
Secondary Findings from Exome Sequencing of a Greek Cohort

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
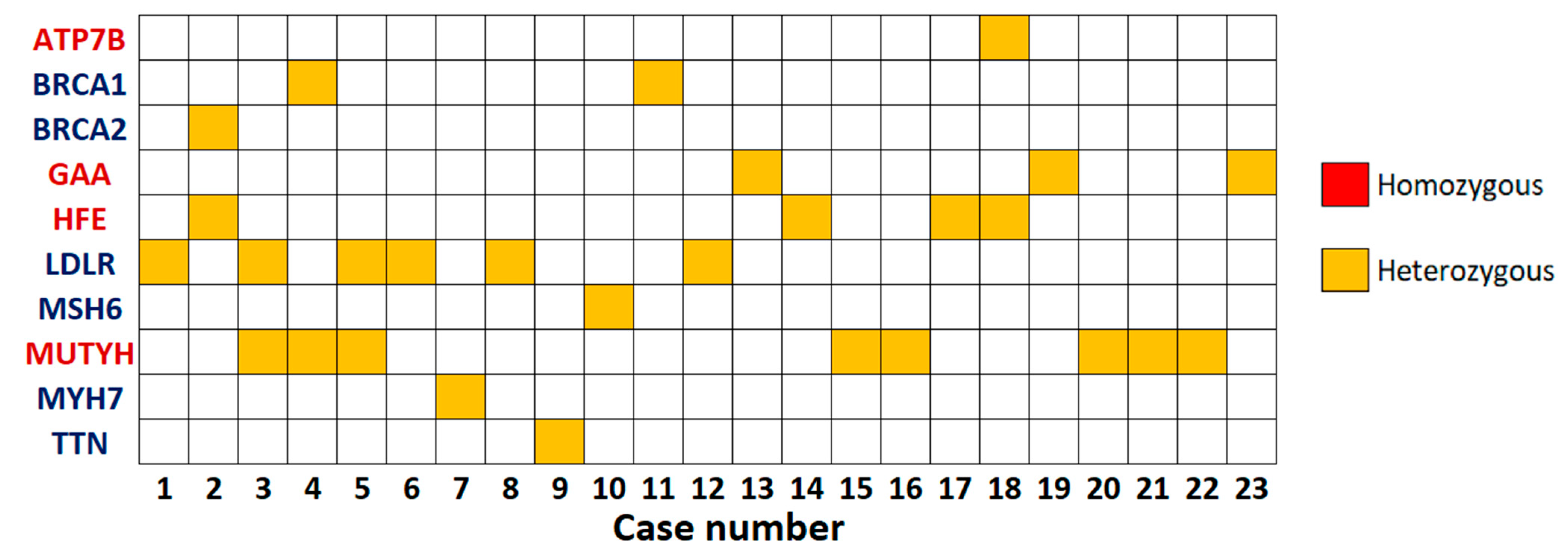
3. Results
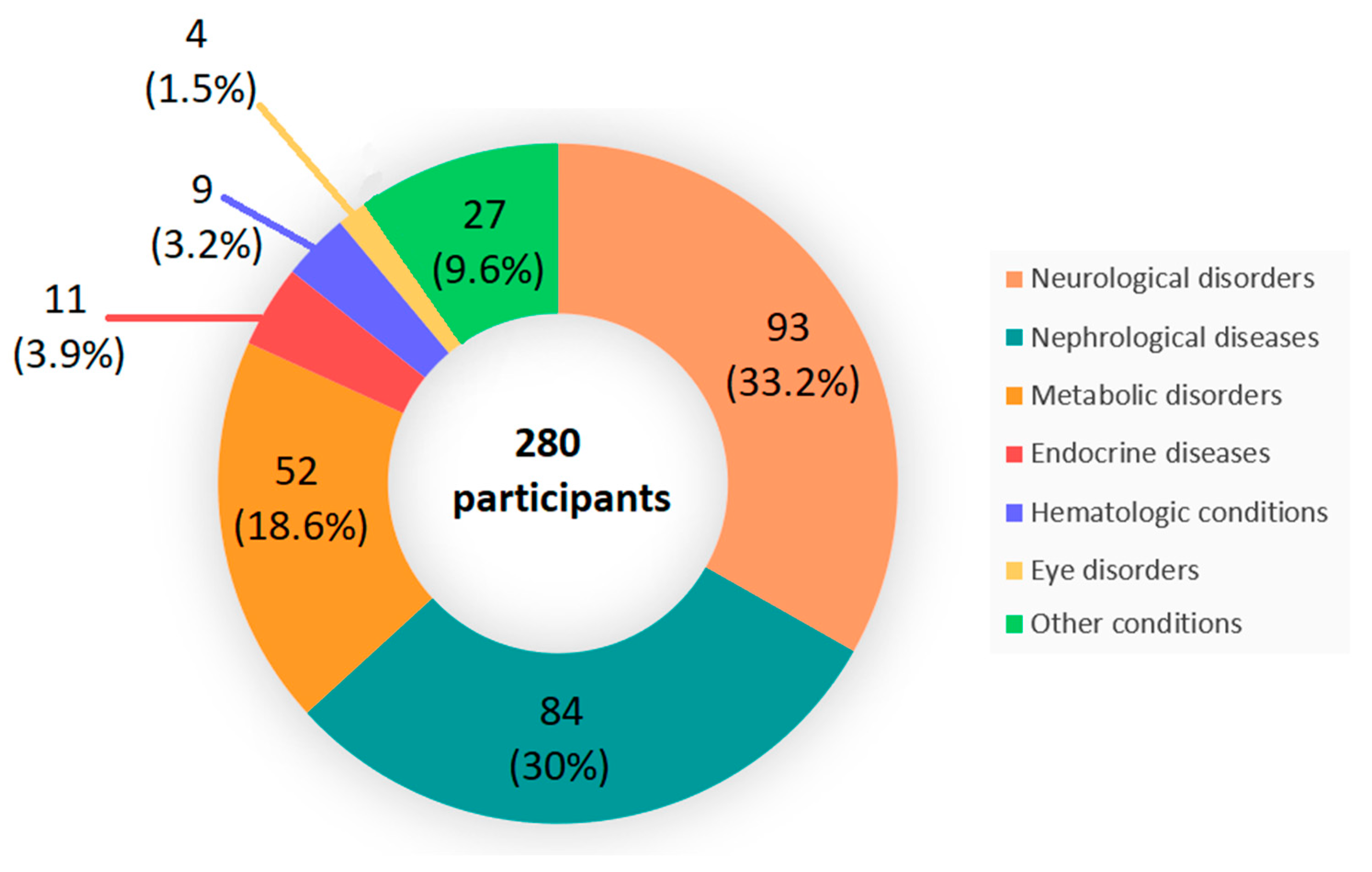
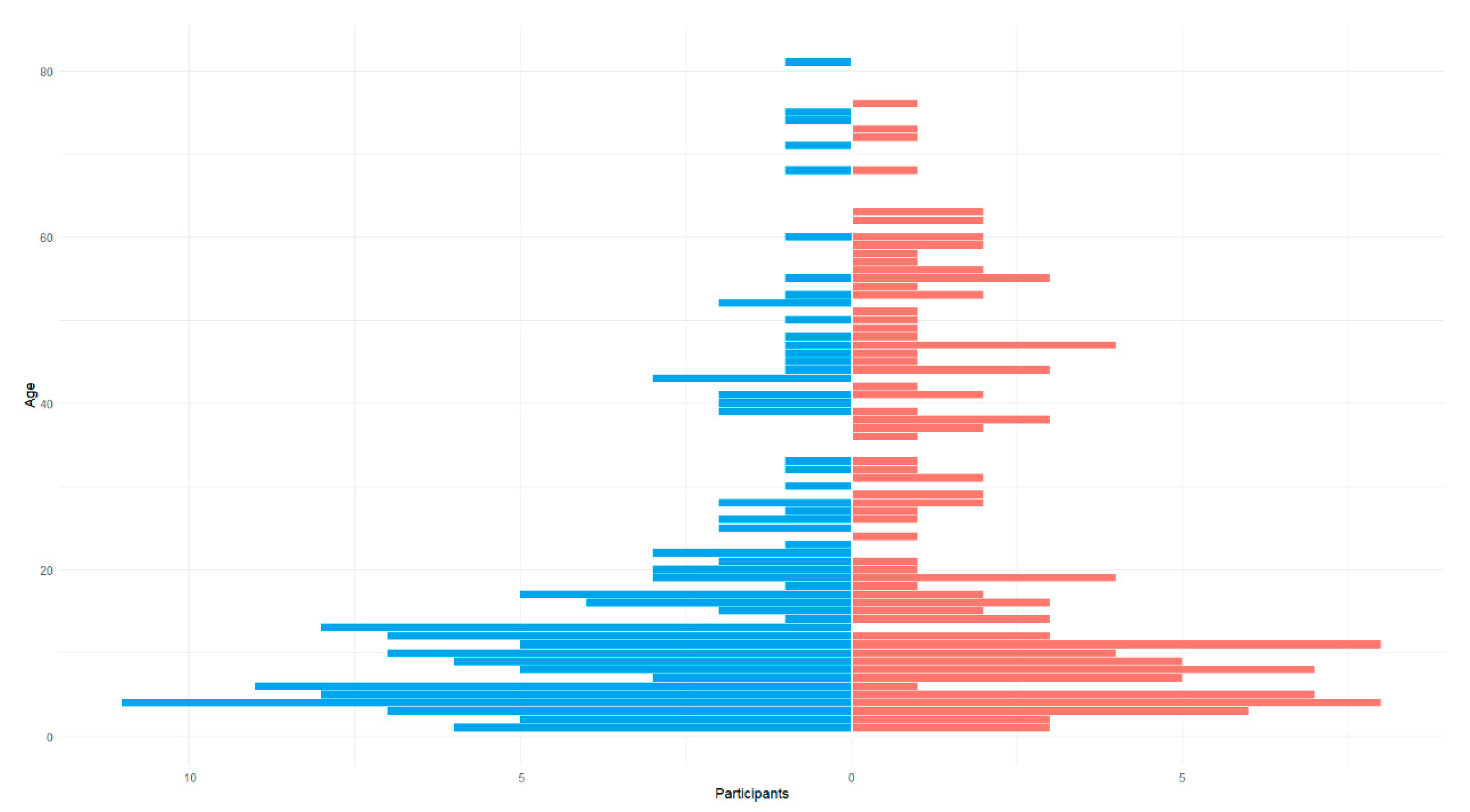
3.1. Population Demographics
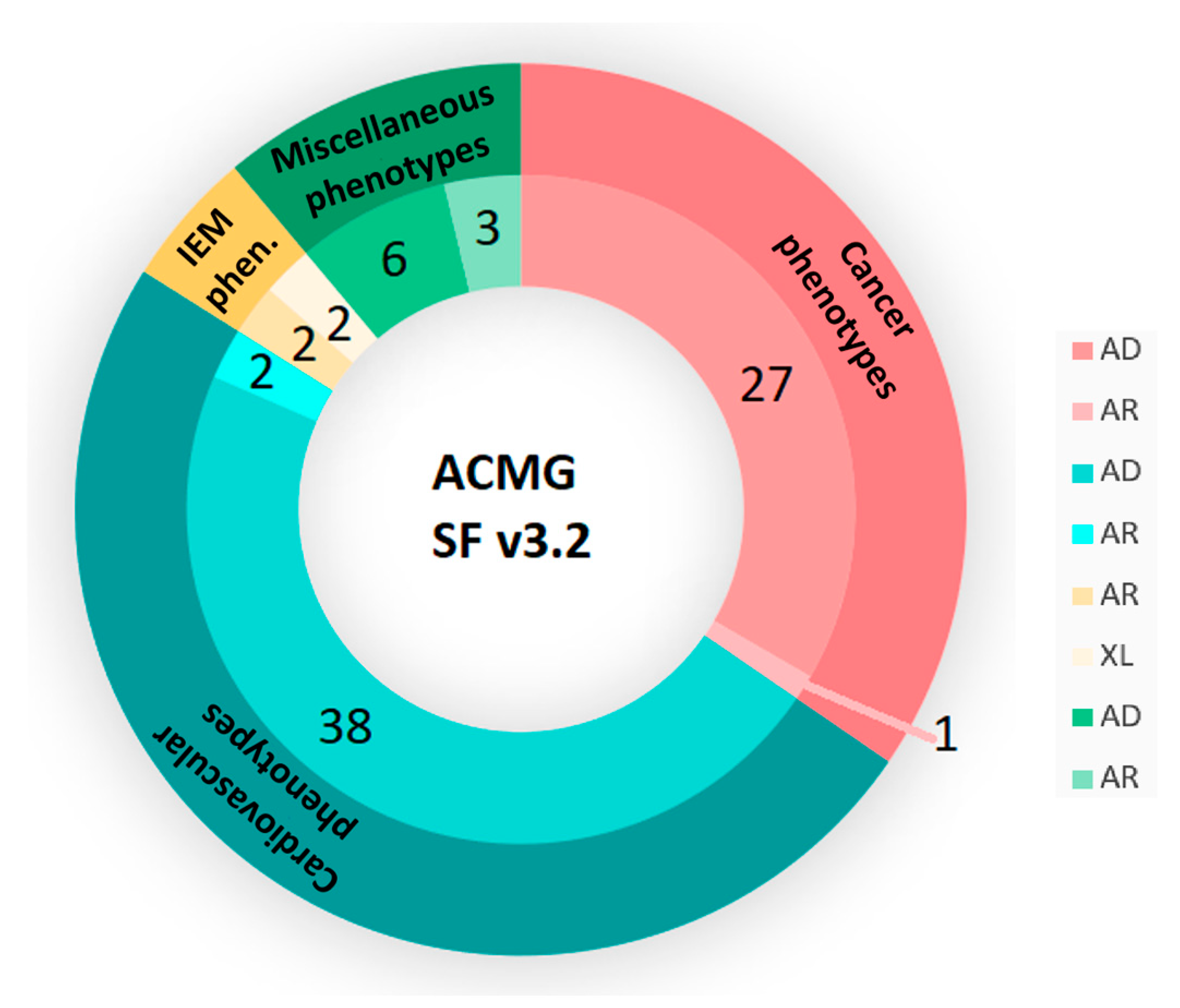
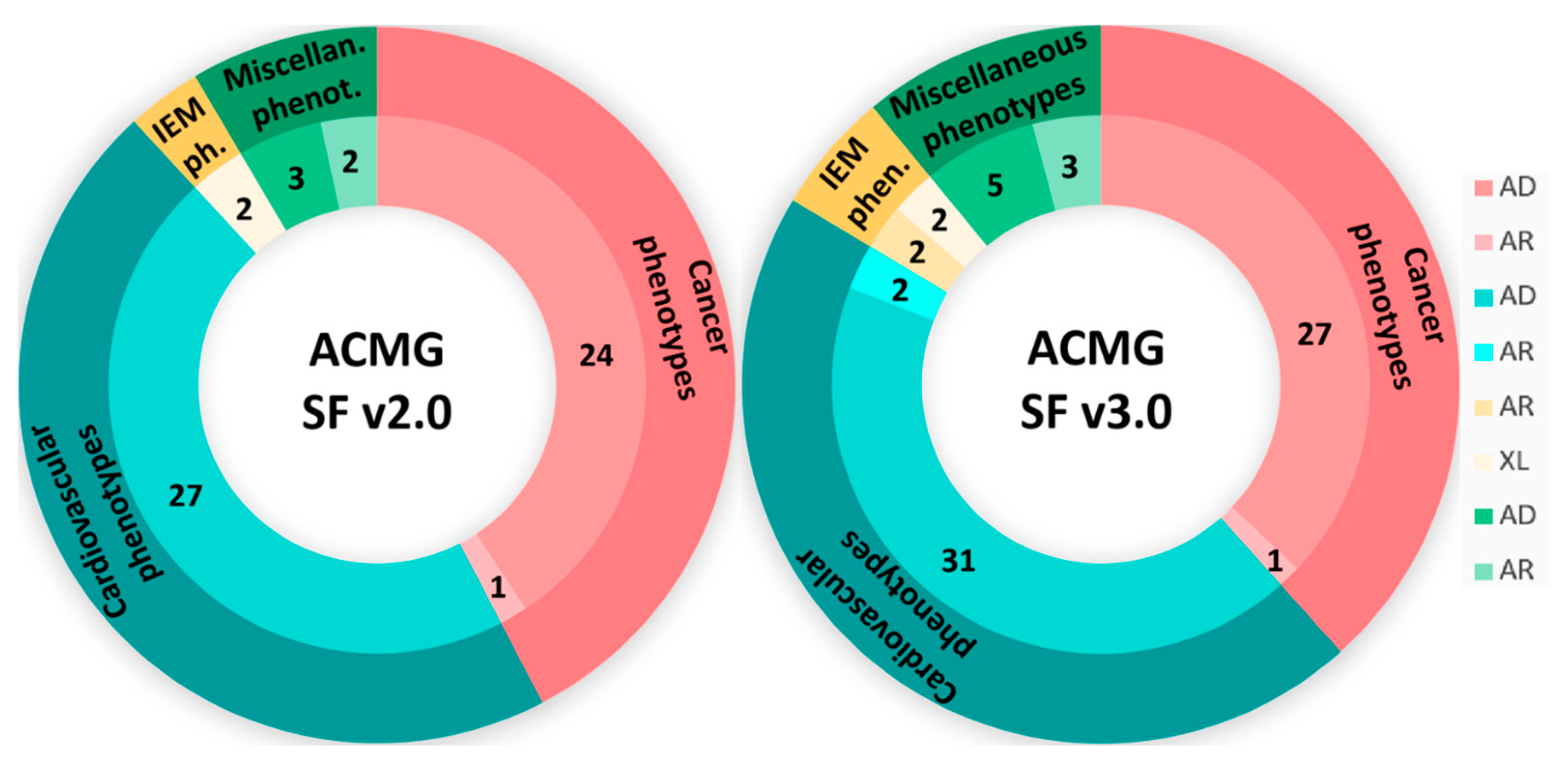
3.2. SFs Analysis
3.2.1. Dominant Actionable Genes
3.2.2. Recessive Actionable Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics and Genomics |
| AD | Autosomal Dominant |
| AMP | Association for Molecular Pathology |
| AR | Autosomal Recessive |
| ES | Exome Sequencing |
| GATK | Genome Analysis Toolkit |
| GS | Genome Sequencing |
| IEM | Inborn Errors of Metabolism |
| IF | Incidental Finding |
| LP | Likely Pathogenic |
| MAF | Minor Allele Frequency |
| NGS | Next-Generation Sequencing |
| P | Pathogenic |
| pLoF | Predicted Loss-of-Function |
| SF | Secondary Finding |
| SNV | Single Nucleotide Variant |
| WES | Whole-Exome Sequencing |
| XL | X-Linked |
References
- Tang, C.S.; Dattani, S.; So, M.; Cherny, S.S.; Tam, P.K.H.; Sham, P.C.; Garcia-Barcelo, M.-M. Actionable Secondary Findings from Whole-Genome Sequencing of 954 East Asians. Hum. Genet. 2018, 137, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG Recommendations for Reporting of Incidental Findings in Clinical Exome and Genome Sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for Reporting of Secondary Findings in Clinical Exome and Genome Sequencing, 2016 Update (ACMG SF v2.0): A Policy Statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 List for Reporting of Secondary Findings in Clinical Exome and Genome Sequencing: A Policy Statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Abul-Husn, N.S.; Amendola, L.M.; Brothers, K.; Chung, W.K.; Gollob, M.H.; Gordon, A.S.; Harrison, S.M.; Hershberger, R.E.; et al. ACMG SF v3.1 List for Reporting of Secondary Findings in Clinical Exome and Genome Sequencing: A Policy Statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2022, 24, 1407–1414. