atherton, M.J. Applications and opportunities for immune cell CAR engineering in comparative oncology. Clin. Cancer Res. 2024, 30, 2359–2369. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, S.; Zhang, W.; Liang, Z.; Fang, Y.; Sun, T.; Wan, Y.; Ma, X.; Zhang, S.; Xu, Y.; et al. Identification of genetic modifiers enhancing B7-H3-targeting CAR T cell therapy against glioblastoma through large-scale CRISPRi screening. J. Exp. Clin. Cancer Res. 2024, 43, 95. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Wang, Y.; Liu, P.; Geng, Y.; Liu, N.; Chen, J.; Yang, J. Gastrointestinal infections and gastrointestinal haemorrhage are underestimated but serious adverse events in chimeric antigen receptor T-cell recipients: A real-world study. Cancer Gene Ther. 2024, 31, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Dobersberger, M.; Sumesgutner, D.; Zajc, C.U.; Salzer, B.; Laurent, E.; Emminger, D.; Sylvander, E.; Lehner, E.; Teufl, M.; Seigner, J.; et al. An engineering strategy to target activated EGFR with CAR T cells. Cell Rep. Methods 2024, 4, 100728. [Google Scholar] [CrossRef] [PubMed]
- Shishido, S.N.; Hart, O.; Jeong, S.; Moriarty, A.; Heeke, D.; Rossi, J.; Bot, A.; Kuhn, P. Liquid biopsy approach to monitor the efficacy and response to CAR-T cell therapy. J. Immunother. Cancer 2024, 12, e007329. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, G.; Fraietta, J.A.; Gerson, J.N.; Van Deerlin, V.M.; Morrissette, J.J.D.; Caponetti, G.C.; Paruzzo, L.; Harris, J.C.; Chong, E.A.; Susanibar Adaniya, S.P.; et al. T cell lymphoma and secondary primary malignancy risk after commercial CAR T cell therapy. Nat. Med. 2024, 30, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Dhawale, T.; Johnson, P.C.; Boateng, K.; Barata, A.; Traeger, L.; Nelson, A.M.; Lavoie, M.W.; Holmbeck, K.; Choe, J.; Nabily, A.; et al. Communication About Chimeric Antigen Receptor T-Cell (CAR-T) Therapy. Transplant. Cell Ther. 2024, 30, 402.e1–402.e12. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.K.; Uricoli, B.; Freeman, R.; Hossian, A.N.; He, Z.; Anderson, J.Y.L.; Neffling, M.; Legier, J.M.; Blake, D.A.; Doxie, D.B.; et al. Engineering Improved CAR T Cell Products with A Multi-Cytokine Particle Platform for Hematologic and Solid Tumors. Adv. Healthc. Mater. 2024, 13, e2302425. [Google Scholar] [CrossRef] [PubMed]
- Yangjie, L.; Peng, C.; Ahad, F.; Ali Zaidi, S.A.; Muluh, T.A.; Fu, Q. Advanced Strategies of CAR-T Cell Therapy in Solid Tumors and Hematological Malignancies. Recent Pat. Anticancer Drug Discov. 2024, 19, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Faramand, R.G.; Lee, S.B.; Jain, M.D.; Cao, B.; Wang, X.; Rejeski, K.; Subklewe, M.; Fahrmann, J.F.; Saini, N.Y.; Hanash, S.M.; et al. Baseline Serum Inflammatory Proteins Predict Poor CAR T Outcomes in Diffuse Large B-cell Lymphoma. Blood Cancer Discov. 2024, 5, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.B.; Rubin, M.; Szabo, A.; Longo, W.; Fenske, T.S.; McCoy, C.; Lorge, A.; Abedin, S.; D’Souza, A.; Dhakal, B.; et al. Efficacy of Multiple SARS-CoV-2 Vaccine Doses in Patients with B Cell Hematologic Malignancies Receiving Chimeric Antigen Receptor T Cell Therapy: A Contemporary Cohort Analysis. Transplant. Cell Ther. 2024, 30, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Deng, L.; Lu, H.; Zhang, W. Bibliometric analysis of research trends and active research areas in chimeric antigen receptor T cell therapy for hematologic malignancies. Int. J. Clin. Pharm. 2024, 46, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Thoidingjam, S.; Sriramulu, S.; Freytag, S.; Brown, S.L.; Kim, J.H.; Chetty, I.J.; Siddiqui, F.; Movsas, B.; Nyati, S. Oncolytic virus-based suicide gene therapy for cancer treatment: A perspective of the clinical trials conducted at Henry Ford Health. Transl. Med. Commun. 2023, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Wu, Y.; Albers, A.; Fang, M.; Qian, X. Strategies for Advanced Oncolytic Virotherapy: Current Technology Innovations and Clinical Approaches. Pharmaceutics 2022, 14, 1811. [Google Scholar] [CrossRef] [PubMed]
- Muigai, A.W.T. Expanding global access to genetic therapies. Nat. Biotechnol. 2022, 40, 20–21. [Google Scholar] [CrossRef]
- Subica, A.M. CRISPR in Public Health: The Health Equity Implications and Role of Community in Gene-Editing Research and Applications. Am. J. Public Health 2023, 113, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, L.; Jiang, J.; Wu, M.; Lin, P. Applications and challenges of CRISPR-Cas gene-editing to disease treatment in clinics. Precis. Clin. Med. 2021, 4, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Cesur-Ergün, B.; Demir-Dora, D. Gene therapy in cancer. J. Gene Med. 2023, 25, e3550. [Google Scholar] [CrossRef]
- Tian, Y.; Xie, D.; Yang, L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 117. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Shen, Y.; Liang, T. Oncolytic virotherapy: Basic principles, recent advances and future directions. Signal Transduct. Target. Ther. 2023, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Abudoureyimu, M.; Lai, Y.; Tian, C.; Wang, T.; Wang, R.; Chu, X. Oncolytic Adenovirus-A Nova for Gene-Targeted Oncolytic Viral Therapy in HCC. Front. Oncol. 2019, 9, 1182. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chard Dunmall, L.S.; Cheng, Z.; Wang, Y. Remodeling the tumor microenvironment by oncolytic viruses: Beyond oncolysis of tumor cells for cancer treatment. J. Immunother. Cancer 2022, 10, e004167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, X.; Cheng, P. Remodeling of Tumor Immune Microenvironment by Oncolytic Viruses. Front. Oncol. 2021, 10, 561372. [Google Scholar] [CrossRef] [PubMed]
- de Vries, C.R.; Kaufman, H.L.; Lattime, E.C. Oncolytic viruses: Focusing on the tumor microenvironment. Cancer Gene Ther. 2015, 22, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Berkey, S.E.; Thorne, S.H.; Bartlett, D.L. Oncolytic Virotherapy and the Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Shin, D.H.; Sohoni, S.; Singh, S.K.; Rivera-Molina, Y.; Jiang, H.; Fan, X.; Gumin, J.; Lang, F.F.; Alvarez-Breckenridge, C.; et al. Reshaping the tumor microenvironment with oncolytic viruses, positive regulation of the immune synapse, and blockade of the immunosuppressive oncometabolic circuitry. J. Immunother. Cancer 2022, 10, e004935. [Google Scholar] [CrossRef] [PubMed]
- Bastin, D.; Aitken, A.S.; Pelin, A.; Pikor, L.A.; Crupi, M.J.F.; Huh, M.S.; Bourgeois-Daigneault, M.C.; Bell, J.C.; Ilkow, C.S. Enhanced susceptibility of cancer cells to oncolytic rhabdo-virotherapy by expression of Nodamura virus protein B2 as a suppressor of RNA interference. J. Immunother. Cancer 2018, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Lowy, D.R. An Introduction to Virus Infections and Human Cancer. Recent Results Cancer Res. 2021, 217, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.B.; Bartee, E. Decreasing the Susceptibility of Malignant Cells to Infection Does Not Impact the Overall Efficacy of Myxoma Virus-Based Oncolytic Virotherapy. Mol. Ther. Oncolytics 2020, 19, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Davola, M.E.; Vito, A.; Wei, J.; El-Sayes, N.; Workenhe, S.; Mossman, K.L. Genetic modification of oncolytic viruses to enhance antitumor immunity. Methods Enzymol. 2020, 635, 231–250. [Google Scholar] [CrossRef]
