
The Hallmarks of Ageing in Human Immunodeficiency Virus Infection and the Impact of Antiretroviral Therapy on Telomeres: A Molecular Perspective

 , ,
, ,  ,
, 
 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Current Perspectives on HIV Infection
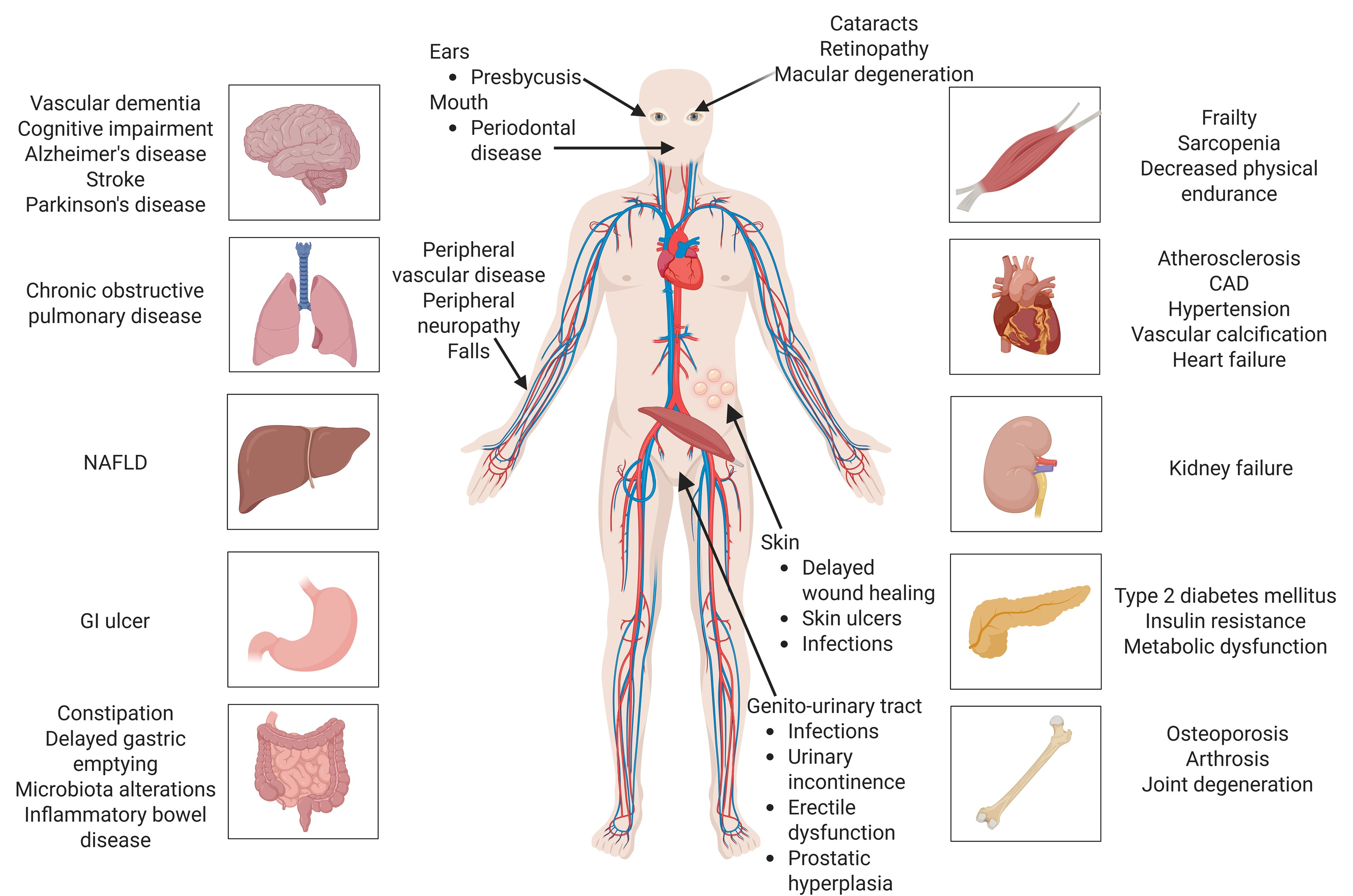
3. The Hallmarks of Ageing in HIV Infection
3.1. Primary Hallmarks of Ageing
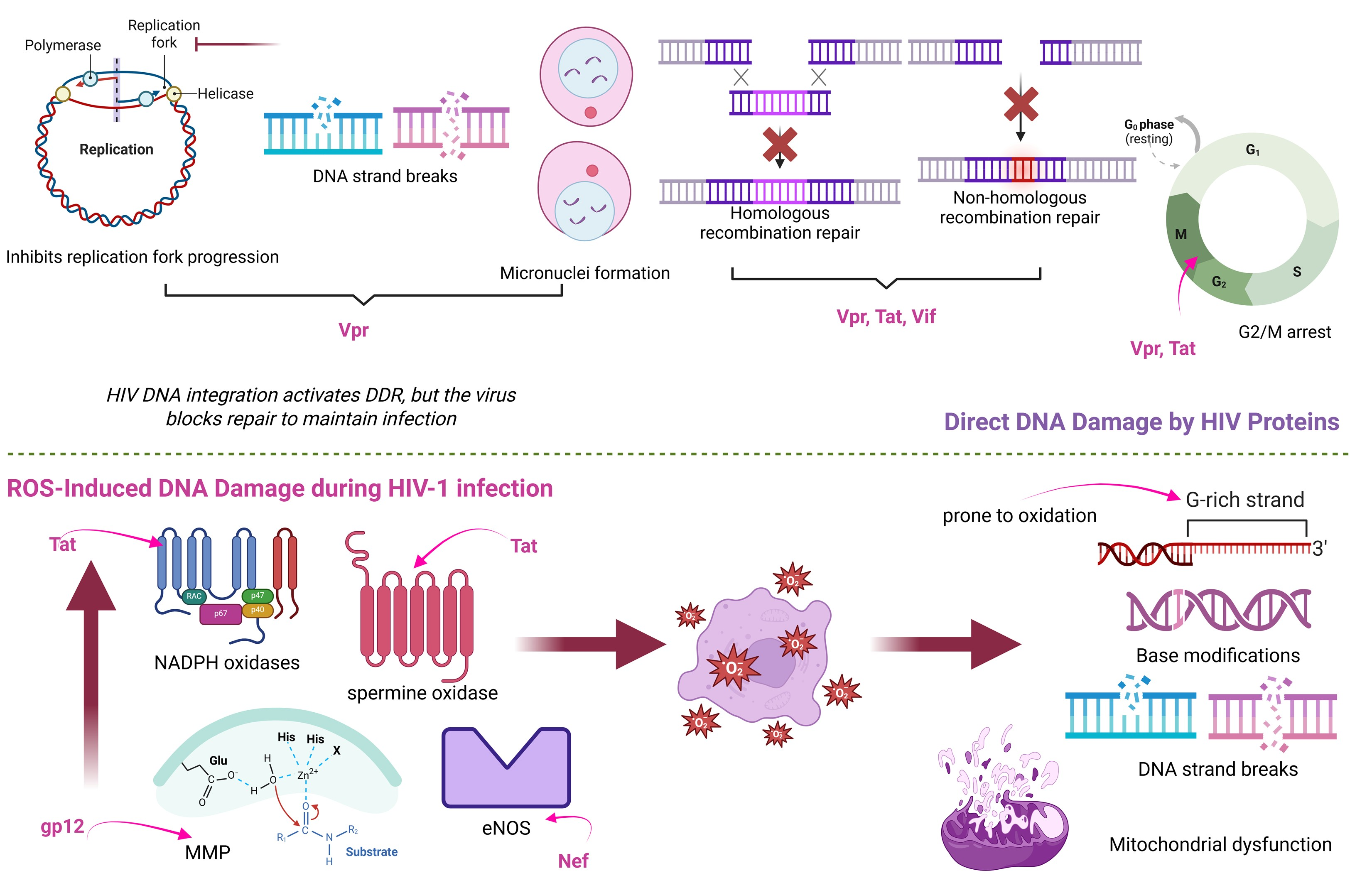
3.1.1. Genomic Instability
3.1.2. Telomere Attrition
3.1.3. Epigenetic Alterations
3.1.4. Loss of Proteostasis
3.1.5. Disabled Macroautophagy
3.2. Antagonistic Hallmarks of Ageing
3.2.1. Cellular Senescence
3.2.2. Mitochondrial Dysfunction
3.2.3. Deregulated Nutrient-Sensing
3.3. Integrative Hallmarks of Ageing
3.3.1. Stem Cell Exhaustion
3.3.2. Altered Intercellular Communication
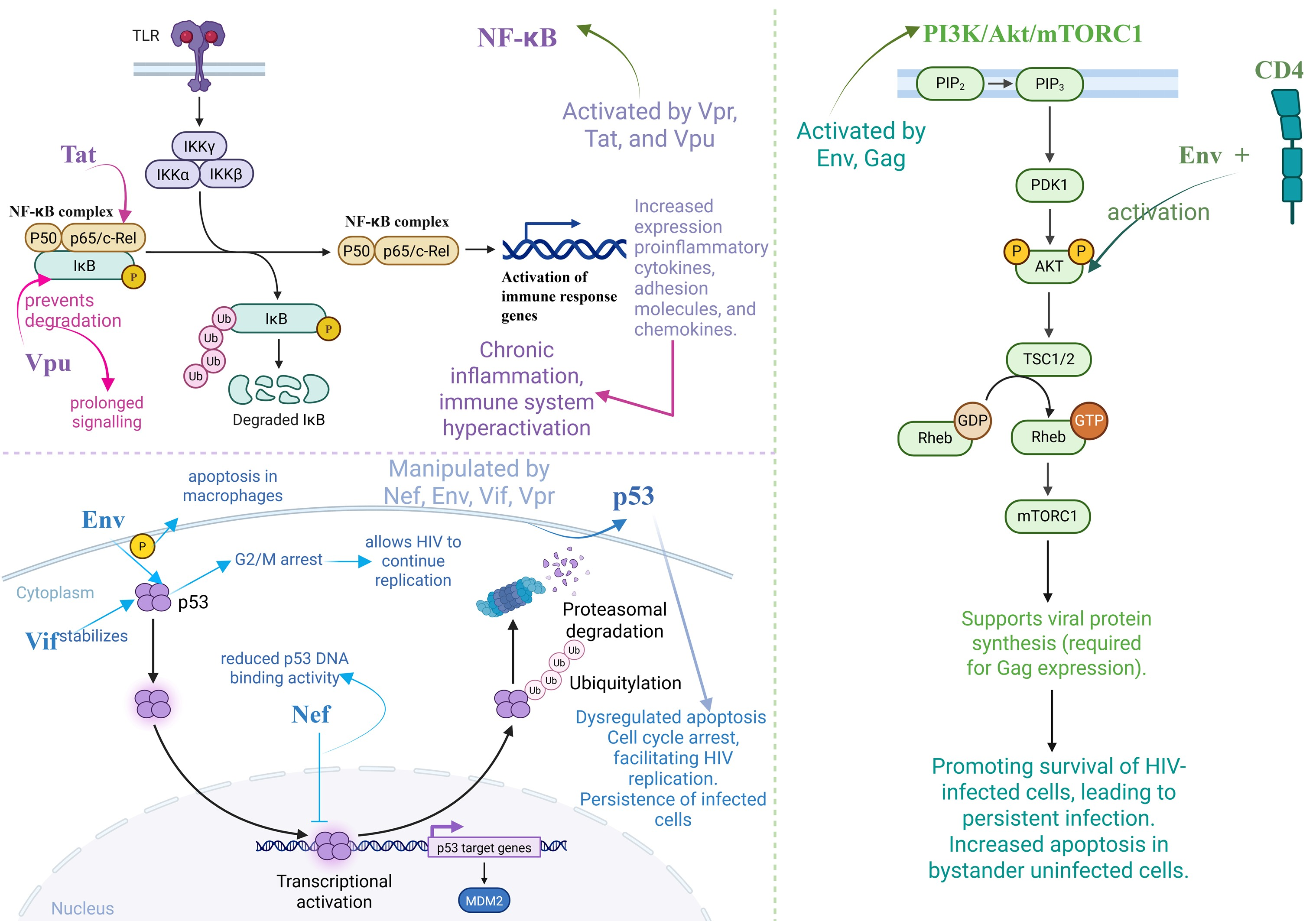
3.3.3. Chronic Inflammation
3.3.4. Dysbiosis
4. The Impact of ART on Telomere Biology
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, Y.; Tian, X.; Luo, J.; Bao, T.; Wang, S.; Wu, X. Molecular Mechanisms of Aging and Anti-Aging Strategies. Cell Commun. Signal. 2024, 22, 285. [Google Scholar] [CrossRef] [PubMed]
- Ariestanti, D.; Elya, B.; Hartrianti, P.; Arifin, V.; Hana, C.; Lovina, P.; Fadhila, R. The Anti-Ageing Potential of Litsea Oppositifolia Stem Extract: Evidence from in Vitro and Ex Vivo Study on Skin Cell Lines. Farmacia 2024, 72, 521–531. [Google Scholar] [CrossRef]
- Oliveros, A.; Poleschuk, M.; Cole, P.D.; Boison, D.; Jang, M.-H. Chemobrain: An Accelerated Aging Process Linking Adenosine A2A Receptor Signaling in Cancer Survivors. Int. Rev. Neurobiol. 2023, 170, 267–305. [Google Scholar] [PubMed]
- Falzone, L.; Candido, S.; Docea, A.O.; Calina, D. Editorial: Inflammation and Aging in Chronic and Degenerative Diseases: Current and Future Therapeutic Strategies. Front. Pharmacol. 2023, 13, 1122786. [Google Scholar] [CrossRef]
- Hamczyk, M.R.; Nevado, R.M.; Barettino, A.; Fuster, V.; Andrés, V. Biological Versus Chronological Aging. J. Am. Coll. Cardiol. 2020, 75, 919–930. [Google Scholar] [CrossRef]
- Statsenko, Y.; Kuznetsov, N.V.; Morozova, D.; Liaonchyk, K.; Simiyu, G.L.; Smetanina, D.; Kashapov, A.; Meribout, S.; Gorkom, K.N.-V.; Hamoudi, R.; et al. Reappraisal of the Concept of Accelerated Aging in Neurodegeneration and Beyond. Cells 2023, 12, 2451. [Google Scholar] [CrossRef] [PubMed]
- Kalmykova, A. Telomere Checkpoint in Development and Aging. Int. J. Mol. Sci. 2023, 24, 15979. [Google Scholar] [CrossRef] [PubMed]
- Frydrychová, R.Č.; Konopová, B.; Peska, V.; Brejcha, M.; Sábová, M. Telomeres and Telomerase: Active but Complex Players in Life-History Decisions. Biogerontology 2024, 25, 205–226. [Google Scholar] [CrossRef]
- Monsen, R.C.; Chakravarthy, S.; Dean, W.L.; Chaires, J.B.; Trent, J.O. The Solution Structures of Higher-Order Human Telomere G-Quadruplex Multimers. Nucleic Acids Res. 2021, 49, 1749–1768. [Google Scholar] [CrossRef]
- Wu, S.; Jiang, L.; Lei, L.; Fu, C.; Huang, J.; Hu, Y.; Dong, Y.; Chen, J.; Zeng, Q. Crosstalk between G-Quadruplex and ROS. Cell Death Dis. 2023, 14, 37. [Google Scholar] [CrossRef]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere Dysfunction in Ageing and Age-Related Diseases. Nat. Cell Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and Aging-Related Diseases: From Molecular Mechanisms to Interventions and Treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef] [PubMed]