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Abul-Husn, N.S.; Amendola, L.M.; Brothers, K.; Chung, W.K.; Gollob, M.H.; Gordon, A.S.; Harrison, S.M.; Hershberger, R.E.; et al. ACMG SF v3.2 List for Reporting of Secondary Findings in Clinical Exome and Genome Sequencing: A Policy Statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2023, 25, 100866. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ben-Shachar, R.; Svenson, A.; Goldberg, J.D.; Muzzey, D. A Data-Driven Evaluation of the Size and Content of Expanded Carrier Screening Panels. Genet. Med. 2019, 21, 1931–1939. [Google Scholar] [CrossRef]
- Guo, M.H.; Gregg, A.R. Estimating Yields of Prenatal Carrier Screening and Implications for Design of Expanded Carrier Screening Panels. Genet. Med. 2019, 21, 1940–1947. [Google Scholar] [CrossRef]
- Abifadel, M.; Boileau, C. Genetic and Molecular Architecture of Familial Hypercholesterolemia. J. Intern. Med. 2023, 293, 144–165. [Google Scholar] [CrossRef]
- Xiang, Z.; Li, J.-R.; Wan, W.-M.; Li, S.-H.; Wu, J. Familial Hypercholesterolemia: Current Limitations and Future Breakthroughs. World J. Exp. Med. 2024, 14, 99968. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.M.; Lorenzini, M.; Cicerchia, M.; Ochoa, J.P.; Hey, T.M.; Sabater Molina, M.; Restrepo-Cordoba, M.A.; Dal Ferro, M.; Stolfo, D.; Johnson, R.; et al. Clinical Phenotypes and Prognosis of Dilated Cardiomyopathy Caused by Truncating Variants in the TTN Gene. Circ. Heart Fail. 2020, 13, e006832. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Mestroni, L.; Taylor, M.R.G. Genetics of Dilated Cardiomyopathy. Curr. Opin. Cardiol. 2021, 36, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Molecular Genetic Basis of Hypertrophic Cardiomyopathy. Circ. Res. 2021, 128, 1533–1553. [Google Scholar] [CrossRef]
- Gao, Y.; Peng, L.; Zhao, C. MYH7 in Cardiomyopathy and Skeletal Muscle Myopathy. Mol. Cell Biochem. 2024, 479, 393–417. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian Cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast Cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Lee, Y.-L.; Li, C.-Y. BRCA Genes and Related Cancers: A Meta-Analysis from Epidemiological Cohort Studies. Medicina 2021, 57, 905. [Google Scholar] [CrossRef]