- Davola, M.E.; Cormier, O.; Vito, A.; El-Sayes, N.; Collins, S.; Salem, O.; Revill, S.; Ask, K.; Wan, Y.; Mossman, K. Oncolytic BHV-1 Is Sufficient to Induce Immunogenic Cell Death and Synergizes with Low-Dose Chemotherapy to Dampen Immunosuppressive T Regulatory Cells. Cancers 2023, 15, 1295. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Barber, G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; McFadden, G. Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy. Cancers 2021, 13, 5452. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Laffranchi, M.; Tomaipitinca, L.; Del Prete, A.; Santoni, A.; Sozzani, S.; Bernardini, G. NK Cell Antitumor Surveillance in a Myeloid Cell-Shaped Environment. Front. Immunol. 2021, 12, 787116. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Shi, Y.; Yin, B. Macrophage barrier in the tumor microenvironment and potential clinical applications. Cell Commun. Signal. 2024, 22, 74. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Salvi, V.; Soriani, A.; Laffranchi, M.; Sozio, F.; Bosisio, D.; Sozzani, S. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell Mol. Immunol. 2023, 20, 432–447. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Yang, F.; Huang, Z.Q.; Li, Y.Y.; Shi, H.Y.; Sun, Q.; Ma, Y.; Wang, Y.; Zhang, Y.; Yang, S.; et al. T cells, NK cells, and tumor-associated macrophages in cancer immunotherapy and the current state of the art of drug delivery systems. Front. Immunol. 2023, 14, 1199173. [Google Scholar] [CrossRef] [PubMed]
- Śledź, M.; Wojciechowska, A.; Zagożdżon, R.; Kaleta, B. In Situ Programming of CAR-T Cells: A Pressing Need in Modern Immunotherapy. Arch. Immunol. Ther. Exp. 2023, 71, 18. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; He, H.; Wang, H. Oncolytic herpes simplex virus and immunotherapy. BMC Immunol. 2018, 19, 40. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.; Haddad, D.; Bressman, M.; Fong, Y. Oncolytic Herpes Simplex Virus 1 (HSV-1) Vectors: Increasing Treatment Efficacy and Range Through Strategic Virus Design. Drugs Future 2010, 35, 183–195. [Google Scholar] [CrossRef]
- Tang, G.; Wang, D.; Zhao, X.; Feng, Z.; Chen, Q.; Shen, Y. The Dilemma of HSV-1 Oncolytic Virus Delivery: The Method Choice and Hurdles. Int. J. Mol. Sci. 2023, 24, 3681. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, H.; Coffman, Z.; Bettencourt, J.; Shipley, T.; Bramblett, D.E. Herpes simplex virus 1 as an oncolytic viral therapy for refractory cancers. Front. Oncol. 2022, 12, 940019. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, K.; Iwai, M.; Yajima, S.; Tanaka, M.; Yanagihara, K.; Seto, Y.; Todo, T. Efficacy of a Third-Generation Oncolytic Herpes Virus G47Δ in Advanced Stage Models of Human Gastric Cancer. Mol. Ther. Oncolytics 2020, 17, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Nakatake, R.; Kaibori, M.; Nakamura, Y.; Tanaka, Y.; Matushima, H.; Okumura, T.; Murakami, T.; Ino, Y.; Todo, T.; Kon, M. Third-generation oncolytic herpes simplex virus inhibits the growth of liver tumors in mice. Cancer Sci. 2018, 109, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Hatta, M.; Kaibori, M.; Matsushima, H.; Yoshida, T.; Okumura, T.; Hayashi, M.; Yoshii, K.; Todo, T.; Sekimoto, M. Efficacy of a third-generation oncolytic herpes simplex virus in refractory soft tissue sarcoma xenograft models. Mol. Ther. Oncolytics 2022, 25, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Puig-Saus, C.; Ribas, A. Gene editing: Towards the third generation of adoptive T-cell transfer therapies. Immunooncol. Technol. 2019, 1, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P.F.; Yu, T. CRISPR/Cas9 therapeutics: Progress and prospects. Signal Transduct. Target. Ther. 2023, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.L.; Schmitt, A.; Hückelhoven-Krauss, A.; Neuber, B.; Kunz, A.; Waldhoff, P.; Vonficht, D.; Yousefian, S.; Jopp-Saile, L.; Wang, L.; et al. Treatment of adult ALL patients with third-generation CD19-directed CAR T cells: Results of a pivotal trial. J. Hematol. Oncol. 2023, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Taghi Khani, A.; Sanchez Ortiz, A.; Swaminathan, S. GM-CSF: A Double-Edged Sword in Cancer Immunotherapy. Front. Immunol. 2022, 13, 901277. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, P.F.; Pala, L.; Conforti, F.; Cocorocchio, E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. [Google Scholar] [CrossRef]
- Rong, Q.X.; Wang, F.; Guo, Z.X.; Hu, Y.; An, S.N.; Luo, M.; Zhang, H.; Wu, S.C.; Huang, H.Q.; Fu, L.W. GM-CSF mediates immune evasion via upregulation of PD-L1 expression in extranodal natural killer/T cell lymphoma. Mol. Cancer 2021, 20, 80. [Google Scholar] [CrossRef] [PubMed]
- Tijtgat, J.; De Munck, J.; Dufait, I.; Schwarze, J.K.; Van Riet, I.; Franceschini, L.; Breckpot, K.; Aerts, J.L.; Neyns, B.; Tuyaerts, S. Unraveling the Effects of a Talimogene Laherparepvec (T-VEC)-Induced Tumor Oncolysate on Myeloid Dendritic Cells. Front. Immunol. 2021, 12, 733506. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Teserpaturev/G47Δ: First Approval. BioDrugs 2022, 36, 667–672. [Google Scholar] [CrossRef]
- Ghouse, S.M.; Nguyen, H.M.; Bommareddy, P.K.; Guz-Montgomery, K.; Saha, D. Oncolytic Herpes Simplex Virus Encoding IL12 Controls Triple-Negative Breast Cancer Growth and Metastasis. Front. Oncol. 2020, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Guz-Montgomery, K.; Saha, D. Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy. Cells 2020, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Sato, Y.T.; Hou, J.; Iwai, M.; Todo, T. Fusion peptide is superior to co-expressing subunits for arming oncolytic herpes virus with interleukin 12. Commun. Med. 2023, 3, 40. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, J.; Feng, K.; Wang, S.; Chen, L.; Niu, S.; Lu, Q.; Fang, Y. Efficacy and safety of oncolytic virus combined with chemotherapy or immune checkpoint inhibitors in solid tumor patients: A meta-analysis. Front. Pharmacol. 2022, 13, 1023533. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Ding, G.; Wang, L. The efficacy and safety of oncolytic viruses in the treatment of intermediate to advanced solid tumors: A systematic review and meta-analysis. Transl. Cancer Res. 2021, 10, 4290–4302. [Google Scholar] [CrossRef] [PubMed]
- Santos Apolonio, J.; Lima de Souza Gonçalves, V.; Cordeiro Santos, M.L.; Silva Luz, M.; Silva Souza, J.V.; Rocha Pinheiro, S.L.; de Souza, W.R.; Sande Loureiro, M.; de Melo, F.F. Oncolytic virus therapy in cancer: A current review. World J. Virol. 2021, 10, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Benmebarek, M.R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, J.; Zheng, H.; Yang, S.; Hua, Y.; Huang, N.; Kleeff, J.; Liao, Q.; Wu, W. Adoptive cellular immunotherapy for solid neoplasms beyond CAR-T. Mol. Cancer 2023, 22, 28. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, K.J.; Lee, S.W. Cancer immunotherapy with T-cell targeting cytokines: IL-2 and IL-7. BMB Rep. 2021, 54, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Dafni, U.; Michielin, O.; Lluesma, S.M.; Tsourti, Z.; Polydoropoulou, V.; Karlis, D.; Besser, M.J.; Haanen, J.; Svane, I.M.; Ohashi, P.S.; et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: A systematic review and meta-analysis. Ann. Oncol. 2019, 30, 1902–1913. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, J.; Yoshida, T.; Isser, A.; Laino, A.S.; Vassallo, M.; Woods, D.; Kim, S.; Oelke, M.; Jones, K.; Schneck, J.P.; et al. Rapid Expansion of Highly Functional Antigen-Specific T Cells from Patients with Melanoma by Nanoscale Artificial Antigen-Presenting Cells. Clin. Cancer Res. 2020, 26, 3384–3396. [Google Scholar] [CrossRef] [PubMed]