- Dascalu, A.; Alexandrescu, C.; Vancea, G.; Stana, D.; Costea, D.; Zgura, A.; Serban, D.; Dumitrescu, D.; Tudosie, M.; Serboiu, C.; et al. The Relationship between Statin Therapy and Age-Related Macular Degeneration—A Systematic Review. Farmacia 2024, 72, 285–293. [Google Scholar] [CrossRef]
- Fragkiadaki, P.; Apetroaei, M.-M.; Kouvidi, E.; Vakonaki, E.; Renieri, E.; Fragkiadoulaki, I.; Spanakis, M.; Baliou, S.; Alegakis, A.; Tsatsakis, A. The Association between Short Telomere Length and Cardiovascular Disease. Cytogenet. Genome Res. 2024, 164, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Tsoukalas, D.; Buga, A.; Docea, A.; Sarandi, E.; Mitrut, R.; Renieri, E.; Spandidos, D.; Rogoveanu, I.; Cercelaru, L.; Niculescu, M.; et al. Reversal of Brain Aging by Targeting Telomerase: A Nutraceutical Approach. Int. J. Mol. Med. 2021, 48, 199. [Google Scholar] [CrossRef]
- Tsatsakis, A.; Oikonomopoulou, T.; Nikolouzakis, T.; Vakonaki, E.; Tzatzarakis, M.; Flamourakis, M.; Renieri, E.; Fragkiadaki, P.; Iliaki, E.; Bachlitzanaki, M.; et al. Role of Telomere Length in Human Carcinogenesis (Review). Int. J. Oncol. 2023, 63, 78. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.-J.; Semeraro, M.D.; Renner, W.; Herrmann, M. Telomeres and Age-Related Diseases. Biomedicines 2021, 9, 1335. [Google Scholar] [CrossRef] [PubMed]
- Seitz, R. Human Immunodeficiency Virus (HIV). Transfus. Med. Hemotherapy 2016, 43, 203–222. [Google Scholar] [CrossRef]
- Sever, B.; Otsuka, M.; Fujita, M.; Ciftci, H. A Review of FDA-Approved Anti-HIV-1 Drugs, Anti-Gag Compounds, and Potential Strategies for HIV-1 Eradication. Int. J. Mol. Sci. 2024, 25, 3659. [Google Scholar] [CrossRef]
- Huber, A.; Baas, F.S.; van der Ven, A.J.A.M.; Dos Santos, J.C. Innate Immune Cell Functions Contribute to Spontaneous HIV Control. Curr. HIV/AIDS Rep. 2024, 22, 6. [Google Scholar] [CrossRef]
- Tekeste, S.S.; Wilkinson, T.A.; Weiner, E.M.; Xu, X.; Miller, J.T.; Le Grice, S.F.J.; Clubb, R.T.; Chow, S.A. Interaction between Reverse Transcriptase and Integrase Is Required for Reverse Transcription during HIV-1 Replication. J. Virol. 2015, 89, 12058–12069. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Maggirwar, N.S.; Marsden, M.D. HIV Persistence, Latency, and Cure Approaches: Where Are We Now? Viruses 2024, 16, 1163. [Google Scholar] [CrossRef]
- Apetroaei, M.-M.; Velescu, B. Ștefan; Nedea, M.I.; Dinu-Pîrvu, C.E.; Drăgănescu, D.; Fâcă, A.I.; Udeanu, D.I.; Arsene, A.L. The Phenomenon of Antiretroviral Drug Resistance in the Context of Human Immunodeficiency Virus Treatment: Dynamic and Ever Evolving Subject Matter. Biomedicines 2024, 12, 915. [Google Scholar] [CrossRef]
- Borrajo, A. Breaking Barriers to an HIV-1 Cure: Innovations in Gene Editing, Immune Modulation, and Reservoir Eradication. Life 2025, 15, 276. [Google Scholar] [CrossRef]
- van Schalkwyk, C.; Mahy, M.; Johnson, L.F.; Imai-Eaton, J.W. Updated Data and Methods for the 2023 UNAIDS HIV Estimates. JAIDS J. Acquir. Immune Defic. Syndr. 2024, 95, e1–e4. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS Global HIV & AIDS Statistics—Fact Sheet. 2023. Available online: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (accessed on 12 December 2024).