- Cerretelli, G.; Ager, A.; Arends, M.J.; Frayling, I.M. Molecular Pathology of Lynch Syndrome. J. Pathol. 2020, 250, 518–531. [Google Scholar] [CrossRef]
- Ene, C.V.; Bulai, C.T.L.A.; Geavlete, P.; Popescu, R.I.; Vacaroiu, I.A.; Georgescu, D.E.; Isaconi, I.V.; da Lina Andreea Munteanu, M.; Ene, C.D.; Militaru, A.; et al. New Insights into Lynch Syndrome: A Narrative Review. Chirurgia 2023, 118, 584–595. [Google Scholar] [CrossRef]
- Ma, H.; Brosens, L.A.A.; Offerhaus, G.J.A.; Giardiello, F.M.; de Leng, W.W.J.; Montgomery, E.A. Pathology and Genetics of Hereditary Colorectal Cancer. Pathology 2018, 50, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Aelvoet, A.S.; Buttitta, F.; Ricciardiello, L.; Dekker, E. Management of Familial Adenomatous Polyposis and MUTYH-Associated Polyposis; New Insights. Best Pract. Res. Clin. Gastroenterol. 2022, 58–59, 101793. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, G.S.; Paynton, B.V.; DiStefano, J.K. Identification of Genes for Hereditary Hemochromatosis. Methods Mol. Biol. 2018, 1706, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, M.-S.; Papasavva, M.; Latsi, R.; Drakoulis, N. Hemochromatosis: Hereditary Hemochromatosis and HFE Gene. Vitam. Horm. 2019, 110, 201–222. [Google Scholar] [CrossRef]
- Członkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson Disease. Nat. Rev. Dis. Primers 2018, 4, 21. [Google Scholar] [CrossRef]
- Mulligan, C.; Bronstein, J.M. Wilson Disease: An Overview and Approach to Management. Neurol. Clin. 2020, 38, 417–432. [Google Scholar] [CrossRef]
- Taverna, S.; Cammarata, G.; Colomba, P.; Sciarrino, S.; Zizzo, C.; Francofonte, D.; Zora, M.; Scalia, S.; Brando, C.; Curto, A.L.; et al. Pompe Disease: Pathogenesis, Molecular Genetics and Diagnosis. Aging 2020, 12, 15856–15874. [Google Scholar] [CrossRef]
- Davison, J.E. Advances in Diagnosis and Management of Pompe Disease. J. Mother Child 2020, 24, 3–8. [Google Scholar] [CrossRef]