- Hont, A.B.; Powell, A.B.; Sohai, D.K.; Valdez, I.K.; Stanojevic, M.; Geiger, A.E.; Chaudhary, K.; Dowlati, E.; Bollard, C.M.; Cruz, C.R.Y. The generation and application of antigen-specific T cell therapies for cancer and viral-associated disease. Mol. Ther. 2022, 30, 2130–2152. [Google Scholar] [CrossRef] [PubMed]
- Eerkens, A.L.; Vledder, A.; van Rooij, N.; Foijer, F.; Nijman, H.W.; de Bruyn, M. Rapid and efficient generation of antigen-specific isogenic T cells from cryopreserved blood samples. Immunol. Cell Biol. 2022, 100, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Sheih, A.; Voillet, V.; Hanafi, L.A.; DeBerg, H.A.; Yajima, M.; Hawkins, R.; Gersuk, V.; Riddell, S.R.; Maloney, D.G.; Wohlfahrt, M.E.; et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 2020, 11, 219. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.; Pahl, J.; Schottelius, A.; Carter, P.J.; Koch, J. Reimagining antibody-dependent cellular cytotoxicity in cancer: The potential of natural killer cell engagers. Trends Immunol. 2022, 43, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.U.; Shi, J.; Li, P.; Wang, H.; Jia, Y.; Deng, C.; Jiang, L.; Wong, A.H. Assessment of chimeric antigen receptor T cytotoxicity by droplet microfluidics in vitro. Antib. Ther. 2022, 5, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Brakel, B.A.; Chokshi, C.R.; Salim, S.K.; Venugopal, C.; Singh, S. In vitro evaluation of CAR-T cells in patient-derived glioblastoma models. STAR Protoc. 2021, 2, 100920. [Google Scholar] [CrossRef] [PubMed]
- Kiesgen, S.; Messinger, J.C.; Chintala, N.K.; Tano, Z.; Adusumilli, P.S. Comparative analysis of assays to measure CAR T-cell-mediated cytotoxicity. Nat. Protoc. 2021, 16, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Espie, D.; Donnadieu, E. New insights into CAR T cell-mediated killing of tumor cells. Front. Immunol. 2022, 13, 1016208. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Pascua, E.; Lindquist, K.C.; Kimberlin, C.; Deng, X.; Mak, Y.S.L.; Melton, Z.; Johnson, T.O.; Lin, R.; Boldajipour, B.; et al. Direct control of CAR T cells through small molecule-regulated antibodies. Nat. Commun. 2021, 12, 710. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.; Chowdhury, A.; Karulkar, A.; Jaiswal, A.K.; Banik, A.; Asija, S.; Purwar, R. Immunogenicity of CAR-T Cell Therapeutics: Evidence, Mechanism and Mitigation. Front. Immunol. 2022, 13, 886546. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Maruta, M.; Tanimoto, K.; Kondo, F.; Yamamoto, T.; Kurata, M.; Fujiwara, H.; Masumoto, J.; Takenaka, K.; Yasukawa, M. A single-chain antibody generation system yielding CAR-T cells with superiorantitumor function. Commun. Biol. 2021, 4, 273. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhang, J.S.; Zhou, J.; Zhou, K.S.; Xu, B.L.; Li, L.L.; Fang, B.J.; Yin, Q.S.; Zhu, X.H.; Zhou, H.; et al. Single VHH-directed BCMA CAR-T cells cause remission of relapsed/refractory multiple myeloma. Leukemia 2021, 35, 3002–3006. [Google Scholar] [CrossRef]
- Lam, N.; Trinklein, N.D.; Buelow, B.; Patterson, G.H.; Ojha, N.; Kochenderfer, J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat. Commun. 2020, 11, 283. [Google Scholar] [CrossRef] [PubMed]
- Pomaznoy, M.; Sethi, A.; Greenbaum, J.; Peters, B. Identifying inaccuracies in gene expression estimates from unstranded RNA-seq data. Sci. Rep. 2019, 9, 16342. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Al-Haidari, A.; Sun, J.; Kazi, J.U. T cell receptor (TCR) signaling in health and disease. Signal Transduct. Target. Ther. 2021, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Birley, K.; Leboreiro-Babe, C.; Rota, E.M.; Buschhaus, M.; Gavriil, A.; Vitali, A.; Alonso-Ferrero, M.; Hopwood, L.; Parienti, L.; Ferry, G.; et al. A novel anti-B7-H3 chimeric antigen receptor from a single-chain antibody library for immunotherapy of solid cancers. Mol. Ther. Oncolytics 2022, 26, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Staehr, M.; Masakayan, R.; Tatake, I.J.; Purdon, T.J.; Wang, X.; Wang, P.; Liu, H.; Xu, Y.; Garrett-Thomson, S.C.; et al. Development and Evaluation of an Optimal Human Single-Chain Variable Fragment-Derived BCMA-Targeted CAR T Cell Vector. Mol. Ther. 2018, 26, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh Kozani, P.; Naseri, A.; Mirarefin, S.M.J.; Salem, F.; Nikbakht, M.; Evazi Bakhshi, S.; Safarzadeh Kozani, P. Nanobody-based CAR-T cells for cancer immunotherapy. Biomark. Res. 2022, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wei, Q.; Brzostek, J.; Gascoigne, N.R.J. Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cell Mol. Immunol. 2020, 17, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2018, 12, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Daei Sorkhabi, A.; Mohamed Khosroshahi, L.; Sarkesh, A.; Mardi, A.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Baradaran, B. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front. Immunol. 2023, 14, 1113882. [Google Scholar] [CrossRef] [PubMed]
- Asmamaw Dejenie, T.; Tiruneh, G.; Medhin, M.; Dessie Terefe, G.; Tadele Admasu, F.; Wale Tesega, W.; Chekol Abebe, E. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum. Vaccin. Immunother. 2022, 18, 2114254. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.G.; Xiao, B.F.; Zhang, J.T.; Cui, X.R.; Lu, Z.M.; Wu, N. Genetically Modified T Cells for Esophageal Cancer Therapy: A Promising Clinical Application. Front. Oncol. 2021, 11, 763806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, S.; Deng, W.; Li, R.; Zhou, H.; Xiong, H. The landscape of chimeric antigen receptor T cell therapy in breast cancer: Perspectives and outlook. Front. Immunol. 2022, 13, 887471. [Google Scholar] [CrossRef] [PubMed]
- Tomasik, J.; Jasiński, M.; Basak, G.W. Next generations of CAR-T cells—New therapeutic opportunities in hematology? Front. Immunol. 2022, 13, 1034707. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Shiina, S.; Iriguchi, S.; Terakura, S.; Kawai, Y.; Kabai, R.; Sakamoto, S.; Watanabe, A.; Ohara, K.; Wang, B.; et al. Optimization of the proliferation and persistency of CAR T cells derived from human induced pluripotent stem cells. Nat. Biomed. Eng. 2023, 7, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Kawana-Tachikawa, A.; Kitayama, S.; Ueda, T.; Miki, S.; Watanabe, A.; Kaneko, S. Generation of highly proliferative, rejuvenated cytotoxic T cell clones through pluripotency reprogramming for adoptive immunotherapy. Mol. Ther. 2021, 29, 3027–3041. [Google Scholar] [CrossRef] [PubMed]
- Wakao, R.; Fukaya-Shiba, A. In vivo CAR T cells and targeted gene delivery: A theme for the Pharmaceuticals and Medical Devices Agency Science Board to address. Front. Med. 2023, 10, 1141880. [Google Scholar] [CrossRef] [PubMed]
- Mhaidly, R.; Verhoeyen, E. The Future: In Vivo CAR T Cell Gene Therapy. Mol. Ther. 2019, 27, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Jogalekar, M.P.; Rajendran, R.L.; Khan, F.; Dmello, C.; Gangadaran, P.; Ahn, B.C. CAR T-Cell-Based gene therapy for cancers: New perspectives, challenges, and clinical developments. Front. Immunol. 2022, 13, 925985. [Google Scholar] [CrossRef] [PubMed]