- Hull, M.W.; Montaner, J.S.G. HIV Treatment as Prevention: The Key to an AIDS-Free Generation. J. Food Drug Anal. 2013, 21, S95–S101. [Google Scholar] [CrossRef]
- Taramasso, L.; Andreoni, M.; Antinori, A.; Bandera, A.; Bonfanti, P.; Bonora, S.; Borderi, M.; Castagna, A.; Cattelan, A.M.; Celesia, B.M.; et al. Pillars of Long-Term Antiretroviral Therapy Success. Pharmacol. Res. 2023, 196, 106898. [Google Scholar] [CrossRef]
- Bertagnolio, S.; Hermans, L.; Jordan, M.R.; Avila-Rios, S.; Iwuji, C.; Derache, A.; Delaporte, E.; Wensing, A.; Aves, T.; Borhan, A.S.M.; et al. Clinical Impact of Pretreatment Human Immunodeficiency Virus Drug Resistance in People Initiating Nonnucleoside Reverse Transcriptase Inhibitor–Containing Antiretroviral Therapy: A Systematic Review and Meta-Analysis. J. Infect. Dis. 2021, 224, 377–388. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, T.; Wu, C.; Deng, K.; Wang, M.; Qi, H.; Wang, N.; Li, Y.; Deng, Y.; Cao, G.; et al. Therapeutic Strategies and Use of Lopinavir/Ritonavir in Patients with Highly Resistant Human Immunodeficiency Virus. Farmacia 2024, 72, 588–596. [Google Scholar] [CrossRef]
- Payagala, S.; Pozniak, A. The Global Burden of HIV. Clin. Dermatol. 2024, 42, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Karris, M.Y.; Dubé, K.; Moore, A.A. What Lessons It Might Teach Us? Community Engagement in HIV Research. Curr. Opin. HIV AIDS 2020, 15, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Chahine, E.B. Fostemsavir: The First Oral Attachment Inhibitor for Treatment of HIV-1 Infection. Am. J. Health Pharm. 2021, 78, 376–388. [Google Scholar] [CrossRef]
- Beccari, M.V.; Mogle, B.T.; Sidman, E.F.; Mastro, K.A.; Asiago-Reddy, E.; Kufel, W.D. Ibalizumab, a Novel Monoclonal Antibody for the Management of Multidrug-Resistant HIV-1 Infection. Antimicrob. Agents Chemother. 2019, 63, e00110-19. [Google Scholar] [CrossRef]
- Temereanca, A.; Ruta, S. Strategies to Overcome HIV Drug Resistance-Current and Future Perspectives. Front. Microbiol. 2023, 14, 1133407. [Google Scholar] [CrossRef]
- Günthard, H.F.; Saag, M.S.; Benson, C.A.; del Rio, C.; Eron, J.J.; Gallant, J.E.; Hoy, J.F.; Mugavero, M.J.; Sax, P.E.; Thompson, M.A.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults. JAMA 2016, 316, 191. [Google Scholar] [CrossRef]
- Lungu, G.N.; Diaconescu, G.I.; Dumitrescu, F.; Docea, O.O.; Mitrut, R.; Giubelan, L.; Zlatian, O.; Mitrut, P. Liver Damage During Treatment with Reverse-Transcriptase Inhibitors in HIV Patients. Curr. Health Sci. J. 2024, 50, 181–197. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Tartiere, A.G.; Freije, J.M.P.; López-Otín, C. The Hallmarks of Aging as a Conceptual Framework for Health and Longevity Research. Front. Aging 2024, 5, 1334261. [Google Scholar] [CrossRef]
- Biga, P.R.; Duan, J.E.; Young, T.E.; Marks, J.R.; Bronikowski, A.; Decena, L.P.; Randolph, E.C.; Pavuluri, A.G.; Li, G.; Fang, Y.; et al. Hallmarks of Aging: A User’s Guide for Comparative Biologists. Ageing Res. Rev. 2025, 104, 102616. [Google Scholar] [CrossRef]
- Premeaux, T.A.; Ndhlovu, L.C. Decrypting Biological Hallmarks of Aging in People with HIV. Curr. Opin. HIV AIDS 2023, 18, 237–245. [Google Scholar] [CrossRef]
- Montano, M.; Oursler, K.K.; Xu, K.; Sun, Y.V.; Marconi, V.C. Biological Ageing with HIV Infection: Evaluating the Geroscience Hypothesis. Lancet Health Longev. 2022, 3, e194–e205. [Google Scholar] [CrossRef]
- Vijg, J.; Montagna, C. Genome Instability and Aging: Cause or Effect? Transl. Med. Aging 2017, 1, 5–11. [Google Scholar] [CrossRef]
- Popovic, M.; Kahl, V.; Hoch, N.C. Editorial: Genome Instability: Old Problem, New Solutions. Front. Cell Dev. Biol. 2022, 10, 868038. [Google Scholar] [CrossRef]
- Lopez, A.; Nichols Doyle, R.; Sandoval, C.; Nisson, K.; Yang, V.; Fregoso, O.I. Viral Modulation of the DNA Damage Response and Innate Immunity: Two Sides of the Same Coin. J. Mol. Biol. 2022, 434, 167327. [Google Scholar] [CrossRef]
- Luftig, M.A. Viruses and the DNA Damage Response: Activation and Antagonism. Annu. Rev. Virol. 2014, 1, 605–625. [Google Scholar] [CrossRef]
- Ellwanger, J.H.; Kulmann-Leal, B.; Ziliotto, M.; Chies, J.A.B. HIV Infection, Chromosome Instability, and Micronucleus Formation. Viruses 2023, 15, 155. [Google Scholar] [CrossRef]
- Anisenko, A.; Nefedova, A.; Agapkina, Y.; Gottikh, M. Both ATM and DNA-PK Are the Main Regulators of HIV-1 Post-Integrational DNA Repair. Int. J. Mol. Sci. 2023, 24, 2797. [Google Scholar] [CrossRef]
- Mdletshe, N.; Nel, A.; Shires, K.; Mowla, S. HIV Nef Enhances the Expression of Oncogenic C-MYC and Activation-Induced Cytidine Deaminase in Burkitt Lymphoma Cells, Promoting Genomic Instability. Infect. Agent. Cancer 2020, 15, 54. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Bunting, S.; Feldhahn, N.; Bothmer, A.; Camps, J.; Deroubaix, S.; McBride, K.M.; Klein, I.A.; Stone, G.; Eisenreich, T.R.; et al. AID Produces DNA Double-Strand Breaks in Non-Ig Genes and Mature B Cell Lymphomas with Reciprocal Chromosome Translocations. Mol. Cell 2009, 36, 631–641. [Google Scholar] [CrossRef]
- Poetsch, A.R. The Genomics of Oxidative DNA Damage, Repair, and Resulting Mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Isaguliants, M.; Bayurova, E.; Avdoshina, D.; Kondrashova, A.; Chiodi, F.; Palefsky, J. Oncogenic Effects of HIV-1 Proteins, Mechanisms Behind. Cancers 2021, 13, 305. [Google Scholar] [CrossRef]
- Louboutin, J.-P.; Agrawal, L.; Reyes, B.A.S.; Van Bockstaele, E.J.; Strayer, D.S. HIV-1 Gp120-Induced Injury to the Blood-Brain Barrier: Role of Metalloproteinases 2 and 9 and Relationship to Oxidative Stress. J. Neuropathol. Exp. Neurol. 2010, 69, 801–816. [Google Scholar] [CrossRef] [PubMed]
- Mangino, G.; Famiglietti, M.; Capone, C.; Veroni, C.; Percario, Z.A.; Leone, S.; Fiorucci, G.; Lülf, S.; Romeo, G.; Agresti, C.; et al. HIV-1 Myristoylated Nef Treatment of Murine Microglial Cells Activates Inducible Nitric Oxide Synthase, NO2 Production and Neurotoxic Activity. PLoS ONE 2015, 10, e0130189. [Google Scholar] [CrossRef]
- Gutiérrez-Sevilla, J.E.; Cárdenas-Bedoya, J.; Escoto-Delgadillo, M.; Zúñiga-González, G.M.; Pérez-Ríos, A.M.; Gómez-Meda, B.C.; González-Enríquez, G.V.; Figarola-Centurión, I.; Chavarría-Avila, E.; Torres-Mendoza, B.M. Genomic Instability in People Living with HIV. Mutat. Res. Toxicol. Environ. Mutagen. 2021, 865, 503336. [Google Scholar] [CrossRef]
- Lindberg, H.K.; Wang, X.; Järventaus, H.; Falck, G.C.-M.; Norppa, H.; Fenech, M. Origin of Nuclear Buds and Micronuclei in Normal and Folate-Deprived Human Lymphocytes. Mutat. Res. Mol. Mech. Mutagen. 2007, 617, 33–45. [Google Scholar] [CrossRef]
- Zanet, D.L.; Thorne, A.; Singer, J.; Maan, E.J.; Sattha, B.; Le Campion, A.; Soudeyns, H.; Pick, N.; Murray, M.; Money, D.M.; et al. Association between Short Leukocyte Telomere Length and HIV Infection in a Cohort Study: No Evidence of a Relationship with Antiretroviral Therapy. Clin. Infect. Dis. 2014, 58, 1322–1332. [Google Scholar] [CrossRef]
- Gonzalez-Serna, A.; Ajaykumar, A.; Gadawski, I.; Muñoz-Fernández, M.A.; Hayashi, K.; Harrigan, P.R.; Côté, H.C.F. Rapid Decrease in Peripheral Blood Mononucleated Cell Telomere Length After HIV Seroconversion, but Not HCV Seroconversion. J. Acquir. Immune Defic. Syndr. 2017, 76, e29–e32. [Google Scholar] [CrossRef] [PubMed]
- Pathai, S.; Lawn, S.D.; Gilbert, C.E.; McGuinness, D.; McGlynn, L.; Weiss, H.A.; Port, J.; Christ, T.; Barclay, K.; Wood, R.; et al. Accelerated Biological Ageing in HIV-Infected Individuals in South Africa: A Case-Control Study. AIDS 2013, 27, 2375–2384. [Google Scholar] [CrossRef]
- Yang, N.Y.; Hsieh, A.Y.Y.; Chen, Z.; Campbell, A.R.; Gadawska, I.; Kakkar, F.; Sauve, L.; Bitnun, A.; Brophy, J.; Murray, M.C.M.; et al. Chronic and Latent Viral Infections and Leukocyte Telomere Length across the Lifespan of Female and Male Individuals Living with or without HIV. Viruses 2024, 16, 755. [Google Scholar] [CrossRef]