- Dorschner, M.O.; Amendola, L.M.; Turner, E.H.; Robertson, P.D.; Shirts, B.H.; Gallego, C.J.; Bennett, R.L.; Jones, K.L.; Tokita, M.J.; Bennett, J.T.; et al. Actionable, Pathogenic Incidental Findings in 1,000 Participants’ Exomes. Am. J. Hum. Genet. 2013, 93, 631–640. [Google Scholar] [CrossRef]
- Amendola, L.M.; Dorschner, M.O.; Robertson, P.D.; Salama, J.S.; Hart, R.; Shirts, B.H.; Murray, M.L.; Tokita, M.J.; Gallego, C.J.; Kim, D.S.; et al. Actionable Exomic Incidental Findings in 6503 Participants: Challenges of Variant Classification. Genome Res. 2015, 25, 305–315. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Hart, S.N.; Vijai, J.; Schrader, K.A.; Slavin, T.P.; Thomas, T.; Wubbenhorst, B.; Ravichandran, V.; Moore, R.M.; Hu, C.; et al. Evaluation of ACMG-Guideline-Based Variant Classification of Cancer Susceptibility and Non-Cancer-Associated Genes in Families Affected by Breast Cancer. Am. J. Hum. Genet. 2016, 98, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Branković, M.; Han, H.; Janković, M.; Marjanović, A.; Andrejic, N.; Gunjić, I.; Virić, V.; Palibrk, A.; Lee, H.; Peric, S. Secondary Findings in 443 Exome Sequencing Data. Ann. Hum. Genet. 2024, ahg.12586. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, B.; Shi, J.; Zhao, S.; Xu, K.; Sun, L.; Chen, N.; Tian, W.; Zhang, J.; Wu, N. Landscape of Secondary Findings in Chinese Population: A Practice of ACMG SF v3.0 List. J. Pers. Med. 2022, 12, 1503. [Google Scholar] [CrossRef] [PubMed]
- Jensson, B.O.; Arnadottir, G.A.; Katrinardottir, H.; Fridriksdottir, R.; Helgason, H.; Oddsson, A.; Sveinbjornsson, G.; Eggertsson, H.P.; Halldorsson, G.H.; Atlason, B.A.; et al. Actionable Genotypes and Their Association with Life Span in Iceland. N. Engl. J. Med. 2023, 389, 1741–1752. [Google Scholar] [CrossRef]
- Martone, S.; Buonagura, A.T.; Marra, R.; Rosato, B.E.; Del Giudice, F.; Bonfiglio, F.; Capasso, M.; Iolascon, A.; Andolfo, I.; Russo, R. Clinical Exome-Based Panel Testing for Medically Actionable Secondary Findings in a Cohort of 383 Italian Participants. Front. Genet. 2022, 13, 956723. [Google Scholar] [CrossRef]