- Mehrabadi, A.Z.; Ranjbar, R.; Farzanehpour, M.; Shahriary, A.; Dorostkar, R.; Hamidinejad, M.A.; Ghaleh, H.E.G. Therapeutic potential of CAR T cell in malignancies: A scoping review. Biomed. Pharmacother. 2022, 146, 112512. [Google Scholar] [CrossRef] [PubMed]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef]
- Dai, Q.; Han, P.; Qi, X.; Li, F.; Li, M.; Fan, L.; Zhang, H.; Zhang, X.; Yang, X. 4-1BB Signaling Boosts the Antitumor Activity of CD28-Incorporated 2nd Generation Chimeric Antigen Receptor-Modified T Cells. Front. Immunol. 2020, 11, 539654. [Google Scholar] [CrossRef] [PubMed]
- Murthy, H.; Iqbal, M.; Chavez, J.C.; Kharfan-Dabaja, M.A. Cytokine Release Syndrome: Current Perspectives. Immunotargets Ther. 2019, 8, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [PubMed]
- Quintarelli, C.; Orlando, D.; Boffa, I.; Guercio, M.; Polito, V.A.; Petretto, A.; Lavarello, C.; Sinibaldi, M.; Weber, G.; Del Bufalo, F.; et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology 2018, 7, e1433518. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Shen, R. Complexities in comparing the impact of costimulatory domains on approved CD19 CAR functionality. J. Transl. Med. 2023, 21, 515. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.L.; Fritsche, E.; Pulsipher, M.A.; Ahmed, N.; Hamieh, M.; Hegde, M.; Ruella, M.; Savoldo, B.; Shah, N.N.; Turtle, C.J.; et al. Immunogenicity of CAR T cells in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, Z.; Wan, R.; Huang, W. Clinical Strategies for Enhancing the Efficacy of CAR T-Cell Therapy for Hematological Malignancies. Cancers 2022, 14, 4452. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, S.; Keyes, S.; Kilgour, M.K.; Smazynski, J.; Chan, V.; Sudderth, J.; Turcotte, T.; Devlieger, A.; Yu, J.; Huggler, K.S.; et al. Clinically relevant T cell expansion media activate distinct metabolic programs uncoupled from cellular function. Mol. Ther. Methods Clin. Dev. 2022, 24, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guan, X.Y.; Jiang, P. Cytokine and Chemokine Signals of T-Cell Exclusion in Tumors. Front. Immunol. 2020, 11, 594609. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.; Gottschalk, S. Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front. Immunol. 2021, 12, 684642. [Google Scholar] [CrossRef] [PubMed]
- Herda, S.; Heimann, A.; Obermayer, B.; Ciraolo, E.; Althoff, S.; Ruß, J.; Grunert, C.; Busse, A.; Bullinger, L.; Pezzutto, A.; et al. Long-term in vitro expansion ensures increased yield of central memory T cells as perspective for manufacturing challenges. Int. J. Cancer 2021, 148, 3097–3110. [Google Scholar] [CrossRef] [PubMed]
- Waugh, R.E.; Lomakina, E.; Amitrano, A.; Kim, M. Activation effects on the physical characteristics of T lymphocytes. Front. Bioeng. Biotechnol. 2023, 11, 1175570. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Qi, T.; Milner, J.J.; Lu, Y.; Cao, Y. Speed and Location Both Matter: Antigen Stimulus Dynamics Controls CAR-T Cell Response. Front. Immunol. 2021, 12, 748768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Q.; Hostetler, A.; Chen, L.E.; Mukkamala, V.; Abraham, W.; Padilla, L.T.; Wolff, A.N.; Maiorino, L.; Backlund, C.M.; Aung, A.; et al. Universal redirection of CAR T cells against solid tumours via membrane-inserted ligands for the CAR. Nat. Biomed. Eng. 2023, 7, 1113–1128. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, X.; Zhou, W.L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.M.; Melenhorst, J.J.; Tan, K. B cell targeting in CAR T cell therapy: Side effect or driver of CAR T cell function? Sci. Transl. Med. 2022, 14, eabn3353. [Google Scholar] [CrossRef] [PubMed]
- Fousek, K.; Watanabe, J.; Joseph, S.K.; George, A.; An, X.; Byrd, T.T.; Morris, J.S.; Luong, A.; Martínez-Paniagua, M.A.; Sanber, K.; et al. CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia 2021, 35, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.T.; Chen, B.; Liu, Y.Y.; Lan, H.U.; Yan, J.P. Monoclonal antibodies and chimeric antigen receptor (CAR) T cells in the treatment of colorectal cancer. Cancer Cell Int. 2021, 21, 83. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, L.; Masoumi, E.; Fallah-Mehrjardi, K.; Mirzaei, H.R.; Hadjati, J. Prolonged Persistence of Chimeric Antigen Receptor (CAR) T Cell in Adoptive Cancer Immunotherapy: Challenges and Ways Forward. Front. Immunol. 2020, 11, 702. [Google Scholar] [CrossRef] [PubMed]
- López-Cantillo, G.; Urueña, C.; Camacho, B.A.; Ramírez-Segura, C. CAR-T Cell Performance: How to Improve Their Persistence? Front. Immunol. 2022, 13, 878209. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Engel, N.W.; Uslu, U.; Wellhausen, N.; June, C.H. Next-Generation CAR T-cell Therapies. Cancer Discov. 2022, 12, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Obergfell, K.; Topp, S.; Panier, V.; Wu, J. The next-generation CAR-T therapy landscape. Nat. Rev. Drug Discov. 2023, 22, 776–777. [Google Scholar] [CrossRef] [PubMed]
- Labanieh, L.; Mackall, C.L. CAR immune cells: Design principles, resistance and the next generation. Nature 2023, 614, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Halim, L.; Maher, J. CAR T-cell immunotherapy of B-cell malignancy: The story so far. Ther. Adv. Vaccines Immunother. 2020, 8, 2515135520927164. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Jiang, T.; Liu, D. BCMA-targeted immunotherapy for multiple myeloma. J. Hematol. Oncol. 2020, 13, 125. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef] [PubMed]
- Kleber, M.; Ntanasis-Stathopoulos, I.; Terpos, E. BCMA in Multiple Myeloma-A Promising Key to Therapy. J. Clin. Med. 2021, 10, 4088. [Google Scholar] [CrossRef] [PubMed]
- Baulu, E.; Gardet, C.; Chuvin, N.; Depil, S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci. Adv. 2023, 9, eadf3700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z.; Wei, W.; Li, Y. TCR engineered T cells for solid tumor immunotherapy. Exp. Hematol. Oncol. 2022, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Nidadavolu, L.S.; Walston, J.D. Underlying Vulnerabilities to the Cytokine Storm and Adverse COVID-19 Outcomes in the Aging Immune System. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, e13–e18. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Soper, B.; Shopland, L. Cytokine release syndrome and cancer immunotherapies—Historical challenges and promising futures. Front. Immunol. 2023, 14, 1190379. [Google Scholar] [CrossRef] [PubMed]
- Hines, M.R.; Knight, T.E.; McNerney, K.O.; Leick, M.B.; Jain, T.; Ahmed, S.; Frigault, M.J.; Hill, J.A.; Jain, M.D.; Johnson, W.T.; et al. Immune Effector Cell-Associated Hemophagocytic Lymphohistiocytosis-Like Syndrome. Transplant. Cell Ther. 2023, 29, 438.e1–438.e16. [Google Scholar] [CrossRef] [PubMed]
- Fugere, T.; Baltz, A.; Mukherjee, A.; Gaddam, M.; Varma, A.; Veeraputhiran, M.; Gentille Sanchez, C.G. Immune Effector Cell-Associated HLH-like Syndrome: A Review of the Literature of an Increasingly Recognized Entity. Cancers 2023, 15, 5149. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Sheng, Z.; Lu, D.R.; Ellwanger, D.C.; Li, C.M.; Homann, O.; Wang, S.; Yin, H.; Ren, R. Blinatumomab-induced T cell activation at single cell transcriptome resolution. BMC Genom. 2021, 22, 145. [Google Scholar] [CrossRef] [PubMed]