- Macamo, E.D.; Mkhize-Kwitshana, Z.L.; Mthombeni, J.; Naidoo, P. The Impact of HIV and Parasite Single Infection and Coinfection on Telomere Length: A Systematic Review. Curr. Issues Mol. Biol. 2024, 46, 7258–7290. [Google Scholar] [CrossRef] [PubMed]
- Macamo, E.D.; Mkhize-Kwitshana, Z.L.; Duma, Z.; Mthombeni, J.; Naidoo, P. Telomere Length in a South African Population Co-Infected with HIV and Helminths. Curr. Issues Mol. Biol. 2024, 46, 6853–6867. [Google Scholar] [CrossRef]
- Schoepf, I.C.; Thorball, C.W.; Ledergerber, B.; Kootstra, N.A.; Reiss, P.; Raffenberg, M.; Engel, T.; Braun, D.L.; Hasse, B.; Thurnheer, C.; et al. Telomere Length Declines in Persons With Human Immunodeficiency Virus Before Antiretroviral Therapy Start but Not After Viral Suppression: A Longitudinal Study Over >17 Years. J. Infect. Dis. 2022, 225, 1581–1591. [Google Scholar] [CrossRef]
- Toljić, B.; Milašin, J.; De Luka, S.R.; Dragović, G.; Jevtović, D.; Maslać, A.; Ristić-Djurović, J.L.; Trbovich, A.M. HIV-Infected Patients as a Model of Aging. Microbiol. Spectr. 2023, 11, e0053223. [Google Scholar] [CrossRef]
- Grosso, T.M.; Alcamí, J.; Arribas, J.R.; Martín, M.; Sereti, I.; Tarr, P.; Cahn, P.; Clotet, B.; Sued, O.; Negredo, E. HIV and Aging, Biological Mechanisms, and Therapies: What Do We Know? Aids Rev. 2022, 24, 79–86. [Google Scholar] [CrossRef]
- Mehta, S.R.; Iudicello, J.E.; Lin, J.; Ellis, R.J.; Morgan, E.; Okwuegbuna, O.; Cookson, D.; Karris, M.; Saloner, R.; Heaton, R.; et al. Telomere Length Is Associated with HIV Infection, Methamphetamine Use, Inflammation, and Comorbid Disease Risk. Drug Alcohol Depend. 2021, 221, 108639. [Google Scholar] [CrossRef]
- Schoepf, I.C.; Thorball, C.W.; Ledergerber, B.; Engel, T.; Raffenberg, M.; Kootstra, N.A.; Reiss, P.; Hasse, B.; Marzolini, C.; Thurnheer, C.; et al. Coronary Artery Disease-Associated and Longevity-Associated Polygenic Risk Scores for Prediction of Coronary Artery Disease Events in Persons Living With Human Immunodeficiency Virus: The Swiss HIV Cohort Study. Clin. Infect. Dis. 2021, 73, 1597–1604. [Google Scholar] [CrossRef]
- Kurashova, N.A.; Vanyarkina, A.S.; Petrova, A.G.; Rychkova, L.V.; Kolesnikov, S.I.; Darenskaya, M.A.; Moskaleva, E.V.; Kolesnikova, L.I. Length of Leukocyte Telomeres in Newborns of HIV-Infected Mothers. Bull. Exp. Biol. Med. 2023, 175, 260–264. [Google Scholar] [CrossRef]
- Lu, A.T.; Seeboth, A.; Tsai, P.-C.; Sun, D.; Quach, A.; Reiner, A.P.; Kooperberg, C.; Ferrucci, L.; Hou, L.; Baccarelli, A.A.; et al. DNA Methylation-Based Estimator of Telomere Length. Aging 2019, 11, 5895–5923. [Google Scholar] [CrossRef]
- Liang, X.; Aouizerat, B.E.; So-Armah, K.; Cohen, M.H.; Marconi, V.C.; Xu, K.; Justice, A.C. DNA Methylation-Based Telomere Length Is Associated with HIV Infection, Physical Frailty, Cancer, and All-Cause Mortality. Aging Cell 2024, 23, e14174. [Google Scholar] [CrossRef]
- Hu, H.; Li, B.; Duan, S. The Alteration of Subtelomeric DNA Methylation in Aging-Related Diseases. Front. Genet. 2018, 9, 697. [Google Scholar] [CrossRef]
- Mender, I.; Shay, J.W. Telomere Restriction Fragment (TRF) Analysis. Bio-Protocol 2015, 5, e1658. [Google Scholar] [CrossRef]
- Pearce, E.E.; Horvath, S.; Katta, S.; Dagnall, C.; Aubert, G.; Hicks, B.D.; Spellman, S.R.; Katki, H.; Savage, S.A.; Alsaggaf, R.; et al. DNA-Methylation-Based Telomere Length Estimator: Comparisons with Measurements from Flow FISH and QPCR. Aging 2021, 13, 14675–14686. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic Modifications. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef]
- Titanji, B.K.; Gwinn, M.; Marconi, V.C.; Sun, Y.V. Epigenome-Wide Epidemiologic Studies of Human Immunodeficiency Virus Infection, Treatment, and Disease Progression. Clin. Epigenetics 2022, 14, 8. [Google Scholar] [CrossRef]
- Lange, U.C.; Verdikt, R.; Ait-Ammar, A.; Van Lint, C. Epigenetic Crosstalk in Chronic Infection with HIV-1. Semin. Immunopathol. 2020, 42, 187–200. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramphal, U.; Adimulam, T.; Chinniah, R.; Ramsuran, V. Deciphering DNA Methylation in HIV Infection. Front. Immunol. 2021, 12, 795121. [Google Scholar] [CrossRef]
- Nunes, J.M.; Furtado, M.N.; de Morais Nunes, E.R.; Sucupira, M.C.A.; Diaz, R.S.; Janini, L.M.R. Modulation of Epigenetic Factors during the Early Stages of HIV-1 Infection in CD4+ T Cells in Vitro. Virology 2018, 523, 41–51. [Google Scholar] [CrossRef]
- Abdel-Hameed, E.A.; Ji, H.; Sherman, K.E.; Shata, M.T.M. Epigenetic Modification of FOXP3 in Patients With Chronic HIV Infection. JAIDS J. Acquir. Immune Defic. Syndr. 2014, 65, 19–26. [Google Scholar] [CrossRef]
- Pion, M.; Jaramillo-Ruiz, D.; Martínez, A.; Muñoz-Fernández, M.A.; Correa-Rocha, R. HIV Infection of Human Regulatory T Cells Downregulates Foxp3 Expression by Increasing DNMT3b Levels and DNA Methylation in the FOXP3 Gene. AIDS 2013, 27, 2019–2029. [Google Scholar] [CrossRef]
- Abdel-Hameed, E.A.; Ji, H.; Shata, M.T. HIV-Induced Epigenetic Alterations in Host Cells. Adv. Exp. Med. Biol. 2016, 879, 27–38. [Google Scholar] [PubMed]
- Sadowski, I.; Hashemi, F.B. Strategies to Eradicate HIV from Infected Patients: Elimination of Latent Provirus Reservoirs. Cell. Mol. Life Sci. 2019, 76, 3583–3600. [Google Scholar] [CrossRef] [PubMed]
- Battistini, A.; Sgarbanti, M. HIV-1 Latency: An Update of Molecular Mechanisms and Therapeutic Strategies. Viruses 2014, 6, 1715–1758. [Google Scholar] [CrossRef]
- Ma, X.; Chen, T.; Peng, Z.; Wang, Z.; Liu, J.; Yang, T.; Wu, L.; Liu, G.; Zhou, M.; Tong, M.; et al. Histone Chaperone CAF-1 Promotes HIV-1 Latency by Leading the Formation of Phase-separated Suppressive Nuclear Bodies. EMBO J. 2021, 40, e106632. [Google Scholar] [CrossRef] [PubMed]
- Marzio, G.; Tyagi, M.; Gutierrez, M.I.; Giacca, M. HIV-1 Tat Transactivator Recruits P300 and CREB-Binding Protein Histone Acetyltransferases to the Viral Promoter. Proc. Natl. Acad. Sci. USA 1998, 95, 13519–13524. [Google Scholar] [CrossRef]
- Margolis, D.M. Histone Deacetylase Inhibitors and HIV Latency. Curr. Opin. HIV AIDS 2011, 6, 25–29. [Google Scholar] [CrossRef]
- Lenasi, T.; Contreras, X.; Peterlin, B.M. Transcriptional Interference Antagonizes Proviral Gene Expression to Promote HIV Latency. Cell Host Microbe 2008, 4, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Espíndola, M.S.; Soares, L.S.; Galvão-Lima, L.J.; Zambuzi, F.A.; Cacemiro, M.C.; Brauer, V.S.; Marzocchi-Machado, C.M.; de Souza Gomes, M.; Amaral, L.R.; Martins-Filho, O.A.; et al. Epigenetic Alterations Are Associated with Monocyte Immune Dysfunctions in HIV-1 Infection. Sci. Rep. 2018, 8, 5505. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Mainolfi, N.; Rasmusson, T. Targeted Protein Degradation. Annu. Rep. Med. Chem. 2017, 50, 301–334. [Google Scholar]
- Seissler, T.; Marquet, R.; Paillart, J.-C. Hijacking of the Ubiquitin/Proteasome Pathway by the HIV Auxiliary Proteins. Viruses 2017, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Iwatani, Y.; Chan, D.S.B.; Liu, L.; Yoshii, H.; Shibata, J.; Yamamoto, N.; Levin, J.G.; Gronenborn, A.M.; Sugiura, W. HIV-1 Vif-Mediated Ubiquitination/Degradation of APOBEC3G Involves Four Critical Lysine Residues in Its C-Terminal Domain. Proc. Natl. Acad. Sci. USA 2009, 106, 19539–19544. [Google Scholar] [CrossRef]
- Li, Y.-L.; Langley, C.A.; Azumaya, C.M.; Echeverria, I.; Chesarino, N.M.; Emerman, M.; Cheng, Y.; Gross, J.D. The Structural Basis for HIV-1 Vif Antagonism of Human APOBEC3G. Nature 2023, 615, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Ballana, E.; Badia, R.; Terradas, G.; Torres-Torronteras, J.; Ruiz, A.; Pauls, E.; Riveira-Muñoz, E.; Clotet, B.; Martí, R.; Esté, J.A. SAMHD1 Specifically Affects the Antiviral Potency of Thymidine Analog HIV Reverse Transcriptase Inhibitors. Antimicrob. Agents Chemother. 2014, 58, 4804–4813. [Google Scholar] [CrossRef]
- Zheng, Y.-H.; Jeang, K.-T.; Tokunaga, K. Host Restriction Factors in Retroviral Infection: Promises in Virus-Host Interaction. Retrovirology 2012, 9, 112. [Google Scholar] [CrossRef]