- Georgiou, I.; Makis, A.; Chaidos, A.; Bouba, I.; Hatzi, E.; Kranas, V.; Zilidis, C.; Bourantas, K.L. Distribution and Frequency of β -thalassemia Mutations in Northwestern and Central Greece. Eur. J. Haematol. 2003, 70, 75–78. [Google Scholar] [CrossRef]
- De Sanctis, V.; Kattamis, C.; Canatan, D.; Soliman, A.T.; Elsedfy, H.; Karimi, M.; Daar, S.; Wali, Y.; Yassin, M.; Soliman, N.; et al. β-Thalassemia Distribution in the Old World: An Ancient Disease Seen from a Historical Standpoint. Mediterr. J. Hematol. Infect. Dis. 2017, 9, e2017018. [Google Scholar] [CrossRef]
- Kwak, S.H.; Chae, J.; Choi, S.; Kim, M.J.; Choi, M.; Chae, J.-H.; Cho, E.; Hwang, T.J.; Jang, S.S.; Kim, J.-I.; et al. Findings of a 1303 Korean Whole-Exome Sequencing Study. Exp. Mol. Med. 2017, 49, e356. [Google Scholar] [CrossRef]
- Gordon, A.S.; Zouk, H.; Venner, E.; Eng, C.M.; Funke, B.H.; Amendola, L.M.; Carrell, D.S.; Chisholm, R.L.; Chung, W.K.; Denny, J.C.; et al. Frequency of Genomic Secondary Findings among 21,915 eMERGE Network Participants. Genet. Med. 2020, 22, 1470–1477. [Google Scholar] [CrossRef]
- Brunfeldt, M.; Kaare, M.; Saarinen, I.; Koskenvuo, J.; Kääriäinen, H. Opt-in for Secondary Findings as Part of Diagnostic Whole-exome Sequencing: Real-life Experience from an International Diagnostic Laboratory. Mol. Gen. Gen. Med. 2023, 11, e2180. [Google Scholar] [CrossRef]
- Husser, D.; Ueberham, L.; Jacob, J.; Heuer, D.; Riedel-Heller, S.; Walker, J.; Hindricks, G.; Bollmann, A. Prevalence of Clinically Apparent Hypertrophic Cardiomyopathy in Germany-An Analysis of over 5 Million Patients. PLoS ONE 2018, 13, e0196612. [Google Scholar] [CrossRef] [PubMed]
- Miltiadous, G.; Elisaf, M.; Bairaktari, H.; Xenophontos, S.L.; Manoli, P.; Cariolou, M.A. Characterization and Geographic Distribution of the Low Density Lipoprotein Receptor (LDLR) Gene Mutations in Northwestern Greece. Hum. Mutat. 2001, 17, 432–433. [Google Scholar] [CrossRef] [PubMed]