- Adkins, S. CAR T-Cell Therapy: Adverse Events and Management. J. Adv. Pract. Oncol. 2019, 10 (Suppl. S3), 21–28. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Zhang, Z.; Ren, Z.; Li, Y. Reactions Related to CAR-T Cell Therapy. Front. Immunol. 2021, 12, 663201. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Puglianini, O.; Chavez, J.C. Assessing and Management of Neurotoxicity After CAR-T Therapy in Diffuse Large B-Cell Lymphoma. J. Blood Med. 2021, 12, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. Immune effector cell associated neurotoxicity syndrome in chimeric antigen receptor-T cell therapy. Front. Immunol. 2022, 13, 879608. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.H.C.; Mohan, G.S.; Elhaddad, A.; Lehmann, L. Cytokine Release Syndrome and Associated Acute Toxicities in Pediatric Patients Undergoing Immune Effector Cell Therapy or Hematopoietic Cell Transplantation. Front. Oncol. 2022, 12, 841117. [Google Scholar] [CrossRef] [PubMed]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.C.; Fehse, B.; Akyüz, N.; Geffken, M.; Wolschke, C.; Janson, D.; Gagelmann, N.; Luther, M.; Wichmann, D.; Frenzel, C.; et al. Molecular monitoring of T-cell kinetics and migration in severe neurotoxicity after real-world CD19-specific chimeric antigen receptor T cell therapy. Haematologica 2023, 108, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Wang, Y.; Teng, X.; Hu, Y.; Huang, H. Cell subsets and cytokine dynamics in cerebrospinal fluid after CAR-T cell therapy for B-cell acute lymphoblastic leukemia with central nervous system involvement. Bone Marrow Transplant. 2021, 56, 3088–3090. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Harrington, A.M. Hemophagocytosis in cerebrospinal fluid after CAR T-cell therapy. Blood 2022, 139, 1116. [Google Scholar] [CrossRef] [PubMed]
- Lacan, C.; Caron, J.; Tarantino, N.; Fouquet, B.; Cherai, M.; Parizot, C.; Morel, V.; Souchet, L.; Uzunov, M.; Gorochov, G.; et al. CAR T-cell therapy for central nervous system lymphomas: Blood and cerebrospinal fluid biology, and outcomes. Haematologica 2023, 108, 3485–3490. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; van der Veen, S.; Merello, M.; Tijssen, M.A.J.; van de Warrenburg, B. Myoclonus-Ataxia Syndromes: A Diagnostic Approach. Mov. Disord. Clin. Pract. 2020, 8, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.J.; Choi, S.S.; Kim, H.Y.; Yu, I.K. Acute Acquired Metabolic Encephalopathy Based on Diffusion MRI. Korean J. Radiol. 2021, 22, 2034–2051. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Capizzano, A.A.; Moritani, T.; Naganawa, S.; Kurokawa, R.; Srinivasan, A. Comprehensive review of Wernicke encephalopathy: Pathophysiology, clinical symptoms and imaging findings. Jpn. J. Radiol. 2020, 38, 809–820. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, A.M.; Paulino, M.V.; Vieira, A.P.F.; McKinney, A.M.; da Rocha, A.J.; Dos Santos, G.T.; Leite, C.D.C.; Godoy, L.F.S.; Lucato, L.T. Imaging Patterns of Toxic and Metabolic Brain Disorders. Radiographics 2019, 39, 1672–1695. [Google Scholar] [CrossRef] [PubMed]
- Muccioli, L.; Pensato, U.; Cani, I.; Guerra, L.; Provini, F.; Bordin, G.; Riccioli, L.A.; Lodi, R.; Tinuper, P.; Bisulli, F. COVID-19-related encephalopathy presenting with aphasia resolving following tocilizumab treatment. J. Neuroimmunol. 2020, 349, 577400. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Hostetler, A.; Morgan, D.M.; Maiorino, L.; Sulkaj, I.; Whittaker, C.A.; Neeser, A.; Pires, I.S.; Yousefpour, P.; Gregory, J.; et al. Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity. Cell 2023, 186, 3148–3165.e20. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L.; Gardner, R. Mechanisms of and approaches to overcoming resistance to immunotherapy. Hematol. Am. Soc. Hematol. Educ. Program. 2019, 2019, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; Xu, J.; Chen, S.J.; Collins, M.A.; Zhou, J.; Powell, D.J., Jr.; Melenhorst, J.J. Engineering enhanced CAR T-cells for improved cancer therapy. Nat. Cancer 2021, 2, 780–793. [Google Scholar] [CrossRef] [PubMed]
- Zapata, L.; Caravagna, G.; Williams, M.J.; Lakatos, E.; AbdulJabbar, K.; Werner, B.; Chowell, D.; James, C.; Gourmet, L.; Milite, S.; et al. Immune selection determines tumor antigenicity and influences response to checkpoint inhibitors. Nat. Genet. 2023, 55, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Dagar, G.; Gupta, A.; Masoodi, T.; Nisar, S.; Merhi, M.; Hashem, S.; Chauhan, R.; Dagar, M.; Mirza, S.; Bagga, P.; et al. Harnessing the potential of CAR-T cell therapy: Progress, challenges, and future directions in hematological and solid tumor treatments. J. Transl. Med. 2023, 21, 449. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Draper, B.; Chaplin, N.; Philip, B.; Chin, M.; Galas-Filipowicz, D.; Onuoha, S.; Thomas, S.; Baldan, V.; Bughda, R.; et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood 2018, 131, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.C.; Kann, M.C.; Graham, C.; Mount, C.W.; Castano, A.P.; Lee, W.H.; Bouffard, A.A.; Takei, H.N.; Almazan, A.J.; Scarfó, I.; et al. Anti-TACI single and dual-targeting CAR T cells overcome BCMA antigen loss in multiple myeloma. Nat. Commun. 2023, 14, 7509. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Riddell, S.R. Dual Targeting with CAR T Cells to Limit Antigen Escape in Multiple Myeloma. Blood Cancer Discov. 2020, 1, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, A.; Bezerra, E.D.; Hirayama, A.V.; Hill, J.A.; Wu, Q.V.; Voutsinas, J.; Sorror, M.L.; Turtle, C.J.; Maloney, D.G.; Bar, M. Late Events after Treatment with CD19-Targeted Chimeric Antigen Receptor Modified T Cells. Biol. Blood Marrow Transplant. 2020, 26, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Penack, O.; Koenecke, C. Complications after CD19+ CAR T-Cell Therapy. Cancers 2020, 12, 3445. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.X.; Ong, X.J.; D’Souza, C.; Neeson, P.J.; Zhu, J.J. Combining chemotherapy with CAR-T cell therapy in treating solid tumors. Front. Immunol. 2023, 14, 1140541. [Google Scholar] [CrossRef] [PubMed]
- Al-Haideri, M.; Tondok, S.B.; Safa, S.H.; Maleki, A.H.; Rostami, S.; Jalil, A.T.; Al-Gazally, M.E.; Alsaikhan, F.; Rizaev, J.A.; Mohammad, T.A.M.; et al. CAR-T cell combination therapy: The next revolution in cancer treatment. Cancer Cell Int. 2022, 22, 365. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.; Shin, G.; Bae, S. Price and Prejudice? The Value of Chimeric Antigen Receptor (CAR) T-Cell Therapy. Int. J. Environ. Res. Public Health 2022, 19, 12366. [Google Scholar] [CrossRef] [PubMed]
- Cliff, E.R.S.; Kelkar, A.H.; Russler-Germain, D.A.; Tessema, F.A.; Raymakers, A.J.N.; Feldman, W.B.; Kesselheim, A.S. High Cost of Chimeric Antigen Receptor T-Cells: Challenges and Solutions. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e397912. [Google Scholar] [CrossRef] [PubMed]
- Potnis, K.C.; Di, M.; Isufi, I.; Gowda, L.; Seropian, S.E.; Foss, F.M.; Forman, H.P.; Huntington, S.F. Cost-effectiveness of chimeric antigen receptor T-cell therapy in adults with relapsed or refractory follicular lymphoma. Blood Adv. 2023, 7, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Dreger, P.; Dietrich, S.; Schubert, M.L.; Selberg, L.; Bondong, A.; Wegner, M.; Stadtherr, P.; Kimmich, C.; Kosely, F.; Schmitt, A.; et al. CAR T cells or allogeneic transplantation as standard of care for advanced large B-cell lymphoma: An intent-to-treat comparison. Blood Adv. 2020, 4, 6157–6168. [Google Scholar] [CrossRef] [PubMed]