- Hofmann, H.; Logue, E.C.; Bloch, N.; Daddacha, W.; Polsky, S.B.; Schultz, M.L.; Kim, B.; Landau, N.R. The Vpx Lentiviral Accessory Protein Targets SAMHD1 for Degradation in the Nucleus. J. Virol. 2012, 86, 12552–12560. [Google Scholar] [CrossRef]
- Dubé, M.; Paquay, C.; Roy, B.B.; Bego, M.G.; Mercier, J.; Cohen, É.A. HIV-1 Vpu Antagonizes BST-2 by Interfering Mainly with the Trafficking of Newly Synthesized BST-2 to the Cell Surface. Traffic 2011, 12, 1714–1729. [Google Scholar] [CrossRef]
- Lata, S.; Mishra, R.; Banerjea, A.C. Proteasomal Degradation Machinery: Favorite Target of HIV-1 Proteins. Front. Microbiol. 2018, 9, 2738. [Google Scholar] [CrossRef]
- Remoli, A.L.; Marsili, G.; Perrotti, E.; Acchioni, C.; Sgarbanti, M.; Borsetti, A.; Hiscott, J.; Battistini, A. HIV-1 Tat Recruits HDM2 E3 Ligase To Target IRF-1 for Ubiquitination and Proteasomal Degradation. mBio 2016, 7, e01528-16. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Virgilio, L.; Silva-Lucero, M.-C.; Flores-Morelos, D.-S.; Gallardo-Nieto, J.; Lopez-Toledo, G.; Abarca-Fernandez, A.-M.; Zacapala-Gómez, A.-E.; Luna-Muñoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O.; et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11, 2262. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The Machinery of Macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; Hansen, M. Macroautophagy and Aging: The Impact of Cellular Recycling on Health and Longevity. Mol. Aspects Med. 2021, 82, 101020. [Google Scholar] [CrossRef] [PubMed]
- Cheney, L.; Guzik, H.; Macaluso, F.P.; Macian, F.; Cuervo, A.M.; Berman, J.W. HIV Nef and Antiretroviral Therapy Have an Inhibitory Effect on Autophagy in Human Astrocytes That May Contribute to HIV-Associated Neurocognitive Disorders. Cells 2020, 9, 1426. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gonzalez, S.; Shi, Y.; Colomer-Lluch, M.; Song, Y.; Mowery, K.; Almodovar, S.; Bansal, A.; Kirchhoff, F.; Sparrer, K.; Liang, C.; et al. HIV-1 Nef Counteracts Autophagy Restriction by Enhancing the Association between BECN1 and Its Inhibitor BCL2 in a PRKN-Dependent Manner. Autophagy 2021, 17, 553–577. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, M.; Duan, Q.; Bryant, J.L.; Xu, X. The Role of Autophagy in the Progression of HIV Infected Cardiomyopathy. Front. Cell Dev. Biol. 2024, 12, 1372573. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular Senescence in Ageing: From Mechanisms to Therapeutic Opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Ernst, P.; Heidel, F.H. Molecular Mechanisms of Senescence and Implications for the Treatment of Myeloid Malignancies. Cancers 2021, 13, 612. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Duan, X.; Li, L.; Zhou, P.; Zou, C.; Xie, K. Cellular Senescence in Cancer: Molecular Mechanisms and Therapeutic Targets. Med. Comm. 2024, 5, e542. [Google Scholar] [CrossRef]
- Sheekey, E.; Narita, M. P53 in Senescence—It’s a Marathon, Not a Sprint. FEBS J. 2023, 290, 1212–1220. [Google Scholar] [CrossRef]
- Gao, M.; Li, H.; Zhang, J. RB Functions as a Key Regulator of Senescence and Tumor Suppression. Semin. Cancer Biol. 2025, 109, 1–7. [Google Scholar] [CrossRef]
- Talluri, S.; Dick, F.A. Regulation of Transcription and Chromatin Structure by PRB: Here, There and Everywhere. Cell Cycle 2012, 11, 3189–3198. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liang, Q.; Ren, Y.; Guo, C.; Ge, X.; Wang, L.; Cheng, Q.; Luo, P.; Zhang, Y.; Han, X. Immunosenescence: Molecular Mechanisms and Diseases. Signal Transduct. Target. Ther. 2023, 8, 200. [Google Scholar] [CrossRef]
- Desai, S.; Landay, A. Early Immune Senescence in HIV Disease. Curr. HIV/AIDS Rep. 2010, 7, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Lara-Aguilar, V.; Llamas-Adán, M.; Brochado-Kith, Ó.; Crespo-Bermejo, C.; Grande-García, S.; Arca-Lafuente, S.; de los Santos, I.; Prado, C.; Alía, M.; Sainz-Pinós, C.; et al. Low-Level HIV-1 Viremia Affects T-Cell Activation and Senescence in Long-Term Treated Adults in the INSTI Era. J. Biomed. Sci. 2024, 31, 80. [Google Scholar] [CrossRef] [PubMed]
- Pangrazzi, L.; Meryk, A. Molecular and Cellular Mechanisms of Immunosenescence: Modulation Through Interventions and Lifestyle Changes. Biology 2024, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Soares, L.S.; Espíndola, M.S.; Zambuzi, F.A.; Galvão-Lima, L.J.; Cacemiro, M.C.; Soares, M.R.; Santana, B.A.; Calado, R.T.; Bollela, V.R.; Frantz, F.G. Immunosenescence in Chronic HIV Infected Patients Impairs Essential Functions of Their Natural Killer Cells. Int. Immunopharmacol. 2020, 84, 106568. [Google Scholar] [CrossRef]
- Deeks, S.G.; Verdin, E.; McCune, J.M. Immunosenescence and HIV. Curr. Opin. Immunol. 2012, 24, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117–130.e6. [Google Scholar] [CrossRef]
- Apetroaei, M.-M.; Fragkiadaki, P.; Velescu, B. Ștefan; Baliou, S.; Renieri, E.; Dinu-Pirvu, C.E.; Drăgănescu, D.; Vlăsceanu, A.M.; Nedea, M.I. (Ilie); Udeanu, D.I.; et al. Pharmacotherapeutic Considerations on Telomere Biology: The Positive Effect of Pharmacologically Active Substances on Telomere Length. Int. J. Mol. Sci. 2024, 25, 7694. [Google Scholar] [CrossRef]
- Wang, J.; Li, C.; Han, J.; Xue, Y.; Zheng, X.; Wang, R.; Radak, Z.; Nakabeppu, Y.; Boldogh, I.; Ba, X. Reassessing the Roles of Oxidative DNA Base Lesion 8-OxoGua and Repair Enzyme OGG1 in Tumorigenesis. J. Biomed. Sci. 2025, 32, 1. [Google Scholar] [CrossRef]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The Impact of Oxidative DNA Damage and Stress on Telomere Homeostasis. Mech. Ageing Dev. 2019, 177, 37–45. [Google Scholar] [CrossRef]
- Harshithkumar, R.; Shah, P.; Jadaun, P.; Mukherjee, A. ROS Chronicles in HIV Infection: Genesis of Oxidative Stress, Associated Pathologies, and Therapeutic Strategies. Curr. Issues Mol. Biol. 2024, 46, 8852–8873. [Google Scholar] [CrossRef] [PubMed]
- Figarola-Centurión, I.; Escoto-Delgadillo, M.; González-Enríquez, G.V.; Gutiérrez-Sevilla, J.E.; Vázquez-Valls, E.; Torres-Mendoza, B.M. Sirtuins Modulation: A Promising Strategy for HIV-Associated Neurocognitive Impairments. Int. J. Mol. Sci. 2022, 23, 643. [Google Scholar] [CrossRef]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of Mitochondrial Lipid Oxidation: Role of 4-Hydroxynonenal (4-HNE) and Other Bioactive Lipids in Mitochondria. Free Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef] [PubMed]
- Schank, M.; Zhao, J.; Moorman, J.P.; Yao, Z.Q. The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging. Cells 2021, 10, 174. [Google Scholar] [CrossRef]
- Rodriguez, N.R.; Fortune, T.; Hegde, E.; Weinstein, M.P.; Keane, A.M.; Mangold, J.F.; Swartz, T.H. Oxidative Phosphorylation in HIV-1 Infection: Impacts on Cellular Metabolism and Immune Function. Front. Immunol. 2024, 15, 1360342. [Google Scholar] [CrossRef]
- Ostermann, P.N.; Evering, T.H. The Impact of Aging on HIV-1-Related Neurocognitive Impairment. Ageing Res. Rev. 2024, 102, 102513. [Google Scholar] [CrossRef]
- Crater, J.M.; Nixon, D.F.; Furler O’Brien, R.L. HIV-1 Replication and Latency Are Balanced by MTOR-Driven Cell Metabolism. Front. Cell. Infect. Microbiol. 2022, 12, 1068436. [Google Scholar] [CrossRef]
- Akbay, B.; Shmakova, A.; Vassetzky, Y.; Dokudovskaya, S. Modulation of MTORC1 Signaling Pathway by HIV-1. Cells 2020, 9, 1090. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Willig, A.L.; Overton, E.T. Metabolic Complications and Glucose Metabolism in HIV Infection: A Review of the Evidence. Curr. HIV/AIDS Rep. 2016, 13, 289–296. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as Regulators of Metabolism and Healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- Bhutta, M.; Gallo, E.; Borenstein, R. Multifaceted Role of AMPK in Viral Infections. Cells 2021, 10, 1118. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Lawton, A.; Tripodi, N.; Feehan, J. Running on Empty: Exploring Stem Cell Exhaustion in Geriatric Musculoskeletal Disease. Maturitas 2024, 188, 108066. [Google Scholar] [CrossRef] [PubMed]
- Herd, C.L.; Mellet, J.; Mashingaidze, T.; Durandt, C.; Pepper, M.S. Consequences of HIV Infection in the Bone Marrow Niche. Front. Immunol. 2023, 14, 1163012. [Google Scholar] [CrossRef]