- Diakou, M.; Miltiadous, G.; Xenophontos, S.L.; Manoli, P.; Cariolou, M.A.; Elisaf, M. Spectrum of LDLR Gene Mutations, Including a Novel Mutation Causing Familial Hypercholesterolaemia, in North-Western Greece. Eur. J. Intern. Med. 2011, 22, e55–e59. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial Hypercholesterolaemia Is Underdiagnosed and Undertreated in the General Population: Guidance for Clinicians to Prevent Coronary Heart Disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef]
- Rizos, C.V.; Elisaf, M.S.; Skoumas, I.; Tziomalos, K.; Kotsis, V.; Rallidis, L.; Garoufi, A.; Athyros, V.G.; Skalidis, E.; Kolovou, G.; et al. Characteristics and Management of 1093 Patients with Clinical Diagnosis of Familial Hypercholesterolemia in Greece: Data from the Hellenic Familial Hypercholesterolemia Registry (HELLAS-FH). Atherosclerosis 2018, 277, 308–313. [Google Scholar] [CrossRef]
- Rizos, C.V.; Athyros, V.; Bilianou, E.; Chrousos, G.; Garoufi, A.; Kolovou, G.; Kotsis, V.; Rallidis, L.; Skalidis, E.; Skoumas, I.; et al. An Insight into Familial Hypercholesterolemia in Greece: Rationale and Design of the Hellenic Familial Hypercholesterolemia Registry (HELLAS-FH). Hormones 2017, 16, 200–204. [Google Scholar] [CrossRef]
- Konstantopoulou, I.; Tsitlaidou, M.; Fostira, F.; Pertesi, M.; Stavropoulou, A.; Triantafyllidou, O.; Tsotra, E.; Tsiftsoglou, A.; Tsionou, C.; Droufakou, S.; et al. High Prevalence of BRCA1 Founder Mutations in Greek Breast/Ovarian Families. Clin. Genet. 2014, 85, 36–42. [Google Scholar] [CrossRef]
- Papamentzelopoulou, M.; Apostolou, P.; Fostira, F.; Dimitrakakis, C.; Loutradis, D.; Fountzilas, G.; Yannoukakos, D.; Konstantopoulou, I. Prevalence and Founder Effect of the BRCA1 p.(Val1833Met) Variant in the Greek Population, with Further Evidence for Pathogenicity and Risk Modification. Cancer Genet. 2019, 237, 90–96. [Google Scholar] [CrossRef]
- Schiffman, J.D.; Fisher, P.G.; Gibbs, P. Early Detection of Cancer: Past, Present, and Future. Cancer Prev. Genet. Epidemiol. 2015, 35, 57–65. [Google Scholar] [CrossRef]