- Bouziana, S.; Bouzianas, D. Exploring the Dilemma of Allogeneic Hematopoietic Cell Transplantation after Chimeric Antigen Receptor T Cell Therapy: To Transplant or Not? Biol. Blood Marrow Transplant. 2020, 26, e183–e191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.L.; Liu, D.Y.; Sun, R.J.; Zhang, J.P.; Zhou, J.R.; Wei, Z.J.; Xiong, M.; Cao, X.Y.; Lu, Y.; Yang, J.F.; et al. Integrating CAR T-Cell Therapy and Transplantation: Comparisons of Safety and Long-Term Efficacy of Allogeneic Hematopoietic Stem Cell Transplantation After CAR T-Cell or Chemotherapy-Based Complete Remission in B-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2021, 12, 605766. [Google Scholar] [CrossRef] [PubMed]
- Evgin, L.; Kottke, T.; Tonne, J.; Thompson, J.; Huff, A.L.; van Vloten, J.; Moore, M.; Michael, J.; Driscoll, C.; Pulido, J.; et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci. Transl. Med. 2022, 14, eabn2231. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, R.; Esmaeili Gouvarchin Ghaleh, H.; Farzanehpour, M.; Dorostkar, R.; Ranjbar, R.; Bolandian, M.; Mirzaei Nodooshan, M.; Ghorbani Alvanegh, A. Combination therapy with CAR T cells and oncolytic viruses: A new era in cancer immunotherapy. Cancer Gene Ther. 2022, 29, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef] [PubMed]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. 2019, 209, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Barker, T.H.; Gibbons, D.L.; Kurie, J.M. The fibrotic tumor stroma. J. Clin. Investig. 2018, 128, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi Darestani, N.; Gilmanova, A.I.; Al-Gazally, M.E.; Zekiy, A.O.; Ansari, M.J.; Zabibah, R.S.; Jawad, M.A.; Al-Shalah, S.A.J.; Rizaev, J.A.; Alnassar, Y.S.; et al. Mesenchymal stem cell-released oncolytic virus: An innovative strategy for cancer treatment. Cell Commun. Signal. 2023, 21, 43. [Google Scholar] [CrossRef] [PubMed]
- Mahasa, K.J.; de Pillis, L.; Ouifki, R.; Eladdadi, A.; Maini, P.; Yoon, A.R.; Yun, C.O. Mesenchymal stem cells used as carrier cells of oncolytic adenovirus results in enhanced oncolytic virotherapy. Sci. Rep. 2020, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Hadryś, A.; Sochanik, A.; McFadden, G.; Jazowiecka-Rakus, J. Mesenchymal stem cells as carriers for systemic delivery of oncolytic viruses. Eur. J. Pharmacol. 2020, 874, 172991. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Hong, J.A.; Choi, H.J.; Song, J.J. Enhanced tumor targeting and timely viral release of mesenchymal stem cells/oncolytic virus complex due to GRP78 and inducible E1B55K expressions greatly increase the antitumor effect of systemic treatment. Mol. Ther. Oncolytics 2022, 27, 26–47. [Google Scholar] [CrossRef] [PubMed]
- Yoon, A.R.; Rivera-Cruz, C.; Gimble, J.M.; Yun, C.O.; Figueiredo, M.L. Immunotherapy by mesenchymal stromal cell delivery of oncolytic viruses for treating metastatic tumors. Mol. Ther. Oncolytics 2022, 25, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Liu, X.; Fan, G.; Na, J.; Liu, Q.; Lin, F.; Zhang, Z.; Zhong, L. Improving the therapeutic efficacy of oncolytic viruses for cancer: Targeting macrophages. J. Transl. Med. 2023, 21, 842. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Dai, X.; Bianchi, A.; De Castro Silva, I.; Mehra, S.; Garrido, V.T.; Lamichhane, P.; Singh, S.P.; Zhou, Z.; Dosch, A.R.; et al. Combined MEK and STAT3 Inhibition Uncovers Stromal Plasticity by Enriching for Cancer-Associated Fibroblasts With Mesenchymal Stem Cell-Like Features to Overcome Immunotherapy Resistance in Pancreatic Cancer. Gastroenterology 2022, 163, 1593–1612. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Miao, J.M.; Wang, Y.Y.; Fan, Z.; Kong, X.B.; Yang, L.; Cheng, G. Oncolytic viruses combined with immune checkpoint therapy for colorectal cancer is a promising treatment option. Front. Immunol. 2022, 13, 961796. [Google Scholar] [CrossRef] [PubMed]
- Masemann, D.; Meissner, R.; Schied, T.; Lichty, B.D.; Rapp, U.R.; Wixler, V.; Ludwig, S. Synergistic antitumor efficacy of oncolytic influenza viruses and B7-H3 immune- checkpoint inhibitors against IC-resistant lung cancers. Oncoimmunology 2021, 10, 1885778. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.K.; Hong, J.; Yun, C.O. Oncolytic Viruses and Immune Checkpoint Inhibitors: Preclinical Developments to Clinical Trials. Int. J. Mol. Sci. 2020, 21, 8627. [Google Scholar] [CrossRef] [PubMed]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol. Ther. Oncolytics 2019, 13, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; McGray, A.J.R.; Jiang, W.; Lu, B.; Kalinski, P.; Guo, Z.S. Improving cancer immunotherapy by rationally combining oncolytic virus with modulators targeting key signaling pathways. Mol. Cancer 2022, 21, 196. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, Q.; Zhu, X. Mechanisms of CAR T cell exhaustion and current counteraction strategies. Front. Cell Dev. Biol. 2022, 10, 1034257. [Google Scholar] [CrossRef] [PubMed]
- Poorebrahim, M.; Melief, J.; Pico de Coaña, Y.L.; Wickström, S.; Cid-Arregui, A.; Kiessling, R. Counteracting CAR T cell dysfunction. Oncogene 2021, 40, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, C.T.; VanLith, C.J.; Hickey, R.D.; Du, Z.; Hillin, L.G.; Guthman, R.M.; Cao, W.J.; Haugo, B.; Lillegard, A.; Roy, D.; et al. In vivo lentiviral vector gene therapy to cure hereditary tyrosinemia type 1 and prevent development of precancerous and cancerous lesions. Nat. Commun. 2022, 13, 5012. [Google Scholar] [CrossRef] [PubMed]
- Buttery, P.C.; Barker, R.A. Gene and Cell-Based Therapies for Parkinson’s Disease: Where Are We? Neurotherapeutics 2020, 17, 1539–1562. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Gao, J.; Li, G.; Hu, H.; Zhou, L.; Lu, S.; Chen, X. Gene and cell therapies in China: Booming landscape under dual-track regulation. J. Hematol. Oncol. 2022, 15, 139. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q. Spotlight on gene therapy in China. Gene Ther. 2020, 27, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhu, Q.; Ga, M.; Wu, D.; Meng, X.; Wang, S.; Fang, H.; Tang, Y.; Li, N. Availability and Affordability of Oncology Drugs in 2012-2021 in China and the United States. Front. Oncol. 2022, 12, 930846. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Lauer, U.M.; Beil, J. Oncolytic viruses: Challenges and considerations in an evolving clinical landscape. Future Oncol. 2022, 18, 2713–2732. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Chen, Y.Y.; Spencer, M.J. Successes and challenges in clinical gene therapy. Gene Ther. 2023, 30, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Cornetta, K.; Bonamino, M.; Mahlangu, J.; Mingozzi, F.; Rangarajan, S.; Rao, J. Gene therapy access: Global challenges, opportunities, and views from Brazil, South Africa, and India. Mol. Ther. 2022, 30, 2122–2129. [Google Scholar] [CrossRef] [PubMed]