- Bogeska, R.; Mikecin, A.-M.; Kaschutnig, P.; Fawaz, M.; Büchler-Schäff, M.; Le, D.; Ganuza, M.; Vollmer, A.; Paffenholz, S.V.; Asada, N.; et al. Inflammatory Exposure Drives Long-Lived Impairment of Hematopoietic Stem Cell Self-Renewal Activity and Accelerated Aging. Cell Stem Cell 2022, 29, 1273–1284.e8. [Google Scholar] [CrossRef]
- Zhao, H.G.; Deininger, M.W. Always stressed but never exhausted: How stem cells in myeloid neoplasms avoid extinction in inflammatory conditions. Blood 2023, 141, 2797–2812. [Google Scholar] [CrossRef]
- Abbas, W.; Herbein, G. T-Cell Signaling in HIV-1 Infection. Open Virol. J. 2013, 7, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Gras, G.; Khan, K.A.; Abbas, W. Macrophage Signaling in HIV-1 Infection. Retrovirology 2010, 7, 34. [Google Scholar] [CrossRef]
- Liu, R.; Lin, Y.; Jia, R.; Geng, Y.; Liang, C.; Tan, J.; Qiao, W. HIV-1 Vpr Stimulates NF-ΚB and AP-1 Signaling by Activating TAK1. Retrovirology 2014, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Fiume, G.; Vecchio, E.; De Laurentiis, A.; Trimboli, F.; Palmieri, C.; Pisano, A.; Falcone, C.; Pontoriero, M.; Rossi, A.; Scialdone, A.; et al. Human Immunodeficiency Virus-1 Tat Activates NF-ΚB via Physical Interaction with IκB-α and P65. Nucleic Acids Res. 2012, 40, 3548–3562. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; McFadden, G. Modulation of NF-ΚB Signalling by Microbial Pathogens. Nat. Rev. Microbiol. 2011, 9, 291–306. [Google Scholar] [CrossRef]
- Cafaro, A.; Schietroma, I.; Sernicola, L.; Belli, R.; Campagna, M.; Mancini, F.; Farcomeni, S.; Pavone-Cossut, M.R.; Borsetti, A.; Monini, P.; et al. Role of HIV-1 Tat Protein Interactions with Host Receptors in HIV Infection and Pathogenesis. Int. J. Mol. Sci. 2024, 25, 1704. [Google Scholar] [CrossRef] [PubMed]
- Horie, R.; Ishida, T.; Maruyama-Nagai, M.; Ito, K.; Watanabe, M.; Yoneyama, A.; Higashihara, M.; Kimura, S.; Watanabe, T. TRAF Activation of C/EBPβ (NF-IL6) via P38 MAPK Induces HIV-1 Gene Expression in Monocytes/Macrophages. Microbes Infect. 2007, 9, 721–728. [Google Scholar] [CrossRef]
- Muthumani, K.; Choo, A.Y.; Shedlock, D.J.; Laddy, D.J.; Sundaram, S.G.; Hirao, L.; Wu, L.; Thieu, K.P.; Chung, C.W.; Lankaraman, K.M.; et al. Human Immunodeficiency Virus Type 1 Nef Induces Programmed Death 1 Expression through a P38 Mitogen-Activated Protein Kinase-Dependent Mechanism. J. Virol. 2008, 82, 11536–11544. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.A.; Johnson, A.L.; Pawlak, E.N.; Dirk, B.S.; Van Nynatten, L.R.; Haeryfar, S.M.M.; Dikeakos, J.D. The Interaction between HIV-1 Nef and Adaptor Protein-2 Reduces Nef-Mediated CD4+ T Cell Apoptosis. Virology 2017, 509, 1–10. [Google Scholar] [CrossRef]
- Pasquereau, S.; Herbein, G. CounterAKTing HIV: Toward a “Block and Clear” Strategy? Front. Cell. Infect. Microbiol. 2022, 12, 827717. [Google Scholar] [CrossRef]
- Markle, T.J.; Mwimanzi, P.; Brockman, M.A. HIV-1 Nef and T-Cell Activation: A History of Contradictions. Future Virol. 2013, 8, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Kuo, A.M.-S.; Bertrand, L.; Toborek, M. HIV Alters Gap Junction-Mediated Intercellular Communication in Human Brain Pericytes. Front. Mol. Neurosci. 2017, 10, 410. [Google Scholar] [CrossRef]
- Bakhanashvili, M. The Role of Tumor Suppressor P53 Protein in HIV–Host Cell Interactions. Cells 2024, 13, 1512. [Google Scholar] [CrossRef]
- Periyasamy, P.; Thangaraj, A.; Bendi, V.S.; Buch, S. HIV-1 Tat-Mediated Microglial Inflammation Involves a Novel MiRNA-34a-NLRC5-NFκB Signaling Axis. Brain Behav. Immun. 2019, 80, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Bartz, S.R.; Emerman, M. Human Immunodeficiency Virus Type 1 Tat Induces Apoptosis and Increases Sensitivity to Apoptotic Signals by Up-Regulating FLICE/Caspase-8. J. Virol. 1999, 73, 1956–1963. [Google Scholar] [CrossRef]
- Conti, L.; Rainaldi, G.; Matarrese, P.; Varano, B.; Rivabene, R.; Columba, S.; Sato, A.; Belardelli, F.; Malorni, W.; Gessani, S. The HIV-1 Vpr Protein Acts as a Negative Regulator of Apoptosis in a Human Lymphoblastoid T Cell Line: Possible Implications for the Pathogenesis of AIDS. J. Exp. Med. 1998, 187, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Ali, A.; Arora, S.; Banerjea, A.C. Inhibition of β-TrcP–Dependent Ubiquitination of P53 by HIV-1 Vpu Promotes P53–Mediated Apoptosis in Human T Cells. Blood 2011, 117, 6600–6607. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Bian, Q.; Rong, D.; Wang, L.; Song, J.; Huang, H.-S.; Zeng, J.; Mei, J.; Wang, P.-Y. JAK/STAT Pathway: Extracellular Signals, Diseases, Immunity, and Therapeutic Regimens. Front. Bioeng. Biotechnol. 2023, 11, 1110765. [Google Scholar] [CrossRef]
- Wang, L.; Yukselten, Y.; Nuwagaba, J.; Sutton, R.E. JAK/STAT Signaling Pathway Affects CCR5 Expression in Human CD4 + T Cells. Sci. Adv. 2024, 10, adl0368. [Google Scholar] [CrossRef]
- Gargan, S.; Ahmed, S.; Mahony, R.; Bannan, C.; Napoletano, S.; O’Farrelly, C.; Borrow, P.; Bergin, C.; Stevenson, N.J. HIV-1 Promotes the Degradation of Components of the Type 1 IFN JAK/STAT Pathway and Blocks Anti-Viral ISG Induction. eBioMedicine 2018, 30, 203–216. [Google Scholar] [CrossRef]
- Fan, Y.; Timani, K.; He, J. STAT3 and Its Phosphorylation Are Involved in HIV-1 Tat-Induced Transactivation of Glial Fibrillary Acidic Protein. Curr. HIV Res. 2015, 13, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.V.; Tran, J.T.; Sanchez, D.J. HIV Blocks Type I IFN Signaling through Disruption of STAT1 Phosphorylation. Innate Immun. 2018, 24, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Perdaens, O.; van Pesch, V. Molecular Mechanisms of Immunosenescene and Inflammaging: Relevance to the Immunopathogenesis and Treatment of Multiple Sclerosis. Front. Neurol. 2022, 12, 811518. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Pratico, D.; Tang, D.; Zhou, S.; Franceschi, C.; Ren, J. Immunosenescence and Inflammaging: Mechanisms and Role in Diseases. Ageing Res. Rev. 2024, 101, 102540. [Google Scholar] [CrossRef] [PubMed]
- Min, A.K.; Fortune, T.; Rodriguez, N.; Hedge, E.; Swartz, T.H. Inflammasomes as Mediators of Inflammation in HIV-1 Infection. Transl. Res. 2023, 252, 1–8. [Google Scholar] [CrossRef]
- Zevin, A.S.; McKinnon, L.; Burgener, A.; Klatt, N.R. Microbial Translocation and Microbiome Dysbiosis in HIV-Associated Immune Activation. Curr. Opin. HIV AIDS 2016, 11, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Gogokhia, L.; Taur, Y.; Juluru, K.; Yagan, N.; Zhu, Y.-S.; Pamer, E.; Glesby, M.J. Intestinal Dysbiosis and Markers of Systemic Inflammation in Viscerally and Generally Obese Persons Living With HIV. JAIDS J. Acquir. Immune Defic. Syndr. 2020, 83, 81–89. [Google Scholar] [CrossRef]
- Fulcher, J.A.; Li, F.; Tobin, N.H.; Zabih, S.; Elliott, J.; Clark, J.L.; D’Aquila, R.; Mustanski, B.; Kipke, M.D.; Shoptaw, S.; et al. Gut Dysbiosis and Inflammatory Blood Markers Precede HIV with Limited Changes after Early Seroconversion. eBioMedicine 2022, 84, 104286. [Google Scholar] [CrossRef]
- Zaongo, S.D.; Ouyang, J.; Isnard, S.; Zhou, X.; Harypursat, V.; Cui, H.; Routy, J.-P.; Chen, Y. Candida Albicans Can Foster Gut Dysbiosis and Systemic Inflammation during HIV Infection. Gut Microbes 2023, 15, 2167171. [Google Scholar] [CrossRef]
- Martín-Escolano, R.; Virseda-Berdices, A.; Berenguer, J.; González-García, J.; Brochado-Kith, O.; Fernández-Rodríguez, A.; Díez, C.; Hontañon, V.; Resino, S.; Jiménez-Sousa, M.Á. Immune Checkpoint Proteins Are Associated with Persistently High Liver Stiffness after Successful HCV Treatment in People with HIV: A Retrospective Study. Front. Immunol. 2024, 15, 1505864. [Google Scholar] [CrossRef]
- Porichis, F.; Kaufmann, D.E. Role of PD-1 in HIV Pathogenesis and as Target for Therapy. Curr. HIV/AIDS Rep. 2012, 9, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Baroncini, L.; Muller, C.K.S.; Kadzioch, N.P.; Wolfensberger, R.; Russenberger, D.; Bredl, S.; Mlambo, T.; Speck, R.F. Pro-Inflammatory Macrophages Suppress HIV Replication in Humanized Mice and Ex Vivo Co-Cultures. Front. Immunol. 2024, 15, 1439328. [Google Scholar] [CrossRef]