- Hedegaard, B.S.; Bork, C.S.; Kanstrup, H.L.; Thomsen, K.K.; Heitmann, M.; Bang, L.E.; Henriksen, F.L.; Andersen, L.J.; Gohr, T.; Mouridsen, M.R.; et al. Genetic Testing Increases the Likelihood of a Diagnosis of Familial Hypercholesterolaemia among People Referred to Lipid Clinics: Danish National Study. Atherosclerosis 2023, 373, 10–16. [Google Scholar] [CrossRef]
- Medeiros, A.M.; Bourbon, M. Genetic Testing in Familial Hypercholesterolemia: Is It for Everyone? Curr. Atheroscler. Rep. 2023, 25, 127–132. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACMG SF v2.0 | ACMG SF v3.0 | ACMG SF v3.2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| AD | AR | XL | AD | AR | XL | AD | AR | XL | |
| No. of carriers | 11 | 12 | 0 | 12 | 15 | 0 | 12 | 15 | 0 |
| No. of variants | 11 | 13 | 0 | 12 | 16 | 0 | 12 | 16 | 0 |
| Gene | Condition | Transcript | Variant | dbSNP ID | ACMG Classification | Zygosity | Number of Carriers |
|---|---|---|---|---|---|---|---|
| BRCA1 | Hereditary breast and/or ovarian cancer | NM_007294.4 | c.3700_3704del p.(Val1234Glnfs*8) | rs80357609 | P (PVS1, PP5, PM2) | He | 1 |
| BRCA1 | Hereditary breast and/or ovarian cancer | NM_007294.4 | c.5497G>A p.(Val1833Met) | rs80357268 | P (PP5, PM1, PM5, PP3, PM2) | He | 1 |
| BRCA2 | Hereditary breast and/or ovarian cancer | NM_000059.4 | c.8169T>A p.(Asp2723Glu) | rs1060502432 | P (PM1, PM5, PP3, PM2, PP5) | He | 1 |
| LDLR | Familial hypercholesterolemia | NM_000527.5 | c.81C>G p.(Cys27Trp) | rs2228671 | P (PP5, PP3, PM1, PM5) | He | 1 |
| LDLR | Familial hypercholesterolemia | NM_000527.5 | c.858C>A p.(Ser286Arg) | rs140241383 | P (PP5, PS1, PM1, PP3) | He | 3 |
| LDLR | Familial hypercholesterolemia | NM_000527.5 | c.1775G>A p.(Gly592Glu) | rs137929307 | P (PS3, PM5, PP3, PM1, PP5) | He | 2 |
| MSH6 | Lynch syndrome (Hereditary Nonpolyposis Colorectal Cancer; HNPCC) | NM_000179.3 | c.2653A>T p.(Lys885*) | rs587782593 | P (PVS1, PP5, PM2) | He | 1 |
| MYH7 | Hypertrophic cardiomyopathy | NM_000257.4 | c.2609G>T p.(Arg870Leu) | rs36211715 | P (PM1, PM5, PP3, PP5, PM2) | He | 1 |
| TTN | Dilated cardiomyopathy | NM_001267550.2 | c.52903C>T p.(Arg17635*) | rs2154197219 | P (PVS1, PP5, PM2) | He | 1 |