- Lapteva, L.; Purohit-Sheth, T.; Serabian, M.; Puri, R.K. Clinical Development of Gene Therapies: The First Three Decades and Counting. Mol. Ther. Methods Clin. Dev. 2020, 19, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.P.; Ruck, S.; Rafiq, Q.A.; Medcalf, N. Decentralised manufacturing of cell and gene therapy products: Learning from other healthcare sectors. Biotechnol. Adv. 2018, 36, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Gimpel, A.L.; Katsikis, G.; Sha, S.; Maloney, A.J.; Hong, M.S.; Nguyen, T.N.T.; Wolfrum, J.; Springs, S.L.; Sinskey, A.J.; Manalis, S.R.; et al. Analytical methods for process and product characterization of recombinant adeno-associated virus-based gene therapies. Mol. Ther. Methods Clin. Dev. 2021, 20, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Thielen, F.W.; Heine, R.J.S.D.; Berg, S.V.D.; Ham, R.M.T.T.; Groot, C.A.U. Towards sustainability and affordability of expensive cell and gene therapies? Applying a cost-based pricing model to estimate prices for Libmeldy and Zolgensma. Cytotherapy 2022, 24, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Sura, R.; Hutt, J.; Morgan, S. Opinion on the Use of Animal Models in Nonclinical Safety Assessment: Pros and Cons. Toxicol. Pathol. 2021, 49, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Veroniaina, H.; Su, N.; Sha, K.; Jiang, F.; Wu, Z.; Qi, X. Applications and developments of gene therapy drug delivery systems for genetic diseases. Asian J. Pharm. Sci. 2021, 16, 687–703. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Gessler, D.J.; Zhan, W.; Gallagher, T.L.; Gao, G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct. Target. Ther. 2024, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Bulaklak, K.; Gersbach, C.A. The once and future gene therapy. Nat. Commun. 2020, 11, 5820. [Google Scholar] [CrossRef] [PubMed]
- Yates, N.; Hinkel, J. The economics of moonshots: Value in rare disease drug development. Clin. Transl. Sci. 2022, 15, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Farid, S.S.; Baron, M.; Stamatis, C.; Nie, W.; Coffman, J. Benchmarking biopharmaceutical process development and manufacturing cost contributions to R&D. MAbs 2020, 12, 1754999. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Vroom, E.; O’Reilly, D. The Role of Patient Involvement When Developing Therapies. Nucleic Acid. Ther. 2022, 32, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Zvonareva, O.; Craveț, C.; Richards, D.P. Practices of patient engagement in drug development: A systematic scoping review. Res. Involv. Engag. 2022, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Brisse, M.; Vrba, S.M.; Kirk, N.; Liang, Y.; Ly, H. Emerging Concepts and Technologies in Vaccine Development. Front. Immunol. 2020, 11, 583077. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; McKenna, M.K.; Rosewell Shaw, A.; Suzuki, M. Clinical CAR-T Cell and Oncolytic Virotherapy for Cancer Treatment. Mol. Ther. 2021, 29, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Poletti, V.; Charrier, S.; Corre, G.; Gjata, B.; Vignaud, A.; Zhang, F.; Rothe, M.; Schambach, A.; Gaspar, H.B.; Thrasher, A.J.; et al. Preclinical Development of a Lentiviral Vector for Gene Therapy of X-Linked Severe Combined Immunodeficiency. Mol. Ther. Methods Clin. Dev. 2018, 9, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2017, 8, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef] [PubMed]
- Au, H.K.E.; Isalan, M.; Mielcarek, M. Gene Therapy Advances: A Meta-Analysis of AAV Usage in Clinical Settings. Front. Med. 2022, 8, 809118. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Servais, L.; Horton, R.; Saade, D.; Bonnemann, C.; Muntoni, F.; 261st ENMC workshop study group. 261st ENMC International Workshop: Management of safety issues arising following AAV gene therapy. 17th-19th June 2022, Hoofddorp, The Netherlands. Neuromuscul. Disord. 2023, 33, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.; Sarangi, P.; Jayandharan, G.R. Safety of Adeno-associated virus-based vector-mediated gene therapy-impact of vector dose. Cancer Gene Ther. 2022, 29, 1305–1306. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Liu, S.; Ou, L. rAAV immunogenicity, toxicity, and durability in 255 clinical trials: A meta-analysis. Front. Immunol. 2022, 13, 1001263. [Google Scholar] [CrossRef] [PubMed]
- Mondaca, S.P.; Liu, D.; Flynn, J.R.; Badson, S.; Hamaway, S.; Gounder, M.M.; Khalil, D.N.; Drilon, A.E.; Li, B.T.; Jhaveri, K.L.; et al. Clinical implications of drug-induced liver injury in early-phase oncology clinical trials. Cancer 2020, 126, 4967–4974. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Glass, Z.; Huang, M.; Chen, Z.Y.; Xu, Q. Ex vivo cell-based CRISPR/Cas9 genome editing for therapeutic applications. Biomaterials 2020, 234, 119711. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.X.; Loh, S.J.H.; Chan, W.K.; Soh, B.S. In Vivo Genome Editing as a Therapeutic Approach. Int. J. Mol. Sci. 2018, 19, 2721. [Google Scholar] [CrossRef] [PubMed]
- Lubroth, P.; Colasante, G.; Lignani, G. In vivo Genome Editing Therapeutic Approaches for Neurological Disorders: Where Are We in the Translational Pipeline? Front. Neurosci. 2021, 15, 632522. [Google Scholar] [CrossRef] [PubMed]
- Behr, M.; Zhou, J.; Xu, B.; Zhang, H. In vivo delivery of CRISPR-Cas9 therapeutics: Progress and challenges. Acta Pharm. Sin. B 2021, 11, 2150–2171. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Kankel, M.W.; Hana, S.; Lau, S.K.; Zavodszky, M.I.; McKissick, O.; Mastrangelo, N.; Dion, J.; Wang, B.; Ferretti, D.; et al. In vivo genome editing using novel AAV-PHP variants rescues motor function deficits and extends survival in a SOD1-ALS mouse model. Gene Ther. 2023, 30, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Zhai, H.; Shi, Y.; Liu, G.; Lowry, J.; Liu, B.; Ryan, É.B.; Yan, J.; Yang, Y.; Zhang, N.; et al. Efficacy and long-term safety of CRISPR/Cas9 genome editing in the SOD1-linked mouse models of ALS. Commun. Biol. 2021, 4, 396. [Google Scholar] [CrossRef] [PubMed]
- Jayarajan, V.; Kounatidou, E.; Qasim, W.; Di, W.L. Ex vivo gene modification therapy for genetic skin diseases-recent advances in gene modification technologies and delivery. Exp. Dermatol. 2021, 30, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Meneghini, V.; Baldo, G.; Gomez-Ospina, N. Ex-vivo and in-vivo genome engineering for metabolic and neurometabolic diseases. Front. Genome Ed. 2023, 5, 1248904. [Google Scholar] [CrossRef]
- Raguram, A.; Banskota, S.; Liu, D.R. Therapeutic in vivo delivery of gene editing agents. Cell 2022, 185, 2806–2827. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H.; Suh, S.; Sears, A.E.; Hołubowicz, R.; Kedhar, S.R.; Browne, A.W.; Palczewski, K. Genome editing in the treatment of ocular diseases. Exp. Mol. Med. 2023, 55, 1678–1690. [Google Scholar] [CrossRef] [PubMed]
- Kansy, M.; Caron, G. New therapeutic modalities in drug discovery and development: Insights & opportunities. ADMET DMPK 2021, 9, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Landhuis, E. The Definition of Gene Therapy Has Changed. Nature 2021. [Google Scholar] [CrossRef] [PubMed]
- Banoun, H. mRNA: Vaccine or Gene Therapy? The Safety Regulatory Issues. Int. J. Mol. Sci. 2023, 24, 10514. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Webb, E.; Lemoine, N.R.; Wang, Y. CRISPR-Cas9 as a Powerful Tool for Efficient Creation of Oncolytic Viruses. Viruses 2016, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Peters, C.; Kaufman, H.L. Generation and validation of recombinant herpes simplex type 1 viruses (HSV-1) using CRISPR/Cas9 genetic disruption. Methods Enzymol. 2020, 635, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chen, Z.N.; Wang, K. CRISPR/Cas9: A Powerful Strategy to Improve CAR-T Cell Persistence. Int. J. Mol. Sci. 2023, 24, 12317. [Google Scholar] [CrossRef] [PubMed]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- Mulia, G.E.; Picanço-Castro, V.; Stavrou, E.F.; Athanassiadou, A.; Figueiredo, M.L. Advances in the Development and the Applications of Nonviral, Episomal Vectors for Gene Therapy. Hum. Gene Ther. 2021, 2, 1076–1095. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Pietrobon, V.; Peng, M.; Wang, S.; Zhao, L.; Marincola, F.M.; Cai, Q. Current strategies employed in the manipulation of gene expression for clinical purposes. J. Transl. Med. 2022, 20, 535. [Google Scholar] [CrossRef] [PubMed]