- Khansari, N.; Shakiba, Y.; Mahmoudi, M. Chronic Inflammation and Oxidative Stress as a Major Cause of Age- Related Diseases and Cancer. Recent Pat. Inflamm. Allergy Drug Discov. 2009, 3, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Nousis, L.; Kanavaros, P.; Barbouti, A. Oxidative Stress-Induced Cellular Senescence: Is Labile Iron the Connecting Link? Antioxidants 2023, 12, 1250. [Google Scholar] [CrossRef] [PubMed]
- de Magalhães, J.P.; Passos, J.F. Stress, Cell Senescence and Organismal Ageing. Mech. Ageing Dev. 2018, 170, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ishizaka, A.; Koga, M.; Mizutani, T.; Parbie, P.K.; Prawisuda, D.; Yusa, N.; Sedohara, A.; Kikuchi, T.; Ikeuchi, K.; Adachi, E.; et al. Unique Gut Microbiome in HIV Patients on Antiretroviral Therapy (ART) Suggests Association with Chronic Inflammation. Microbiol. Spectr. 2021, 9, e00708-21. [Google Scholar] [CrossRef]
- Russo, E.; Nannini, G.; Sterrantino, G.; Kiros, S.T.; Di Pilato, V.; Coppi, M.; Baldi, S.; Niccolai, E.; Ricci, F.; Ramazzotti, M.; et al. Effects of Viremia and CD4 Recovery on Gut “Microbiome-Immunity” Axis in Treatment-Naïve HIV-1-Infected Patients Undergoing Antiretroviral Therapy. World J. Gastroenterol. 2022, 28, 635–652. [Google Scholar] [CrossRef]
- Xie, Y.; Sun, J.; Wei, L.; Jiang, H.; Hu, C.; Yang, J.; Huang, Y.; Ruan, B.; Zhu, B. Altered Gut Microbiota Correlate with Different Immune Responses to HAART in HIV-Infected Individuals. BMC Microbiol. 2021, 21, 11. [Google Scholar] [CrossRef]
- Ray, S.; Narayanan, A.; Giske, C.G.; Neogi, U.; Sönnerborg, A.; Nowak, P. Altered Gut Microbiome under Antiretroviral Therapy: Impact of Efavirenz and Zidovudine. ACS Infect. Dis. 2021, 7, 1104–1115. [Google Scholar] [CrossRef]
- Pérez-Santiago, J.; Marquine, M.J.; Cookson, D.; Giraud-Colón, R.; Heaton, R.K.; Grant, I.; Ellis, R.J.; Letendre, S.L.; Peterson, S.N. Gut Microbiota Dysbiosis Is Associated with Worse Emotional States in HIV Infection. J. Neurovirol. 2021, 27, 228–238. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Serrano-Villar, S. From Dysbiosis to Defense: Harnessing the Gut Microbiome in HIV/SIV Therapy. Microbiome 2024, 12, 113. [Google Scholar] [CrossRef]
- Montejano, R.; Stella-Ascariz, N.; Monge, S.; Bernardino, J.I.; Pérez-Valero, I.; Montes, M.L.; Valencia, E.; Martín-Carbonero, L.; Moreno, V.; González-García, J.; et al. Impact of Antiretroviral Treatment Containing Tenofovir Difumarate on the Telomere Length of Aviremic HIV-Infected Patients. JAIDS J. Acquir. Immune Defic. Syndr. 2017, 76, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Brummel, S.S.; Beilstein-Wedel, E.; Dagnall, C.L.; Hazra, R.; Kacanek, D.; Chadwick, E.G.; Marsit, C.J.; Chanock, S.J.; Savage, S.A.; et al. Association between Zidovudine-Containing Antiretroviral Therapy Exposure in Utero and Leukocyte Telomere Length at Birth. AIDS 2019, 33, 2091–2096. [Google Scholar] [CrossRef]
- Montejano, R.; Stella-Ascariz, N.; Monge, S.; Bernardino, J.I.; Pérez-Valero, I.; Montes, M.L.; Valencia, E.; Martín-Carbonero, L.; Moreno, V.; González-Garcia, J.; et al. Impact of Nucleos(t)Ide Reverse Transcriptase Inhibitors on Blood Telomere Length Changes in a Prospective Cohort of Aviremic HIV–Infected Adults. J. Infect. Dis. 2018, 218, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Centeno, J.; Esteban-Cantos, A.; Montejano, R.; Stella-Ascariz, N.; De Miguel, R.; Mena-Garay, B.; Saiz-Medrano, G.; Alejos, B.; Jiménez-González, M.; Bernardino, J.I.; et al. Effects of Tenofovir on Telomeres, Telomerase and T Cell Maturational Subset Distribution in Long-Term Aviraemic HIV-Infected Adults. J. Antimicrob. Chemother. 2022, 77, 1125–1132. [Google Scholar] [CrossRef]
- Bollmann, F.M. Telomerase Inhibition May Contribute to Accelerated Mitochondrial Aging Induced by Anti-Retroviral HIV Treatment. Med. Hypotheses 2013, 81, 285–287. [Google Scholar] [CrossRef]
- Hukezalie, K.R.; Thumati, N.R.; Côté, H.C.F.; Wong, J.M.Y. In Vitro and Ex Vivo Inhibition of Human Telomerase by Anti-HIV Nucleoside Reverse Transcriptase Inhibitors (NRTIs) but Not by Non-NRTIs. PLoS ONE 2012, 7, e47505. [Google Scholar] [CrossRef]
- Torres, R.A.; Lewis, W. Aging and HIV/AIDS: Pathogenetic Role of Therapeutic Side Effects. Lab. Investig. 2014, 94, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Leeansyah, E.; Cameron, P.U.; Solomon, A.; Tennakoon, S.; Velayudham, P.; Gouillou, M.; Spelman, T.; Hearps, A.; Fairley, C.; Smit, D.V.; et al. Inhibition of Telomerase Activity by Human Immunodeficiency Virus (HIV) Nucleos(t)Ide Reverse Transcriptase Inhibitors: A Potential Factor Contributing to HIV-Associated Accelerated Aging. J. Infect. Dis. 2013, 207, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Bukic, E.; Milasin, J.; Toljic, B.; Jadzic, J.; Jevtovic, D.; Obradovic, B.; Dragovic, G. Association between Combination Antiretroviral Therapy and Telomere Length in People Living with Human Immunodeficiency Virus. Biology 2023, 12, 1210. [Google Scholar] [CrossRef]
- Rusu, R.; Ababei, D.; Macadan, I.; Ciobîcă, A.; Paraschiv, M.; Bild, W.; Moraru, A.; Nicolae, C.; Bild, V. Factors That Influence Treatment Adherence–Realities, Controversies, Perspectives. Farmacia 2023, 71, 638–647. [Google Scholar] [CrossRef]
- Deeks, S.G. HIV Infection, Inflammation, Immunosenescence, and Aging. Annu. Rev. Med. 2011, 62, 141–155. [Google Scholar] [CrossRef]
- Darcis, G.; Moutschen, M. The Effect of Treatment Simplification on HIV Reservoirs. Lancet HIV 2017, 4, e328–e329. [Google Scholar] [CrossRef] [PubMed]
- Laiho, M.; Saksela, O.; Andreasen, P.A.; Keski-Oja, J. Enhanced Production and Extracellular Deposition of the Endothelial-Type Plasminogen Activator Inhibitor in Cultured Human Lung Fibroblasts by Transforming Growth Factor-Beta. J. Cell Biol. 1986, 103, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Côté, H.C.F.; Soudeyns, H.; Thorne, A.; Alimenti, A.; Lamarre, V.; Maan, E.J.; Sattha, B.; Singer, J.; Lapointe, N.; Money, D.M.; et al. Leukocyte Telomere Length in HIV-Infected and HIV-Exposed Uninfected Children: Shorter Telomeres with Uncontrolled HIV Viremia. PLoS ONE 2012, 7, e39266. [Google Scholar] [CrossRef]
- Minami, R.; Takahama, S.; Yamamoto, M. Correlates of Telomere Length Shortening in Peripheral Leukocytes of HIV-Infected Individuals and Association with Leukoaraiosis. PLoS ONE 2019, 14, e0218996. [Google Scholar] [CrossRef]
- Lombardi, F.; Sanfilippo, A.; Fabbiani, M.; Borghetti, A.; Ciccullo, A.; Tamburrini, E.; Di Giambenedetto, S. Blood Telomere Length Gain in People Living with HIV Switching to Dolutegravir plus Lamivudine versus Continuing Triple Regimen: A Longitudinal, Prospective, Matched, Controlled Study. J. Antimicrob. Chemother. 2023, 78, 2315–2322. [Google Scholar] [CrossRef] [PubMed]
- Molina-Carrión, S.; Brochado-Kith, Ó.; González-García, J.; Berenguer, J.; Díez, C.; Llop, E.; Hontañón, V.; Ibañez-Samaniego, L.; Montes, M.L.; Resino, S.; et al. Telomere Length Increase in HIV/HCV-Coinfected Patients with Cirrhosis after HCV Eradication with Direct-Acting Antivirals. J. Clin. Med. 2020, 9, 2407. [Google Scholar] [CrossRef] [PubMed]
- Monnin, A.; Vizeneux, A.; Nagot, N.; Eymard-Duvernay, S.; Meda, N.; Singata-Madliki, M.; Ndeezi, G.; Tumwine, J.K.; Kankasa, C.; Goga, A.; et al. Longitudinal Follow-Up of Blood Telomere Length in HIV-Exposed Uninfected Children Having Received One Year of Lopinavir/Ritonavir or Lamivudine as Prophylaxis. Children 2021, 8, 796. [Google Scholar] [CrossRef] [PubMed]
- Chalouni, M.; Rodriguez-Centeno, J.; Samri, A.; Blanco, J.; Stella-Ascariz, N.; Wallet, C.; Knobel, H.; Zucman, D.; Alejos Ferreras, B.; Autran, B.; et al. Correlation between Blood Telomere Length and CD4+ CD8+ T-Cell Subsets Changes 96 Weeks after Initiation of Antiretroviral Therapy in HIV-1–Positive Individuals. PLoS ONE 2020, 15, e0230772. [Google Scholar] [CrossRef]