| Gene | Condition | Transcript | Variant | dbSNP ID | ACMG Classification | Zygosity | Number of Carriers |
|---|---|---|---|---|---|---|---|
| ATP7B | Wilson disease | NM_000053.4 | c.2304dup p.(Met769Hisfs*26) | rs137853287 | P (PVS1, PP5, PM2) | He | 1 |
| GAA | Pompe disease | NM_000152.5 | c.-32-13T>G p.? | rs386834236 | P (PVS1, PM3, PP4) | He | 2 |
| GAA | Pompe disease | NM_000152.5 | c.1465G>A p.(Asp489Asn) | rs398123169 | P (PP5, PM5, PP3, PM1, PM2) | He | 1 |
| HFE | Hereditary hemochromatosis | NM_000410.4 | c.845G>A p.(Cys282Tyr) | rs1800562 | P (PS3, PS1, PP5, BP1) | He | 4 |
| MUTYH | MUTYH-associated polyposis (MAP) | NM_001048174.2/ NM_001128425.2 | c.1103G>A p.(Gly368Asp)/c.1187G>A p.(Gly396Asp) | rs36053993 | P (PP5, PP3, PS3, PM5, PP2) | He | 8 |
| Gene | Number of Carriers | Gene Carrier Rate | At-Risk Couple Rate |
|---|---|---|---|
| ATP7B | 1 | 0.0036 | 0.000013 |
| GAA | 3 | 0.0107 | 0.000115 |
| HFE | 4 | 0.0143 | 0.000204 |
| MUTYH | 8 | 0.0286 | 0.000818 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostoulas, C.; Sesse, A.; Bouba, I.; Konitsiotis, S.; Markoula, S.; Georgiou, I. Secondary Findings from Exome Sequencing of a Greek Cohort. Curr. Issues Mol. Biol. 2025, 47, 272. https://doi.org/10.3390/cimb47040272
Kostoulas C, Sesse A, Bouba I, Konitsiotis S, Markoula S, Georgiou I. Secondary Findings from Exome Sequencing of a Greek Cohort. Current Issues in Molecular Biology. 2025; 47(4):272. https://doi.org/10.3390/cimb47040272
Chicago/Turabian StyleKostoulas, Charilaos, Athanasia Sesse, Ioanna Bouba, Spyridon Konitsiotis, Sofia Markoula, and Ioannis Georgiou. 2025. "Secondary Findings from Exome Sequencing of a Greek Cohort" Current Issues in Molecular Biology 47, no. 4: 272. https://doi.org/10.3390/cimb47040272
APA StyleKostoulas, C., Sesse, A., Bouba, I., Konitsiotis, S., Markoula, S., & Georgiou, I. (2025). Secondary Findings from Exome Sequencing of a Greek Cohort. Current Issues in Molecular Biology, 47(4), 272. https://doi.org/10.3390/cimb47040272