- Morshedzadeh, F.; Ghanei, M.; Lotfi, M.; Ghasemi, M.; Ahmadi, M.; Najari-Hanjani, P.; Sharif, S.; Mozaffari-Jovin, S.; Peymani, M.; Abbaszadegan, M.R. An Update on the Application of CRISPR Technology in Clinical Practice. Mol. Biotechnol. 2024, 66, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Tucci, F.; Galimberti, S.; Naldini, L.; Valsecchi, M.G.; Aiuti, A. A systematic review and meta-analysis of gene therapy with hematopoietic stem and progenitor cells for monogenic disorders. Nat. Commun. 2022, 13, 1315. [Google Scholar] [CrossRef] [PubMed]
- Ponterio, E.; Haas, T.L.; De Maria, R. Oncolytic virus and CAR-T cell therapy in solid tumors. Front. Immunol. 2024, 15, 1455163. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Y.; Sun, T.K.; Chen, M.S.; Munir, M.; Liu, H.J. Oncolytic viruses-modulated immunogenic cell death, apoptosis and autophagy linking to virotherapy and cancer immune response. Front. Cell Infect. Microbiol. 2023, 13, 1142172. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.; Dotti, G. Combining Oncolytic Viruses with Chimeric Antigen Receptor T Cell Therapy. Hum. Gene Ther. 2021, 32, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2016, 15, 660. [Google Scholar] [CrossRef] [PubMed]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Ash, S.L.; Orha, R.; Mole, H.; Dinesh-Kumar, M.; Lee, S.P.; Turrell, F.K.; Isacke, C.M. Targeting the activated microenvironment with endosialin (CD248)-directed CAR-T cells ablates perivascular cells to impair tumor growth and metastasis. J. Immunother. Cancer 2024, 12, e008608. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, A.A.; Weiner, L.M. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 2020, 39, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Deng, J.; Gao, L.; Zhou, J. Innovative strategies to advance CAR T cell therapy for solid tumors. Am. J. Cancer Res. 2020, 10, 1979–1992. [Google Scholar] [PubMed]
- Peng, L.; Sferruzza, G.; Yang, L.; Zhou, L.; Chen, S. CAR-T and CAR-NK as cellular cancer immunotherapy for solid tumors. Cell Mol. Immunol. 2024, 21, 1089–1108. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Jin, S.W.; Xu, N.; Liu, W.Y.; Zhao, H.; Zhang, L.; Kang, L.; Tao, Y.; Liu, Y.; Wang, Y.; et al. PD-1 downregulation enhances CAR-T cell antitumor efficiency by preserving a cell memory phenotype and reducing exhaustion. J. Immunother. Cancer 2024, 12, e008429. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Luo, X.; Xie, Z.; Qiu, J.; Yang, J.; Deng, Y.; Long, R.; Tang, G.; Zhang, C.; Zuo, J. Prospects and challenges of CAR-T cell therapy combined with ICIs. Front. Oncol. 2024, 14, 1368732. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J., Jr.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Fogel, D.B. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: A review. Contemp. Clin. Trials Commun. 2018, 11, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Porter, D. Cytokine Release Syndrome with Chimeric Antigen Receptor T Cell Therapy. Biol. Blood Marrow Transplant. 2019, 25, e123–e127. [Google Scholar] [CrossRef] [PubMed]
- Sibbald, B. Death but one unintended consequence of gene-therapy trial. CMAJ 2001, 164, 1612. [Google Scholar] [PubMed]
- Abou-El-Enein, M.; Elsallab, M.; Feldman, S.A.; Fesnak, A.D.; Heslop, H.E.; Marks, P.; Till, B.G.; Bauer, G.; Savoldo, B. Scalable Manufacturing of CAR T cells for Cancer Immunotherapy. Blood Cancer Discov. 2021, 2, 408–422. [Google Scholar] [CrossRef] [PubMed]
- Mikhael, J.; Fowler, J.; Shah, N. Chimeric Antigen Receptor T-Cell Therapies: Barriers and Solutions to Access. JCO Oncol. Pract. 2022, 18, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Maiti, R.; Mohan, P.; Gupta, P. Antigen loss following CAR-T cell therapy: Mechanisms, implications, and potential solutions. Eur. J. Haematol. 2024, 112, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef] [PubMed]
- Levstek, L.; Janžič, L.; Ihan, A.; Kopitar, A.N. Biomarkers for prediction of CAR T therapy outcomes: Current and future perspectives. Front. Immunol. 2024, 15, 1378944. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; DiPersio, J.F. ReCARving the future: Bridging CAR T-cell therapy gaps with synthetic biology, engineering, and economic insights. Front. Immunol. 2024, 15, 1432799. [Google Scholar] [CrossRef] [PubMed]
- Lemos de Matos, A.; Franco, L.S.; McFadden, G. Oncolytic Viruses and the Immune System: The Dynamic Duo. Mol. Ther. Methods Clin. Dev. 2020, 17, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Daeffler, L.; Pozdeev, V.I.; Angelova, A.; Rommelaere, J. Immune Conversion of Tumor Microenvironment by Oncolytic Viruses: The Protoparvovirus H-1PV Case Study. Front. Immunol. 2019, 10, 1848. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Lei, Y.; Huang, J.; An, Y.; Ren, Y.; Chen, L.; Zhao, H.; Zheng, C. Recent advances in oncolytic virus therapy for hepatocellular carcinoma. Front. Oncol. 2023, 13, 1172292. [Google Scholar] [CrossRef] [PubMed]
- Kooti, W.; Esmaeili Gouvarchin Ghaleh, H.; Farzanehpour, M.; Dorostkar, R.; Jalali Kondori, B.; Bolandian, M. Oncolytic Viruses and Cancer, Do You Know the Main Mechanism? Front. Oncol. 2021, 11, 761015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, F. Advances and potential pitfalls of oncolytic viruses expressing immunomodulatory transgene therapy for malignant gliomas. Cell Death Dis. 2020, 11, 485. [Google Scholar] [CrossRef] [PubMed]
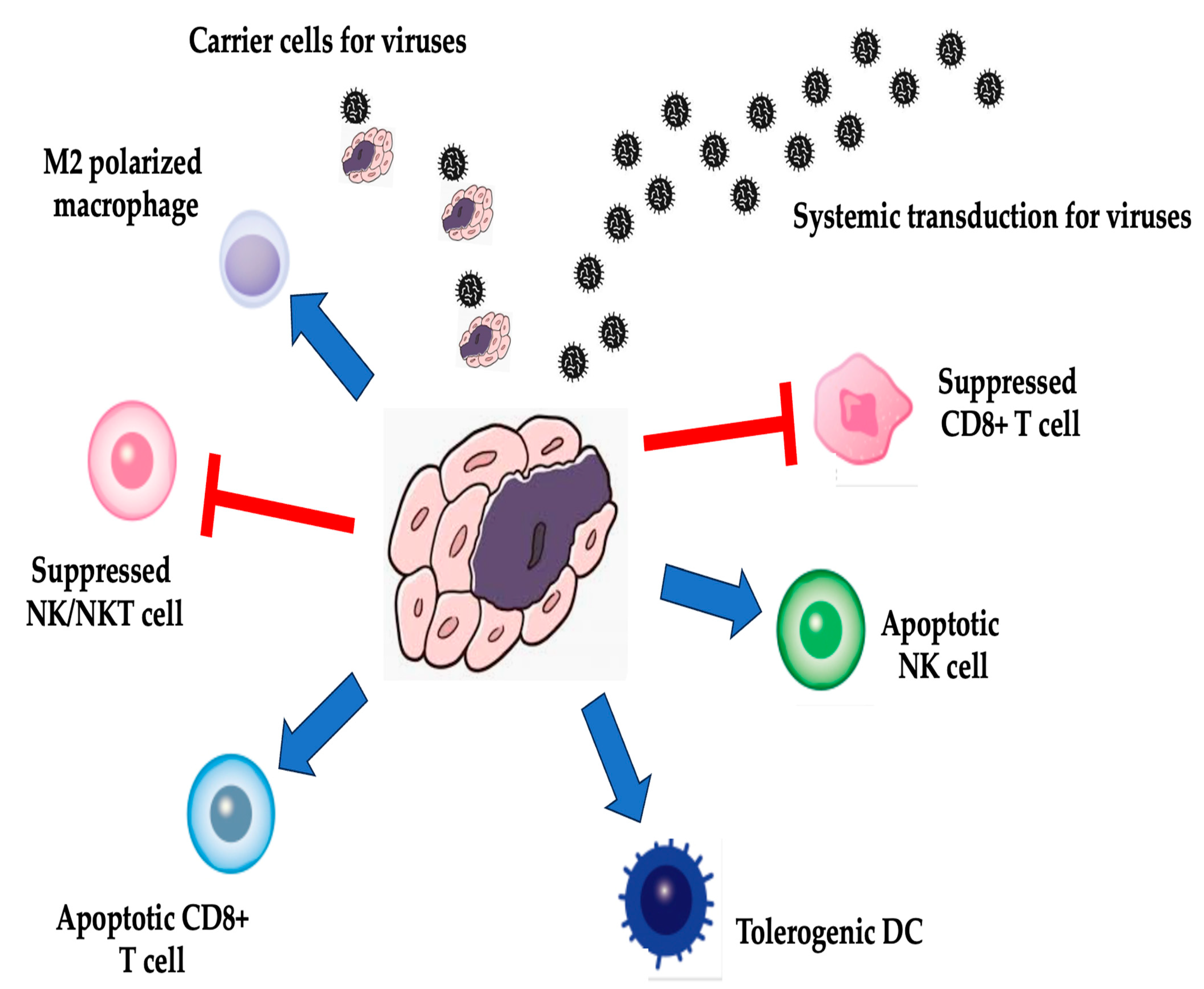

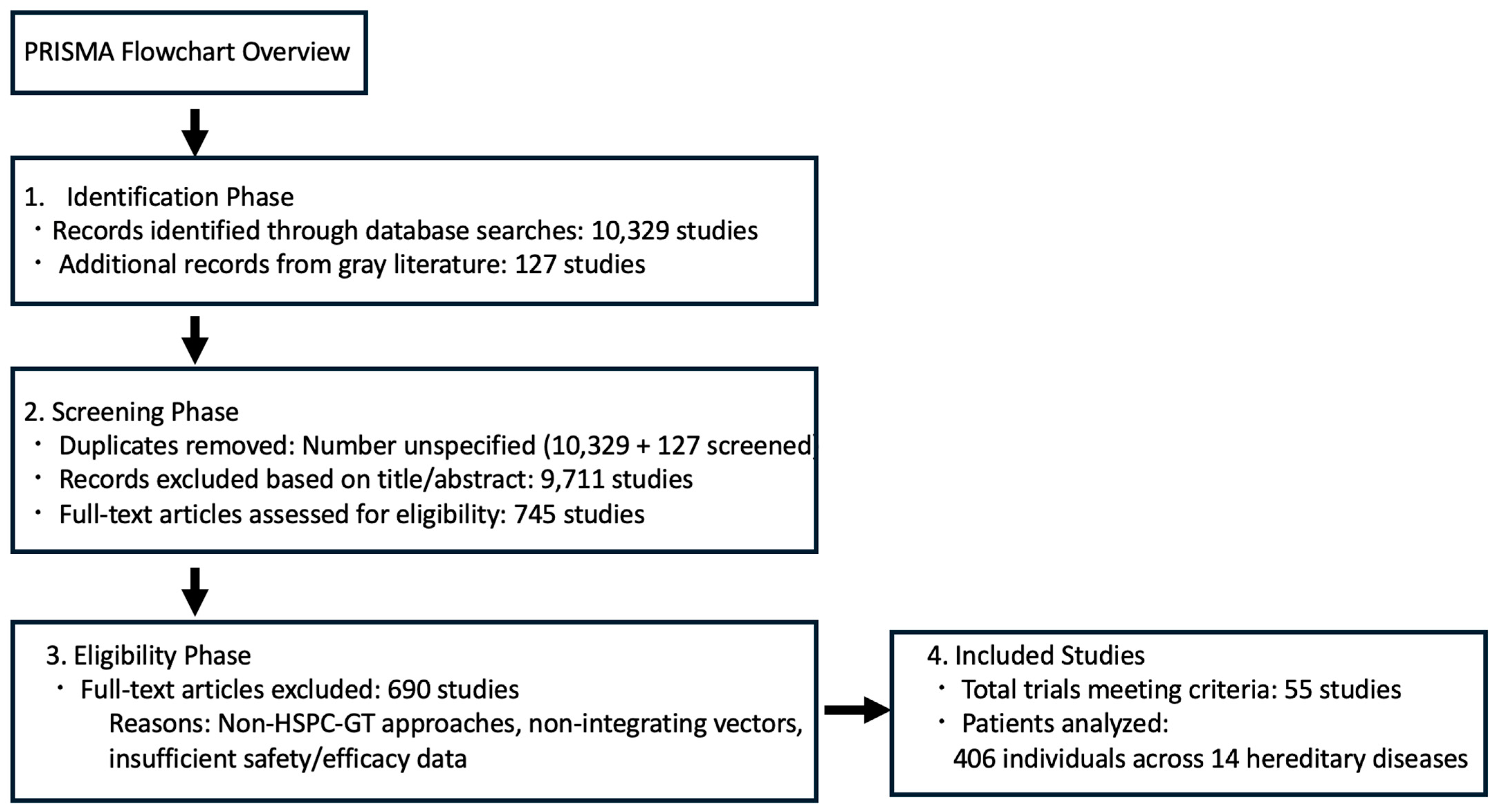

{kind=link}
{kind=link}
{kind=link}
| Virus | Name | Phase | Tumor | Reference |
|---|---|---|---|---|
| Adenovirus | ONYX-015 | III | Squamous cell carcinoma head and neck (SCCHN) | [25] |
| I/II | Pancreatic cancer | [26] | ||
| pilot | Advanced cancers | [27] | ||
| I/II | Advanced sarcoma | [28] | ||
| Oncorine | III | SCCHN or esophageal cancer | [29] | |
| AD5-CD/Tkrep | I | Prostate cancer | [30] | |
| ONCOS-201 | I | Solid tumors | [31] | |
| Herpes simplex virus | Talimogene laherparepvec | I/II | SCCHN | [32] |
| Ib | Melanoma | [33] | ||
| RT3D | I | Glioma | [34] | |
| Reovirus | I/II | Advanced cancers | [35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuzaka, Y.; Yashiro, R. Advances in Gene Therapy with Oncolytic Viruses and CAR-T Cells and Therapy-Related Groups. Curr. Issues Mol. Biol. 2025, 47, 268. https://doi.org/10.3390/cimb47040268
Matsuzaka Y, Yashiro R. Advances in Gene Therapy with Oncolytic Viruses and CAR-T Cells and Therapy-Related Groups. Current Issues in Molecular Biology. 2025; 47(4):268. https://doi.org/10.3390/cimb47040268
Chicago/Turabian StyleMatsuzaka, Yasunari, and Ryu Yashiro. 2025. "Advances in Gene Therapy with Oncolytic Viruses and CAR-T Cells and Therapy-Related Groups" Current Issues in Molecular Biology 47, no. 4: 268. https://doi.org/10.3390/cimb47040268
APA StyleMatsuzaka, Y., & Yashiro, R. (2025). Advances in Gene Therapy with Oncolytic Viruses and CAR-T Cells and Therapy-Related Groups. Current Issues in Molecular Biology, 47(4), 268. https://doi.org/10.3390/cimb47040268