- Saberi, S.; Kalloger, S.E.; Zhu, M.M.T.; Sattha, B.; Maan, E.J.; van Schalkwyk, J.; Money, D.M.; Côté, H.C.F. Dynamics of Leukocyte Telomere Length in Pregnant Women Living with HIV, and HIV-Negative Pregnant Women: A Longitudinal Observational Study. PLoS ONE 2019, 14, e0212273. [Google Scholar] [CrossRef] [PubMed]
- Quiros-Roldan, E.; Properzi, M.; Paghera, S.; Raffetti, E.; Castelli, F.; Imberti, L. Factors Associated with Immunosenescence during Early Adulthood in HIV-Infected Patients after Durable Efficient Combination Antiretroviral Therapy. Sci. Rep. 2020, 10, 10057. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug (Class) | Study Groups | TL Method | Conclusions | Ref. |
|---|---|---|---|---|
| tenofovir | 200 HIV adults: 103 on tenofovir and 97 never exposed to tenofovir | qPCR | No evidence of a significant association between tenofovir exposure and TL when adjusted for confounding variables like age, parental age, race, and duration of HIV infection. | [183] |
| AZT | 179 infants: 94 AZT-exposed; 85 ART-unexposed | qPCR | Infants exposed to AZT during pregnancy had longer telomeres than those unexposed to ART. This may suggest a beneficial effect of maternal ART on neonatal telomere preservation. | [184] |
| TDF, abacavir, lamivudine | 172 HIV+ adults: 67 TDF, 105 non-TDF (69 abacavir, 25 N(t)RTI–sparing regimen, 5 lamivudine | qPCR | Mean TL rose considerably after 2 years. The TDF group gained TL much less than the non-TDF group. TDF exposure had no independent negative impact. Non-TDF patients receiving 2 nucleosides demonstrated considerably less TL improvement compared with N(t)RTI-sparing group or lamivudine. | [185] |
| tenofovir | 128 long-term aviraemic HIV adults: 79 on tenofovir; 49 tenofovir-sparing regimens | qPCR | The inhibitory effect on telomerase by tenofovir in long-term aviraemic HIV adults may account for TL shortening observed in CD8+ T cells. The absence of telomere shortening in the CD4+ compartment, along with the reduction in telomerase activity, can be attributed to both the inhibitory effects of tenofovir and the diminished proportion of recent thymic emigrant CD4+ cells and PD1 marker expression | [186] |
| Study Groups | TL Method | Conclusions | Ref. |
|---|---|---|---|
| 107 HIV+ patients: before and during suppressive ART | qPCR | TL dropped drastically pre-ART. ART stabilised TL without alteration. Early ART initiation may prevent HIV-associated accelerated ageing and preserve telomere integrity. | [195] |
| 375 children: 94 HIV+; 177 HIV-1-exposed uninfected (HEU) exposed to ART; and 104 controls. | qPCR | HIV+, HEU, and HIV− children had similar LTL attrition rates. Linear regression models found no correlation between children’s LTL and perinatal ART exposure or HIV status. The link between a detectable HIV viral load and a shorter LTL shows that uncontrolled HIV viremia, not ART duration, may accelerate blood telomere attrition. | [196] |
| 325 HIV+ patients, stable cART for >1 year: 207 on PIs, 36 on NNRTIs, and 82 on INSTIs; 147 controls. | qPCR | For HIV+ individuals, the rate of decrease in TL with age was more rapid than that of controls. The likelihood of ageing-related neurological disorders in HIV+ may be reduced by the administration of INSTI-based cART and improved viral control, which may delay TL shortening. | [197] |
| 120 HIV+ on stable triple-drug ART: 60 switched to DTG (INSTI) + 3TC (NRTI); 60 continued on TT with 2 NRTIs + an anchor drug. | MMqPCR | A more significant increase in TL over time was associated with a lower basal BTL. The research indicates that the BTL gain is more significant when the simplification is to DTG + 3TC than when the TT is maintained. This could suggest that DTG + 3TC is a potential strategy for mitigating telomere shortening in HIV-treated individuals, as it may reduce telomerase inhibition. | [198] |
| 90 patients with advanced HCV cirrhosis: HIV/HCV group- 60 patients on stable cART + HCV group- 30 patients | MMqPCR | At baseline, patients who were HIV/HCV coinfected exhibited substantially shorter TL than those who were HCV monoinfected. In HIV/HCV-coinfected patients, there was a substantial increase in TL following the eradication of HCV with DAAs. A modest, non-significant increase in TL was observed in HCV-monoinfected patients. Compensated HIV/HCV-coinfected patients exhibited a substantial increase in TL following HCV treatment. | [199] |
| 176 male HIV patients on cART: 53 patients on INSTI-based, 60 patients on PI-based, 63 patients on NNRTI-based, divided into 53 efavirenz and 10 nevirapine. | qPCR | INSTIs, PIs, and NNRTIs did not significantly affect TL in cART groups. Efavirenz (NNRTI) had shorter telomeres than nevirapine (NNRTI), showing that NNRTIs affect cellular ageing indicators differently. Avoiding efavirenz in cART may reduce HIV-related telomere shortening. | [191] |
| 198 HIV-exposed uninfected children (CHEU) on prophylaxis for one year: 0–7 days nevirapine + day 7-week 50–75 LPV/r + 92 3TC | qPCR | TL shortening occurred in 44.3% of CHEU at week 50, regardless of postnatal prophylaxis drugs. Besides motor skills, TL shortening did not affect growth or neuropsychological function at six years. In week 50 and year 6, the LPV/r and 3TC groups experienced similar telomere shortening. | [200] |
| 31 patients: 15 on DRV/r + RAL (PI+INSTI) and 16 on DRV/r + TDF/FTC (PI+NRTI) | qPCR | After 96 weeks of ART, TL rose considerably, more in DRV/r + TDF/FTC than in RAL, but not statistically significant. This TL increase was connected with CD4+ T-cell recovery and inversely associated with effector memory T-cells, suggesting immunological reconstitution changes T-cells with fewer differentiated phenotypes with longer telomeres. Reduced immune activation is linked to TL recovery, as central memory CD8+ T-cells with CD38+ molecules had a negative correlation with TL growth. | [201] |
| 105 pregnant women: 64 HIV+ on cART and 41 HIV-: 39 PI/r; 47 AZT + 3TC, combined with 27 LPV/r or 19 NFV | qPCR | HIV+ mothers who stopped cART post-partum had lower TL. Like both groups, LTL rose during gestation, but it was more evident in women under 35. TL was independently shorter in HIV+ women using ritonavir-boosted protease inhibitors. | [202] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apetroaei, M.-M.; Baliou, S.; Ioannou, P.; Fragkiadaki, P.; Ștefan, G.; Nedea, M.I.; Burcea-Dragomiroiu, G.-T.-A.; Velescu, B.Ș.; Docea, A.O.; Udeanu, D.I.; et al. The Hallmarks of Ageing in Human Immunodeficiency Virus Infection and the Impact of Antiretroviral Therapy on Telomeres: A Molecular Perspective. Curr. Issues Mol. Biol. 2025, 47, 273. https://doi.org/10.3390/cimb47040273
Apetroaei M-M, Baliou S, Ioannou P, Fragkiadaki P, Ștefan G, Nedea MI, Burcea-Dragomiroiu G-T-A, Velescu BȘ, Docea AO, Udeanu DI, et al. The Hallmarks of Ageing in Human Immunodeficiency Virus Infection and the Impact of Antiretroviral Therapy on Telomeres: A Molecular Perspective. Current Issues in Molecular Biology. 2025; 47(4):273. https://doi.org/10.3390/cimb47040273
Chicago/Turabian StyleApetroaei, Miruna-Maria, Stella Baliou, Petros Ioannou, Persefoni Fragkiadaki, Gabriela Ștefan, Marina Ionela (Ilie) Nedea, George-Traian-Alexandru Burcea-Dragomiroiu, Bruno Ștefan Velescu, Anca Oana Docea, Denisa Ioana Udeanu, and et al. 2025. "The Hallmarks of Ageing in Human Immunodeficiency Virus Infection and the Impact of Antiretroviral Therapy on Telomeres: A Molecular Perspective" Current Issues in Molecular Biology 47, no. 4: 273. https://doi.org/10.3390/cimb47040273
APA StyleApetroaei, M.-M., Baliou, S., Ioannou, P., Fragkiadaki, P., Ștefan, G., Nedea, M. I., Burcea-Dragomiroiu, G.-T.-A., Velescu, B. Ș., Docea, A. O., Udeanu, D. I., Tsatsakis, A., & Arsene, A. L. (2025). The Hallmarks of Ageing in Human Immunodeficiency Virus Infection and the Impact of Antiretroviral Therapy on Telomeres: A Molecular Perspective. Current Issues in Molecular Biology, 47(4), 273. https://doi.org/10.3390/cimb47040273